Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Poly(ethylene glycol)-block-poly(ε-caprolactone)– and phospholipid-based stealth nanoparticles with enhanced therapeutic efficacy on murine breast cancer by improved intracellular drug delivery
Authors He X, Li L, Su H, Zhou D, Song H, Wang L ![]() , Jiang X
, Jiang X
Received 30 September 2014
Accepted for publication 13 December 2014
Published 5 March 2015 Volume 2015:10(1) Pages 1791—1804
DOI https://doi.org/10.2147/IJN.S75186
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Thomas Webster
Xiaodan He,1 Li Li,2 Hong Su,1 Dinglun Zhou,3 Hongmei Song,4 Ling Wang,1 Xuehua Jiang1
1West China School of Pharmacy, Sichuan University, Chengdu, Sichuan, People’s Republic of China; 2National Engineering Research Center for Biomaterials, Sichuan University, Chengdu, Sichuan, People’s Republic of China; 3West China School of Public Health, Sichuan University, Chengdu, Sichuan, People’s Republic of China; 4HitGen Ltd., Chengdu, Sichuan, People’s Republic of China
Background: Effective anticancer drug delivery to the tumor site without rapid body clearance is a prerequisite for successful chemotherapy. 1,2-distearoyl-sn-glycero-3-phospho-ethanolamine-N-(methoxy[polyethyleneglycol]-2000) (DSPE-PEG2000) has been widely used in the preparation of stealth liposomes. Although PEG chains can efficiently preserve liposomes from rapid clearance by the reticuloendothelial system (RES), its application has been hindered by poor cellular uptake and unsatisfactory therapeutic effect.
Methods: To address the dilemma, we presented a facile approach to fabricate novel stealth nanoparticles generated by poly(ethylene glycol)-block-poly(ε-caprolactone) (PEG-b-PCL), soybean phosphatidylcholine (SPC), and cholesterol, namely LPPs (L represented lipid and PP represented PEG-b-PCL), for the delivery of anticancer drug paclitaxel (PTX). LPPs were prepared using the thin film hydration method. Two PEG-b-PCL polymers with different molecular weights (MW; PEG2000-b-PCL2000, MW: 4,000 Da and PEG5000-b-PCL5000, MW: 10,000 Da) were used to fabricate stealth nanoparticles. Conventional PEGylated liposome (LDP2000, L represented lipid and DP2000 represented DSPE-PEG2000) composed of SPC, cholesterol, and DSPE-PEG2000 was used as the control. The physical properties, cellular uptake, endocytosis pathway, cytotoxicity, pharmacokinetics, tumor accumulation, and anticancer efficacy of free PTX, PTX-loaded LPPs, and LDP2000 were systemically investigated after injection into 4T1 breast tumor–bearing mice.
Results: LPPs were vesicles around 100 nm in size with negative zeta potential. With enhanced stability, LPPs achieved sustainable release of cancer therapeutics. The cellular uptake level was closely related to the PEG chain length of PEG-b-PCL; a shorter PEG chain resulted in higher cellular uptake. Moreover, the cellular internalization of LPP2000 modified by PEG2000-b-PCL2000 on 4T1 cells was 2.1-fold higher than LDP2000 due to the improved stability of LPP2000. The cytotoxicity of PTX-loaded LPP2000 was also higher than that of LDP2000 and LPP5000 as observed using a WST-8 assay, while blank LPPs showed negligible toxicity. Consistent with the results of the in vitro study, in vivo experiments showed that LPPs allowed significantly improved bioavailability and prolonged T1/2ß as compared to free PTX injection. More importantly, LPPs mainly accumulated at the tumor site, probably due to the enhanced permeation and retention effect (EPR effect). As a nanomedicine, LPP2000 (tumor inhibition rate of 75.1%) significantly enhanced the therapeutic effect of PTX in 4T1 breast tumor–bearing mice by inhibiting tumor growth compared to LDP2000 and LPP5000 (tumor inhibition rates of 56.3% and 49.5%, respectively).
Conclusion: Modification of liposomes with PEG2000-b-PCL2000 can simultaneously improve drug accumulation at the target tumor site and tumor cells, showing great promise for utilization as a PEG modification tool in the fabrication of stealth nanoparticles for cancer chemotherapy.
Keywords: nanoparticles PEG-b-PCL, phospholipid, murine breast cancer chemotherapy, paclitaxel
Introduction
Nanoparticles such as polymeric particles and liposomes have been widely studied for targeted drug delivery, especially for anticancer drugs.1–5 So far, several liposomal anticancer drugs have been approved by the US Food and Drug Administration (FDA) and become commercially available on the market. Typical examples include liposomal paclitaxel (PTX; Lipusu®),6 doxorubicin (Myocet®), daunorubicin (DaunoXome®), and cytarabine (DepoCyt®).3 However, the capture of systemically administered nanoparticles by the reticuloendothelial system (RES), which results in rapid clearance from body, remains one of the major drawbacks in drug delivery. This can be overcome by modifying the nanoparticles with flexible hydrophilic polymers such as polyethylene glycol (PEG), as the PEG chains can form a hydrophilic corona surrounding the nanoparticles. This surface modification reduces clearance of nanoparticles by the cells of the RES and apparently prolongs the duration of nanoparticles remaining in circulation.5,7–9
Lipid–PEGs, especially DSPE-PEG2000, have been extensively used to prepare stealth liposomes.7,8,10,11 Although the PEG-coating brings prolonged circulation time and enhanced accumulation at the tumor site via the EPR effect, the highly hydrophilic surface is unfavorable for the internalization of liposomes by tumor cells.3,6,10 Thus, the anticancer efficiency of stealth liposomes containing DSPE-PEG2000 both in vitro and in vivo is far from what has been expected. To resolve this dilemma, a great deal of work has been devoted to developing cleavable lipid–PEG, which would break and detach the PEG chain after it reaches the tumor site, thereby improving cellular uptake.10,12,13 However, the synthesis of tumor environment–responsive lipid–PEG is quite challenging and is not fit for mass production.
An alternative approach is to modify liposomes with PEG-conjugated polymers that are capable of delivering cargo into cells and can be easily produced. This approach would be simple and easy for large-scale preparation of stealth nanoparticles. Most importantly, we hypothesized that the hybrid nanoparticles may combine the advantages of liposomes and polymeric micelles, such as safety and stability and improve cargo cellular uptake and tumor accumulation. Poly(ethylene glycol)-b-poly(ε-caprolactone) (PEG-b-PCL) diblock copolymer has been widely used to prepare PEG-coated micelles2,4,14–17 and is a promising PEG modification tool for stealth nanoparticles for the following reasons: 1) the synthesis of PEG-b-PCL is facile and is adaptable to mass manufacture;18 2) PEG-b-PCL micelles can stay in the bloodstream for a prolonged time and are capable of efficient cell entry;19,20 and 3) PEG-b-PCL is biocompatible, since PEG and PCL are both FDA-approved for use in humans. Moreover, PEG-b-PCL micelles carrying PTX (Genexol-PM) are approved in several Asian countries for cancer therapy and are in Phase III clinical trials in the US.21–23 However, to our knowledge, few studies have reported the utilization of PEG-b-PCL as a PEG modification tool in the fabrication of stealth nanoparticles.
In this study, we successfully used PEG-b-PCL copolymers with different molecular weights (MW; PEG2000-b-PCL2000, MW: 4,000 and PEG5000-b-PCL5000, MW: 10,000), soybean phosphatidylcholine (SPC), and cholesterol to prepare stealth nanoparticles, namely LPP2000 and LPP5000, respectively. Conventional PEGylated liposomes consisting of DSPE-PEG2000, SPC, and cholesterol were used as the positive control, namely LDP2000. PTX, which has excellent antineoplastic activity against a variety of solid tumors, was used as the model drug. The influence of PEG-b-PCL on the nanoparticles in terms of physical properties, pharmacokinetics, and both in vitro and in vivo anticancer efficacy was also investigated.
Materials and methods
Materials
SPC was purchased from Lipoid GmbH (Ludwigshafen am Rhein, Germany). DSPE-PEG2000 was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). PEG-b-PCL copolymers were kind gifts from Professor Bin He (Sichuan University, People’s Republic of China). 1H nuclear magnetic resonance (1H NMR) spectrums and gel permeation chromatography (GPC) traces of PEG-b-PCL polymers are shown in Figure S1. PTX was purchased from Meilun Biology Technology Company (Dalian, People’s Republic of China). PTX injection (PTX was dissolved in a mixture of ethanol/Cremophor ELP 35 with a volume ratio of 1:1) was purchased from Taiji Pharmaceuticals Company (Sichuan, People’s Republic of China). DiI and DiR were purchased from Fanbo Biochemicals Co. Ltd. (Beijing, People’s Republic of China). Hoechst 33258, RPMI 1640 culture media, fetal bovine serum, and streptomycin were purchased from Thermo Fisher Scientific (Waltham, MA, USA). 2-(2-methoxy-4-ni-trophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H tetrazolium, monosodium salt (WST-8) assay kit was purchased from Dojindo Company (Kumamoto, Japan).
Preparation of PEGylated nanoparticles
The thin film hydration method was selected to prepare the PEGylated nanoparticles. Briefly, the SPC, cholesterol, and PEG modifiers (DSPE-PEG2000, PEG2000-b-PCL2000, and PEG5000-b-PCL5000; molar ratio: 17:2:1) were dissolved in the mixed solvents of chloroform: methanol (3:1, volume per volume [v/v]) in a round-bottom flask. After the organic solvents were removed, the dried lipid films were hydrated in phosphate-buffered saline (PBS, pH 7.4) or ultrapure water at 60°C to achieve a final phospholipid concentration of 2 mg/mL. The crude nanoparticles were sonicated using a sonicator water bath at 60°C (180 W, 10 minutes). Next, the suspension was extruded ten times through polycarbonate membrane filters (EMD Millipore, Billerica, MA, USA) with a pore size of 100 nm using a waterproof Extruder™ (Northern Lipids, Vancouver, BC).
PTX-loaded PEGylated nanoparticles were prepared according to the same protocol as blank PEGylated nanoparticles with the addition of PTX. The weight ratio of drug to lipids was 1:10. The unencapsulated PTX was removed from the nanoparticles by centrifugation at 3,000 rpm for 5 minutes and PTC concentration was determined by high-performance liquid chromatography (HPLC).
The encapsulation efficiency (EE) of PTX-loaded nanoparticles was calculated using the following formula: EE% = (W0–Wfree)/W0 ×100%, where W0 and Wfree were the weight of total drug fed in the formulation and the weight of unentrapped drug as determined by HPLC, respectively. The loading efficiency (LE) of the PTX-loaded nanoparticles was calculated using the following formula: LE% = (W0–Wfree)/Wtotal ×100%, where W0, Wfree, and Wtotal were the weight of total drug fed in the formulation; the weight of unentrapped drug as determined by HPLC; and the total weight of drug and lipids, respectively.
Characterization of PTX-loaded PEGylated nanoparticles
Particle size and zeta potential
The average diameter, polydispersity index (PDI) and zeta potential were measure using a Zetasizer Nano ZS90 system (Malvern Instruments, Malvern, UK). The nanoparticles (100 μL; SPC concentration 2 mg/mL) were diluted in 1 mL of buffer solution at 25°C for dynamic light scattering (DLS) measurements.
Morphology
A drop of freshly prepared PTX loaded LPPs was placed on carbon-coated copper grids and air-dried for 5 min. The rest sample on the grid was absorbed with paper and dried by air. After staining with 1% phosphotungstic acid, samples were visualized with a transmission electron microscope (TEM) (Tecnai G2 F20, FEI, Hillsboro, OR, US) at 200 kV operations.
Stability studies
Stability during storage: The PTX-loaded nanoparticles were stored at 4°C for 1 week under sealed conditions. The mean particle size and drug-loading efficiency were determined at predetermined time intervals (1, 4, and 7 days). The concentration of PTX was determined by HPLC.
Stability in culture media: 1 mL of nanoparticles was mixed with 3 mL of the completed culture media supplemented with 10% fetal bovine serum and 100 μg/mL streptomycin at 37°C. At predetermined time points (24 and 48 hours), samples were pipetted out for measurements of size and drug-loading efficiency. For analysis of PTX content, 400 μL of acetonitrile was added to 100 μL of the sample and the mixture was centrifuged at 3,000 rpm for 10 minutes. The upper layer was filtrated through a 0.22 μm polytetrafluoroethylene filter membrane and the filtrate was used for HPLC analysis.
In vitro drug release study
In vitro release of PTX from nanoparticles was performed using the modified dialysis bag method under sink conditions. Briefly, 0.2 mL of each PTX-loaded liposome was placed into ready-to-use dialysis tubes (MW cut-off [MWCO]: 6,000–8,000). Both ends of the dialysis bags were tightly sealed and immersed in a polypropylene (PP) tube containing 50 mL of release medium. All PP tubes were placed in a water bath maintained at 37°C with a stirring rate of 50 rpm throughout the release study. The release medium was 10 mM PBS buffer (pH 7.4) containing 0.1% (v/v) Tween 80. The sampling time intervals were at 0, 1, 2, 4, 6, 8, 12, 24, and 48 hours; medium (0.5 mL) was withdrawn, and an equal volume of fresh medium was added into the PP tube. The content of PTX was assayed by HPLC.
Cell culture
4T1 mouse mammary tumor cell line, derived from the BALB/c spontaneous mammary carcinoma, was purchased from the Chinese Academy of Science Cell Bank for Type Culture Collection (Shanghai, People’s Republic of China). The cell line was cultured in RPMI 1640 cell culture medium supplemented with 10% fetal bovine serum and 100 μg/mL streptomycin in an incubator under 5% CO2 at 37°C. The cells were harvested with trypsinization and rinsed. The resultant cell suspension was used in the following experiments.
Cellular uptake
The intracellular delivery of nanoparticles in 4T1 cells was evaluated by flow cytometry (FCM; BD, Franklin Lakes, NJ, USA) and confocal laser scanning microscopy (CLSM; TCP SP5; Leica Microsystems, Wetzlar, Germany), respectively.
4T1 cells were seeded at a density of 1×106 cells per well in six-well plates at 37°C and incubated for 24 hours. The DiI-loaded nanoparticles (LDP2000, LPP2000, and LPP5000) were diluted in the culture medium to a DiI concentration of 100 ng/mL. After cell incubation with DiI-loaded nanoparticles at 37°C for 4 hours, the cells were washed with cold PBS three times and harvested by trypsinization and centrifugation at 1,000 rpm for 5 minutes. The cells were resuspended in 1 mL PBS buffer (pH 7.4). The fluorescent intensity of cells was examined by FCM. The cell-associated DiI was excited with an argon laser (549 nm) and the fluorescence was detected at 565 nm.
4T1 cells were seeded with a density of 1×105 cells in each glass-bottomed dish and cultured at 37°C for 24 hours. The cells were incubated with DiI-loaded nanoparticles (DiI concentration of 50 ng/mL). The medium was removed after 4 hours and the cells were washed with PBS three times and stained by Hoechst 33258 solution (10 μg/mL) for 15 minutes. The cells were observed by CLSM. The cell-associated DiI was excited with an argon laser (549 nm) and the fluorescence was detected at 565 nm.
Endocytosis pathway
4T1 cells were seeded at a density of 1×106 cells per well in six-well plates at 37°C and incubated for 24 hours. To estimate the endocytosis pathways of the nanoparticles, the 4T1 cells were pre-incubated with different inhibitors for various kinds of endocytosis: inhibitor of macropinocytosis-mediated endocytosis: amiloride (1.48 mg/mL); inhibitor of clathrin-mediated endocytosis: chlorpromazine (20 mg/mL); inhibitor of caveolin-mediated endocytosis: filipin (5 mg/mL) for 30 minutes at 37°C. To study the effect of temperature on cellular uptake, the cells were incubated at both 37°C and 4°C. Subsequently, the inhibitor-containing culture media was removed and DiI-loaded nanoparticles-containing culture media were added to the cells. After incubation for another 4 hours, the cells were treated as described in the ‘Cell culture’ section and the fluorescence intensity was determined by FCM.
Cytotoxicity in vitro
The in vitro cytotoxicity of blank and PTX-loaded formulations against 4T1 cells was evaluated by WST-8 colorimetric assay. The 4T1 cells were seeded onto 96-well plates at a density of 5×103 cells per well. After culture for 48 hours, the cells were exposed to the fresh medium containing various concentrations of PTX nanoparticles or PTX injection. The control culture was treated with blank nanoparticles and ethanol/Cremophor ELP 35 mixture (v/v: 1/1), which is the same solvent mixture as for the PTX injection. After 48 hours’ incubation, 10 μL of WST-8 solution was added to each well and it was further incubated for 2 hours. The absorbance was measured at 450 nm using a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA). The cell survival rate was calculated using the following formula: cell viability % = (A–A0) ×100/(A100–A0), where A, A0, and A100 were the absorbance of: the treated cells; culture media without cells; and cells without treatment of blank or PTX-loaded formulations.
Animal and tumor transplant models
Female Sprague Dawley (SD) rats, female BALB/c mice, and female BALB/c nude mice (4–6 weeks old) were purchased from the experiment animal center of Sichuan University (Chengdu, People’s Republic of China) and allowed to acclimate for 1 week before use. The animal experiments complied with the requirements of the National Act on the Use of Experimental Animals (People’s Republic of China).
Approximately 5×105 4T1 cells suspended in PBS (50 μL) were subcutaneously injected into the left backs of BALB/c mice under anesthesia. The tumor volume (V) was determined by measuring length (L) and width (W), and calculated as V = L × W2/2. The drug was administrated when the tumor was about 50–100 mm3 in size.
Pharmacokinetic studies in rats
SD rats weighing 200±20 g were randomly divided into four groups (three per group). The free PTX and PTX-loaded PEGylated nanoparticles (LDP2000, LPP2000, and LPP5000) were dissolved in normal saline (0.9% NaCl) and administrated intravenously into the tail vein (equivalent to 10 mg/kg of PTX, 1.0 mL/rat). After a predetermined time (5, 15, and 30 minutes, and 1, 1.5, 2, 4, 7, 10, and 24 hours), blood samples were collected from the postorbital venous plexuses of rats and placed into heparinized test tubes. The plasma samples were harvested after centrifugation (3,000 rpm, 15 minutes) and stored at −20°C until analysis. For analysis, 10 μL of intra-standard norethindrone solution (100 μg/mL) was added to the plasma (100 μL). The mixture was subsequently extracted with 3 mL of diethyl ether. After centrifugation at 3,000 rpm for 10 minutes, the upper organic layer was transferred to a PP tube and evaporated under a stream of nitrogen at 45°C. The residue was dissolved in 200 μL of acetonitrile. After filtration through a 0.22 μm polytetrafluoroethylene filter membrane, the filtrate was used for HPLC analysis. Standard curves for PTX in blood were generated by the addition of free PTX at different concentrations to plasma, followed by extraction and quantification as mentioned above. Pharmacokinetic parameters were acquired with the help of the DAS program (v3.0; Mathematical Pharmacology Professional Committee, People’s Republic of China) by fitting to the two-compartment model.
Fluorescence image studies of model drug accumulation
When the tumor volume was about 50–100 mm3, the female BALB/C nude mice were randomized into three groups (n=3 for each group) and treated with free DiR, DiR-loaded LDP2000, LPP2000, or LPP5000 at a dose of DiR (5 mg/kg) via the tail vein. At 24 hours, the mice were administered chloral hydrate intraperitoneally. Image acquisition was performed on a Maestro In Vivo Imaging System (PerkinElmer Inc., Waltham, MA, USA).
In vivo anticancer efficacy
The tumor-bearing BALB/C mice were randomly assigned into five groups (n=8) and treated with normal saline, free PTX, PTX-loaded LDP2000, LPP2000, and LPP5000 at a dose of PTX (10 mg/kg) via tail vein injection, respectively. Various formulations were injected at 10, 13, 16, and 19 days after the tumor cells injection. The mice were weighed and the tumors were measured every 2 days until day 23. Survival observation of the mice ceased when death occurred due to toxicity resulting from the tumor growth or treatment. The sacrificial date was recorded to calculate the survival time.
PTX quantification by HPLC
The concentration of PTX was determined by HPLC using an HPLC system consisting of a 1,260 pump, and a 1,260 ultraviolet detector (Agilent Technologies, Santa Clara, CA, USA). An ODS3 column (250×4.6 mm, 5 μm; Agilent Technologies) was used for chromatographic separation and the detector wavelength was 227 nm. The eluent was a mixture of acetonitrile: water (50:50, v/v), the flow rate was 1.5 mL/minute, and the column temperature was 30°C.
Statistical analysis
The results were presented as mean ± standard deviation (SD). t-tests or one-way ANOVAs were used to determine statistical significance. Statistical significance was set at P<0.05 and extreme significance was set at P<0.01.
Results and discussion
Characterization of PTX-loaded PEGylated nanoparticles
The particle size, PDI, and zeta potential of PTX-loaded PEGylated nanoparticles were determined by DLS. As shown in Table 1, all the formulations of PTX-loaded nanoparticles had a similar small size (97.7–119.1 nm) and low PDI (<0.2), which suggests a narrow size distribution. Previous articles had reported that small stealth nanoparticles (100 nm) exhibit a higher frequency of encountering permeable capillaries of the tumor and enter into the leaking tumor tissue.6,24 Morphologies of PTX-loaded LPPs were observed by TEM. In the micrographs, well-dispersed and spherical nanoparticles with light color were LPPs. The size of LPPs was in the range of 30 nm to 45 nm (Figure 1A) which was much smaller than the hydrodynamic size of LPPs tested by DLS. This was because LPPs shrunk during dying in the preparation of TEM samples. All these nanoparticles exhibited negative zeta potentials; the LDP2000 showed a relatively high negative value (−17.0 mV) as compared to LPP2000 (−3.0 mV) and LPP5000 (−2.0 mV). Such a physical property favors in vivo drug delivery considering the notion that negatively charged nanoparticles are often more resistant to plasma macromolecular protein adsorption and are easier to disperse in the bloodstream compared to positively charged ones.25 The drug-loading capacities of all the PTX-loaded nanoparticles were around 5.2%–6.3%, and there was no significant difference among them.
 | Table 1 The particle size, zeta potential, drug-loading efficiency, and EE measurements of PTX-loaded nanoparticles |
 | Figure 1 (A) Representative images of LPPs visualized by TEM. (B) Cumulative release of PTX from PEGylated nanoparticles in water containing 0.1% Tween 80 at 37°C. |
The stability of PEGylated nanoparticles stored at 4°C for 1 week is shown in Table 1. No significant variation was found in the size and PDI value of the three stealth nanoparticles. The results revealed that LDP2000 and LPPs were morphologically stable for up to 1 week at 4°C. This is attributable to the PEGylation that stabilized the nanoparticles. However, the drug-loading efficiency of LDP2000 decreased dramatically over 7 days of storage. LDP2000 only retained about 35% of the initial PTX content, which was much lower than for LPP2000 (83%) and LPP5000 (73%). It indicates that LPP2000 and LPP5000 were more stable than LDP2000 in term of drug-loading capacity for 7 days. This was mainly because the larger hydrophobic moiety in PEG-b-PCL could produce much stronger hydrophobic interactions among PEG-b-PCL, SPC, cholesterol, and PTX than DSPE-PEG2000.
Liposomal particle stability against physiological condition is a prerequisite for further applications in vivo; therefore, the formulated cell culture medium (RPMI 1640 culture media supplemented with 10% fetal bovine serum and 100 μg/mL streptomycin) was employed to mimic the in vivo situation. As can be seen in Table 1, the particle sizes had hardly changed for LDP2000 and LPPs over 48 hours and the PDI had only slightly increased, indicating that there was no obvious aggregation in the culture media due to the shielding effect of PEG on the surface of the nanoparticles. The drug-loading efficiency of LDP200 decreased much faster than that of LPPs, which was similar to the results of the storage stability experiment. This result is in good agreement with the former stability test. Many researches have indicated that hydrophobic moiety or block of polymers singingly affect the stability of nanoparticles.29–32 As reported by Cao et al about the hydrophobic components in a copolymer (polycaprolactone–poly(N,N’-bis(2-hydroxyethyl) methylamine ammonium propane sulfonate-polycaprolactone) (PCL-APS-PCL), PCL chain increased, critical micelle concentration (CMC) values decreased. In other words, the longer the PCL hydrophobic segments were, the more easily the NPs could be formed and the more stable the NPs could be.29–30
In vitro drug release
The release experiment was conducted under sink conditions and dynamic dialysis was employed for separation of the free drug from the PTX-loaded liposomes. Figure 1 shows the cumulative PTX release profile over 48 hours from nanoparticles in the aqueous solution containing 0.1% Tween 80. PTX was continuously and slowly released from LPPs and the amount of cumulatively released PTX over 48 hours was around 30%. There was no significant difference between the release profiles of LPP2000 and LPP5000. In contrast, PTX release from LDP2000 (cumulative release of 38%) was a little faster than that of LPP2000 and LPP5000, which was probably because of weaker hydrophobic interactions between PTX and LDP2000 as compared to LPP2000 and LPP5000.
Intracellular delivery and endocytosis pathway
Cellular uptake of nanoparticles labeled with DiI was studied with 4T1 cells using CLSM (Figure 2A). LDP2000 and LPP5000 exhibited similar cellular uptake levels, which were much lower than that of LPP2000, since red florescence in 4T1 cells treated by LDP2000 and LPP5000 was much less than for LPP2000. Consistently, the FCM results (Figures 2B and C) demonstrated that the cellular uptake level of LPP2000 in 4T1 cells was 2.1-fold higher than for LDP2000 and 2.0-fold higher than for LPP5000. Possible explanations for the higher cellular uptake include: 1) LPP2000 was more stable than LDP2000, and capable of carrying more DiI into cells with less leakage into the culture medium; or 2) the hydrophilic, flexible PEG chain can lead to a inhibition of cellular uptake, and longer PEG chains resulted in stronger inhibition as previous articles have suggested.26–28 The cellular uptake level of LPP5000 was much lower than that of LPP2000, and was probably due to the longer PEG chain of LPP5000.
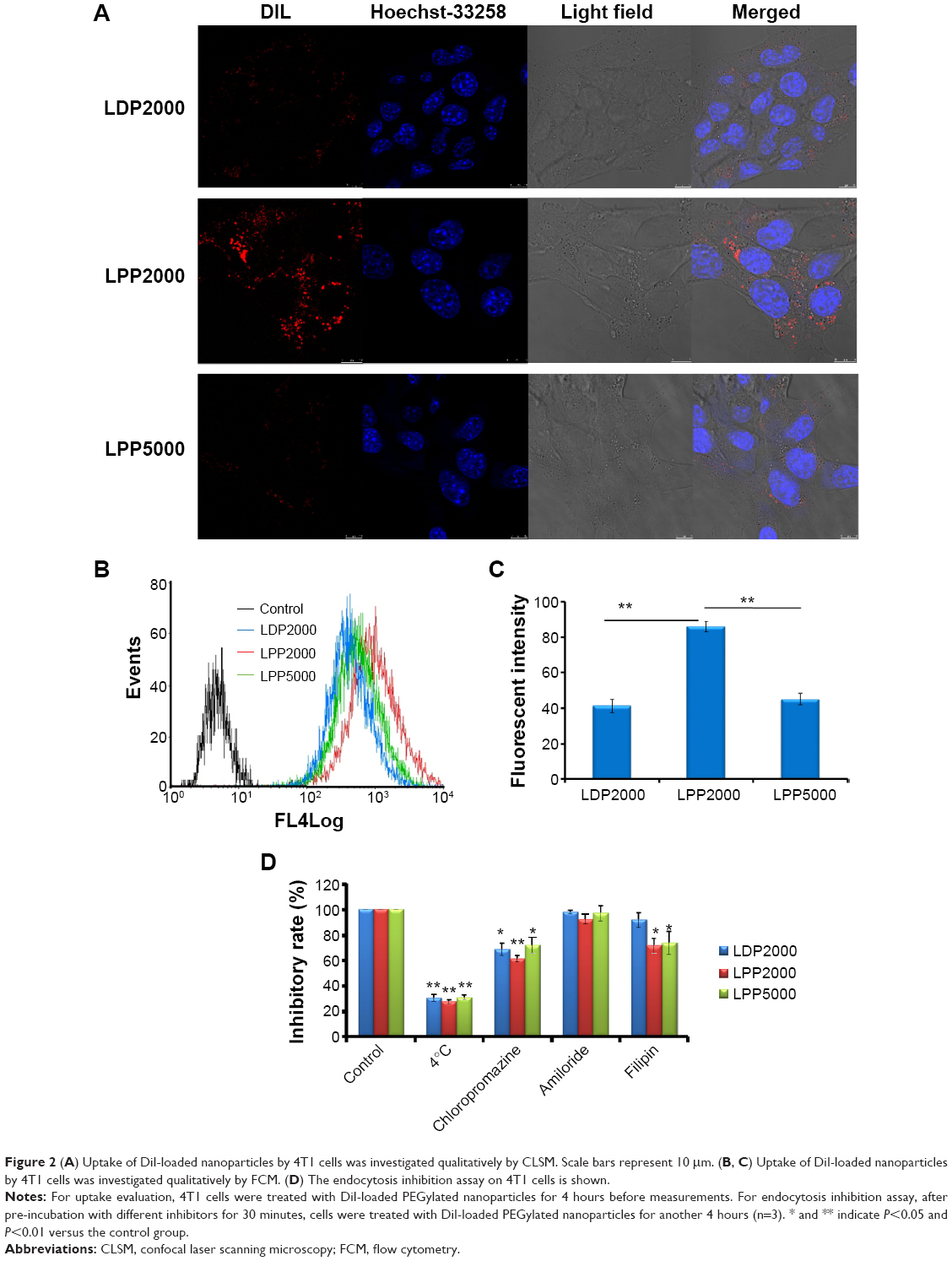 | Figure 2 (A) Uptake of DiI-loaded nanoparticles by 4T1 cells was investigated qualitatively by CLSM. Scale bars represent 10 μm. (B, C) Uptake of DiI-loaded nanoparticles by 4T1 cells was investigated qualitatively by FCM. (D) The endocytosis inhibition assay on 4T1 cells is shown. |
In order to explore the endocytosis pathways for LPPs, an endocytosis inhibition assay was conducted. Chlorpromazine, amiloride, and filipin were employed to block clathrin-, macropinocytosis-, and caveolin-mediated endocytosis, respectively.10,33 The effect of energy on cellular uptake was investigated at 4°C. As shown in Figure 2D, the rate of nanoparticle uptake was inhibited by around 70% at 4°C, and was also inhibited by the treatment of chlorpromazine (decreased to around 61%~72%). Additionally, filipin reduced the cellular uptake of LPP2000 and LPP5000 in both cases. The results showed that LDP2000 was internalized into the cells via energy- and clathrin-dependent endocytosis, while LPP2000 and LPP5000 were entrapped into the cells via energy-, clathrin-, and caveolin-dependent endocytosis pathways. Similarly, several studies reported that clathrin- and caveolin-dependent endocytosis pathways were the main routes for the uptake of nanoparticles such as cross-linked liposomes, pH-sensitive liposomes, PEG-b-PCL micelles, and so on.11,19,34–36
In vitro cytotoxicity
4T1 cells were treated with various doses of PTX formulations (free PTX, LDP2000, LPP2000, and LPP5000) and blank formulations (mixture of ethanol/Cremophor ELP 35 [v/v: 1/1] and the blank nanoparticles). As shown in Figure 3A, the inhibition effects of the PTX formulations were dependent on the dose of PTX. Free PTX exhibited stronger inhibition effects compared to the PEGylated nanoparticles, as free drugs can be quickly transported into cultured cells without the drug release from nanoparticles. In the case of nanoparticles, cells treated with PTX-loaded LPP2000 showed lower viability than those treated with LDP2000 at PTX concentration ranging from 0.01 – 10 μg/mL; than those treated with PTX-loaded LPP5000 at PTX concentration of 0.1 μg/mL. The higher cytotoxicity of PTX-loaded LPP2000 which could be attributed to the improved cellular uptake of PTX delivered by LPP2000, as proved by the CLSM and FCM results. It is worth noting that the solvent of free PTX was toxic when the concentration was above 10 μg/mL, while the blank liposomes were almost non-toxic under the tested concentrations (Figure 3B). Several studies have also reported that dose-dependent cytotoxicity of ethanol-cremophor ELP35 mixture.10
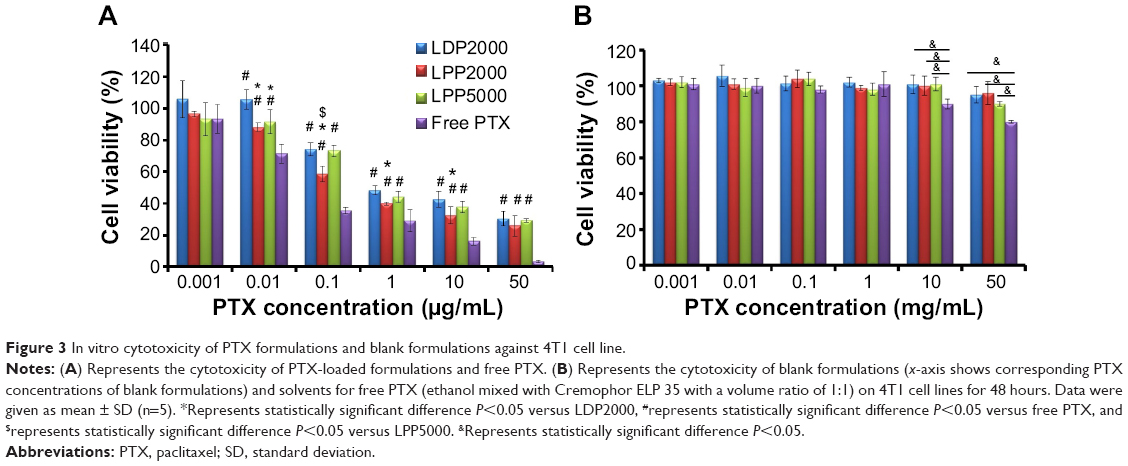 | Figure 3 In vitro cytotoxicity of PTX formulations and blank formulations against 4T1 cell line. |
Pharmacokinetics and tumor distribution
To investigate whether PEGylated formulations improve the bioavailability of the loaded drug, plasma PTX concentrations were analyzed using HPLC after intravenous administration of various formulations into rats, and norethindrone was used as the internal standard. The chromatograms showed stable baselines, as well as good resolution among PTX, norethindrone, and endogenous materials in plasma. Figure 4A shows the mean concentration versus time profiles of PTX and all the plasma concentration–time data were fitted to a two-compartment model. The pharmacokinetic parameters of PTX formulations calculated using DAS 3.0 statistic software are summarized in Table 2. The nanoparticles increased the pharmacokinetic parameters of PTX, including t1/2β, V2, CL2, AUC(0–t), AUC(0–∞), and K12 (P<0.05), but reduced CL1 and K10 (P<0.05). The increased AUC and half-life confirmed slower PTX removal from the plasma compartment and prolonged circulation time of the nanoparticles due to being shielded by the PEG chains. Enhanced escape from RES capture could be a possible explanation. We speculated that there might difference among nanoparticles, since their stability and drug release profile were different. However, contrary to what you might suspect, no significant differences among the PEGylated nanoparticles were observed. The limited detection sensitivity of HPLC analysis might make the difference negligible.
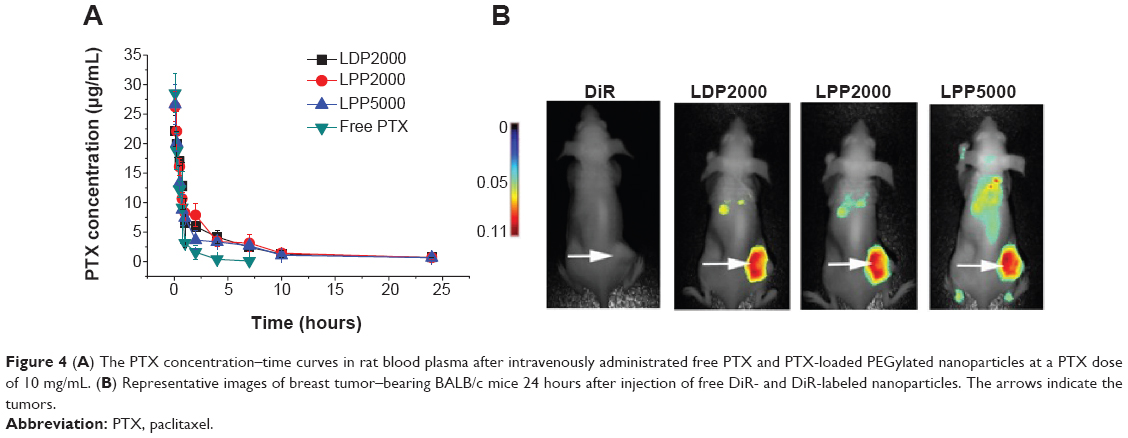 | Figure 4 (A) The PTX concentration–time curves in rat blood plasma after intravenously administrated free PTX and PTX-loaded PEGylated nanoparticles at a PTX dose of 10 mg/mL. (B) Representative images of breast tumor–bearing BALB/c mice 24 hours after injection of free DiR- and DiR-labeled nanoparticles. The arrows indicate the tumors. |
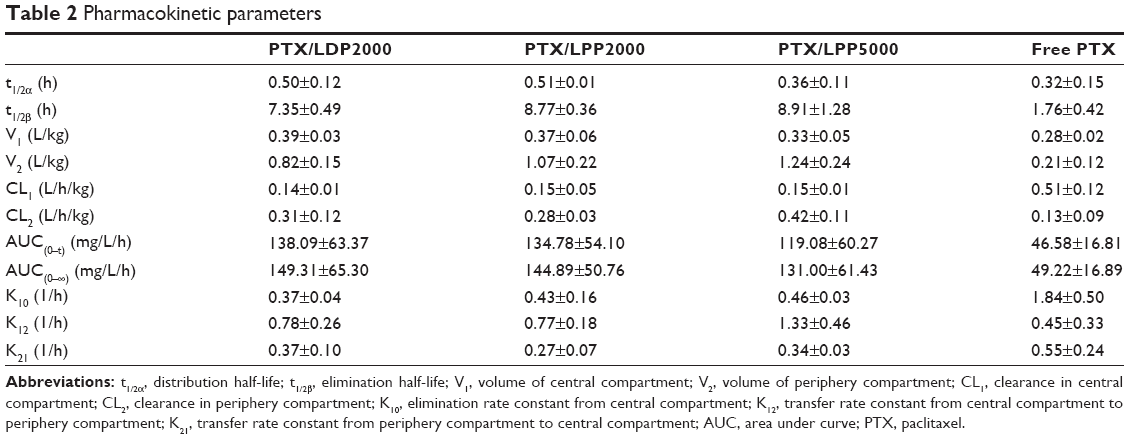 | Table 2 Pharmacokinetic parameters |
Tumor accumulation of the nanoparticles was determined after injection of DiR-loaded liposomes in 4T1 breast tumor–bearing nude mice. The near-infrared spectroscopy images (Figure 4B) show that DiR was mainly localized in tumors when encapsulated into PEGylated nanoparticles as compared to free DiR. Long circulation and slow release of PTX from nanoparticles offers a chance for PTX to accumulate in the tumor through the EPR effect.
In vivo anticancer efficiency
To evaluate the antitumor efficacy of PTX formulations in vivo, we next established the animal transplant tumor models. BALB/c mice bearing 4T1 cells received intravenous administration of PTX formulations and saline every 3 days for 10 days. As shown in Figures 5A and B, a rapid increase in tumor volume was observed in the saline group, while the PTX-treated groups exhibited inhibition effects to different extents. Treatment with free PTX resulted in a tumor inhibition rate of 32.5%, while the PTX nanoparticles groups (LDP2000, LPP2000, and LPP5000) exhibited more potent anticancer effects (inhibition rates of 56.3%, 75.1%, and 49.5%, respectively). This result again confirms the importance of sufficient accumulation of cargo (in this case, PTX) into tumors via the EPR effect by PEGylated nanoparticles. LPP2000 displayed the most significant tumor growth inhibition among the PEGylated nanoparticles, which was mainly due to its highest cellular uptake efficiency and good stability in serum. Mice body weight and survival were monitored to evaluate nanoparticle toxicity. As shown in Figure 5C, no significant changes in mice body weight were found throughout the whole experiment period (Figure 5C). However, some deaths were observed a few minutes after the injection of free PTX, LDP2000, and LPP5000 containing PTX (Figure 5D). This result reveals that PTX formulations except LPP2000 could cause some acute toxicity and even death in mice after intravenous injection, despite the fact that they caused only negligible or a little changes in mice body weight. Therefore, LPP2000 was safer than other three formulations. In addition, the median survival time was greatly prolonged in mice that received PTX-loaded LPP2000 (mean =48 days) compared with other groups (normal saline: 28 days, LDP2000: 41 days, LPP5000: 32 days, free PTX: 31 days).
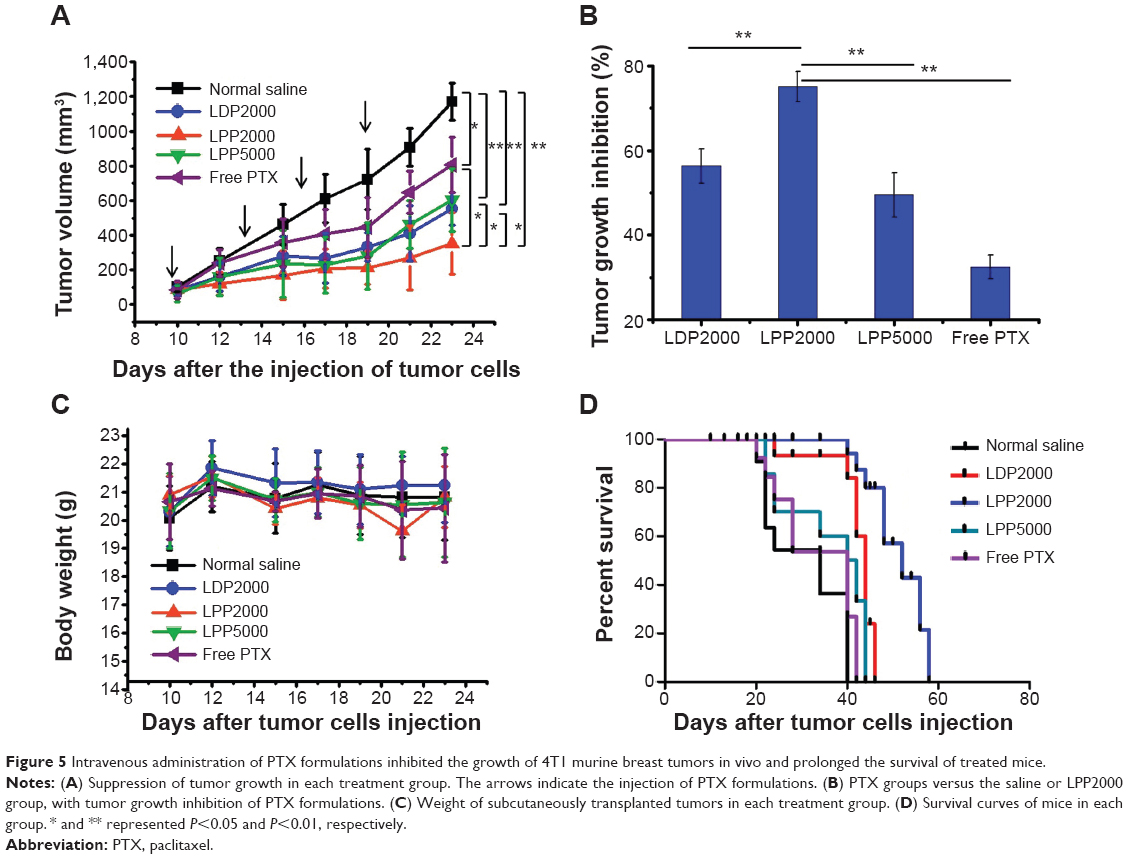 | Figure 5 Intravenous administration of PTX formulations inhibited the growth of 4T1 murine breast tumors in vivo and prolonged the survival of treated mice. |
Conclusion
PEG-b-PCL copolymers were employed as PEG modification tools to fabricate PTX-loaded nanoparticles (LPPs) in the presence of phospholipids and cholesterol. LPP2000 could evade the capture of RES due to PEGylation of its surface and accumulate successfully in tumors by the EPR effect, similar to LDP2000 and LPP5000. The PTX-loaded LPP2000 also exhibited higher cellular uptake efficiency than the other two nanoparticles, leading to greater cytotoxicity. Moreover, the blank LPP2000 was non-cytotoxicity in vitro and PTX-loaded LPP2000 was safe in vivo. All these aspects above contributed to the superior therapeutic efficacy of PTX-loaded LPP2000. Using PEG-b-PCL as a model, the present study suggests that hybrid stealth nanoparticles composed of PEG block polymer and phospholipids could serve as a potent anticancer drug delivery system for future cancer chemotherapy and represents a promising approach for large-scale preparation of stealth nanoparticles.
Acknowledgments
This work was supported by the Science Foundation of China (81371666 and 81473489) and Excellent Scholars Fund of Sichuan University (2013SCU04A09). Professor Bin He offered the PEG-b-PCL as a kind gift.
Disclosure
The authors report no conflicts of interest in this work.
References
Li Z, Yao L, Li J, et al. Celastrol nanoparticles inhibit corneal neovascularization induced by suturing in rats. Int J Nanomedicine. 2012;7:1163–1173. | ||
Zhang Y, Zhang W, Johnston AH, Newman TA, Pyykkö I, Zou J. Targeted delivery of Tet1 peptide functionalized polymersomes to the rat cochlear nerve. Int J Nanomedicine. 2012;7:1015–1022. | ||
Slingerland M, Guchelaar HJ, Gelderblom H. Liposomal drug formulations in cancer therapy: 15 years along the road. Drug Discov Today. 2012;17(3–4):160–166. | ||
Forrest ML, Zhao A, Won CY, Malick AW, Kwon GS. Lipophilic prodrugs of Hsp90 inhibitor geldanamycin for nanoencapsulation in poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles. J Control Release. 2006;116(2):139–149. | ||
Sengupta S, Kulkarni A. Design principles for clinical efficacy of cancer nanomedicine: a look into the basics. ACS Nano. 2013;7(4):2878–2882. | ||
Koudelka S, Turánek J. Liposomal paclitaxel formulations. J Control Release. 2012;163(3):322–334. | ||
Rom J, Bechstein S, Domschke C, et al. Efficacy and toxicity profile of pegylated liposomal doxorubicin (Caelyx) in patients with advanced breast cancer. Anticancer Drugs. 2014;25(2):219–224. | ||
Ichikawa K, Asai T, Shimizu K, et al. Suppression of immune response by antigen-modified liposomes encapsulating model agents: a novel strategy for the treatment of allergy. J Control Release. 2013;167(3):284–289. | ||
Woodle MC, Lasic DD. Sterically stabilized liposomes. Biochim Biophys Acta. 1992;1113(2):171–199. | ||
Zhang Q, Tang J, Fu L, et al. A pH-responsive α-helical cell penetrating peptide-mediated liposomal delivery system. Biomaterials. 2013;34(32):7980–7993. | ||
Joo KI, Xiao L, Liu S, et al. Crosslinked multilamellar liposomes for controlled delivery of anticancer drugs. Biomaterials. 2013;34(12):3098–3109. | ||
Kempegowda GB, Karve S, Bandekar A, Adhikari A, Khaimchayev T, Sofou S. pH-dependent formation of lipid heterogeneities controls surface topography and binding reactivity in functionalized bilayers. Langmuir. 2009;25(14):8144–8151. | ||
Bandekar A, Zhu C, Gomez A, Menzenski MZ, Sempkowski M, Sofou S. Masking and triggered unmasking of targeting ligands on liposomal chemotherapy selectively suppress tumor growth in vivo. Mol Pharm. 2013;10(1):152–160. | ||
Richter A, Olbrich C, Krause M, Kissel T. Solubilization of sagopilone, a poorly water-soluble anticancer drug, using polymeric micelles for parenteral delivery. Int J Pharm. 2010;389(1–2):244–253. | ||
Yang J, Hou Y, Ji G, et al. Targeted delivery of the RGD-labeled biodegradable polymersomes loaded with the hydrophilic drug oxymatrine on cultured hepatic stellate cells and liver fibrosis in rats. Eur J Pharm Sci. 2014;52:180–190. | ||
Hou J, Qian C, Zhang Y, Guo S. Application of poly(ethylene glycol)-b-poly(epsilon-caprolactone) copolymers with different poly(ethylene glycol) contents for the preparation of PEG-coated nanoparticles. J Biomed Nanotechnol. 2013;9(2):231–237. | ||
Cho H, Lai TC, Kwon GS. Poly(ethylene glycol)-block-poly(epsilon-caprolactone) micelles for combination drug delivery: evaluation of paclitaxel, cyclopamine and gossypol in intraperitoneal xenograft models of ovarian cancer. J Control Release. 2013;166(1):1–9. | ||
Mikhail AS, Allen C. Poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles containing chemically conjugated and physically entrapped docetaxel: synthesis, characterization, and the influence of the drug on micelle morphology. Biomacromolecules. 2010;11(5):1273–1280. | ||
Luo L, Tam J, Maysinger D, Eisenberg A. Cellular internalization of poly(ethylene oxide)-b-poly(epsilon-caprolactone) diblock copolymer micelles. Bioconjug Chem. 2002;13(6):1259–1265. | ||
Xiao L, Xiong X, Sun X, et al. Role of cellular uptake in the reversal of multidrug resistance by PEG-b-PLA polymeric micelles. Biomaterials. 2011;32(22):5148–5157. | ||
Kim SC, Kim DW, Shim YH, et al. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release. 2001;72(1–3):191–202. | ||
Kim DW, Kim SY, Kim HK, et al. Multicenter phase II trial of Genexol-PM, a novel Cremophor-free, polymeric micelle formulation of paclitaxel, with cisplatin in patients with advanced non-small-cell lung cancer. Ann Oncol. 2007;18(12):2009–2014. | ||
Lee KS, Chung HC, Im SA, et al. Multicenter phase II trial of Genexol-PM, a Cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast Cancer Res Treat. 2008;108(2):241–250. | ||
Yuan F, Dellian M, Fukumura D, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752–3756. | ||
Mobed M, Chang TM. Comparison of polymerically stabilized PEG-grafted liposomes and physically adsorbed carboxymethylchitin and carboxymethyl/glycolchitin liposomes for biological applications. Biomaterials. 1998;19(13):1167–1177. | ||
Molino NM, Bilotkach K, Fraser DA, Ren D, Wang SW. Complement activation and cell uptake responses toward polymer-functionalized protein nanocapsules. Biomacromolecules. 2012;13(4):974–981. | ||
Zhao J, Feng SS. Effects of PEG tethering chain length of vitamin E TPGS with a Herceptin-functionalized nanoparticle formulation for targeted delivery of anticancer drugs. Biomaterials. 2014;35(10):3340–3347. | ||
Pozzi D, Colapicchioni V, Caracciolo G, et al. Effect of polyethyleneglycol (PEG) chain length on the bio-nano-interactions between PEGylated lipid nanoparticles and biological fluids: from nanostructure to uptake in cancer cells. Nanoscale. 2014;6(5):2782–2792. | ||
Cao J, Xiu KM, Zhu K, et al. Copolymer Nanoparticles Composed of Sulfobetaine and Poly(ε-caprolactone) as Novel Anticancer Drug Carriers. J Biomed Mater Res: Part A, 2012;(8):2079–2087. | ||
Cao J, Chen YW, Chen NC, et al. The preparation of phosphorylcholine-containing poly(L-lactide) nanoparticles with solvent evaporation method. e-Polymers, 2010:074. | ||
Kabanov AV, et al. Micelle formation and solubilization of fluorescent probes in poly(oxyethylene-b-oxypropylene-b-oxyethylene) solutions. Macromolecules. 1995;28:2303–2314. | ||
Jongpa L, Zhou ZH, Wang YZ. Self-association and micelle formation of biodegradable poly(ethylene glycol)-poly(L-lactic acid) amphiphilic di-block co-polymers. J Biomat Sci: Polym Ed. 2006;17:747–763. | ||
Mo R, Sun Q, Li N, Zhang C. Intracellular delivery and antitumor effects of pH-sensitive liposomes based on zwitterionic oligopeptide lipids. Biomaterials. 2013;34(11):2773–2786. | ||
Huth US, Schubert R, Peschka-Süss R. Investigating the uptake and intracellular fate of pH-sensitive liposomes by flow cytometry and spectral bio-imaging. J Control Release. 2006;110(3):490–504. | ||
Pollock S, Antrobus R, Newton L, et al. Uptake and trafficking of liposomes to the endoplasmic reticulum. FASEB J. 2010;24(6):1866–1878. | ||
Tai W, Mo R, Lu Y, Jiang T, Gu Z. Folding graft copolymer with pendant drug segments for co-delivery of anticancer drugs. Biomaterials. 2014;35(25):7194–7203. |
Supplementary material
 | Figure S1 Characterization of PEG-b-PCL. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.