Back to Journals » Hepatic Medicine: Evidence and Research » Volume 14
Polycystic Liver Disease: Pathophysiology, Diagnosis and Treatment
Authors Norcia LF ![]() , Watanabe EM, Hamamoto Filho PT
, Watanabe EM, Hamamoto Filho PT ![]() , Hasimoto CN, Pelafsky L, de Oliveira WK
, Hasimoto CN, Pelafsky L, de Oliveira WK ![]() , Sassaki LY
, Sassaki LY ![]()
Received 6 June 2022
Accepted for publication 7 September 2022
Published 29 September 2022 Volume 2022:14 Pages 135—161
DOI https://doi.org/10.2147/HMER.S377530
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Gerry Lake-Bakaar
Luiz Fernando Norcia,1 Erika Mayumi Watanabe,2 Pedro Tadao Hamamoto Filho,3 Claudia Nishida Hasimoto,1 Leonardo Pelafsky,1 Walmar Kerche de Oliveira,1 Ligia Yukie Sassaki4
1Department of Surgery, São Paulo State University (Unesp), Medical School, Botucatu, São Paulo, Brazil; 2Department of Radiology, São Paulo State University (Unesp), Medical School, Botucatu, São Paulo, Brazil; 3Department of Neurology, Psychology and Psychiatry, São Paulo State University (Unesp), Medical School, Botucatu, São Paulo, Brazil; 4Department of Internal Medicine, São Paulo State University (Unesp), Medical School, Botucatu, São Paulo, Brazil
Correspondence: Luiz Fernando Norcia, Department of Surgery, São Paulo State University (UNESP), Medical School, 783 Pedro Delmanto Street, Botucatu, São Paulo, 18610-303, Brazil, Tel +55 19982840542, Email [email protected]
Abstract: Polycystic liver disease (PLD) is a clinical condition characterized by the presence of more than 10 cysts in the liver. It is a rare disease Of genetic etiology that presents as an isolated disease or assoc\iated with polycystic kidney disease. Ductal plate malformation, ciliary dysfunction, and changes in cell signaling are the main factors involved in its pathogenesis. Most patients with PLD are asymptomatic, but in 2– 5% of cases the disease has disabling symptoms and a significant reduction in quality of life. The diagnosis is based on family history of hepatic and/or renal polycystic disease, clinical manifestations, patient age, and polycystic liver phenotype shown on imaging examinations. PLD treatment has evolved considerably in the last decades. Somatostatin analogues hold promise in controlling disease progression, but liver transplantation remains a unique curative treatment modality.
Keywords: liver, polycystic liver disease, hepatomegaly, liver cysts, therapeutics
Introduction
Polycystic liver disease (PLD) is a rare and debilitating genetic disorder characterized by abnormal bile duct dilatation and maldevelopment of the cholangiocytes lining the bile duct.1
PLD may occur as an isolated manifestation in autosomal dominant PLD (ADPLD), a disease that affects 1/100,000 individuals worldwide, as an extrarenal manifestation of autosomal dominant polycystic kidney disease (ADPKD), which is the most common inherited nephropathy and affects 1/1000 individuals, or, more rarely, autosomal recessive polycystic kidney disease (ARPKD), a disorder that is more common in the pediatric population and has a prevalence ranging from 1/10,000 to 1/40,000.1–3
The clinical presentation, treatment, and prognosis are directly related to the extent of liver involvement. Although most patients are asymptomatic and do not require treatment, in 2–5% of the cases the disease exhibits important symptoms, presenting a wide spectrum of severity.3–7
Knowledge on PLD has increased greatly in recent years, especially regarding the molecular mechanisms involved in hepatic cystogenesis and pharmacological targets for disease treatment. This review aimed to provide an updated summary of the pathophysiological mechanisms and clinical management of PLD.
Pathophysiology
The development of liver cysts is a complex process involving several factors: genetic alterations, ductal plate malformation, ciliary dysfunction, cell signaling abnormalities, action of hormones and growth factors, abnormal proteostasis, increased fluid secretion, fibrosis, change in bile acid metabolism, increased autophagy, modifications in cell-matrix interaction, and epigenetic changes.1,8–11
Genetic Alterations
Various genes are associated with the development of PLD (Table 1). The PRKSCH and SEC63 genes are the main players in ADPLD, whereas PKD1 and PKD2 are the main genes implicated in ADPKD. Most cases of ARPKD are associated with mutations in the PKHD1 gene.8,9 Although ADPLD and ADPKD are inherited in a dominant manner, it is unclear how heterozygous mutations lead to disease development. The most accepted hypothesis is based on Knudson’s “two-hit” theory.32 Individuals carrying a germline mutation (“first hit”) are susceptible to but do not develop PLD; therefore, the occurrence of a second mutation (“second hit”) affecting the somatic cells and promoting the loss of heterozygosity causes the expression of the polycystic liver phenotype. The compromise of the healthy allele, resulting in a recessive cell genotype, would be the triggering factor of hepatic cystogenesis. Although PLD is associated with dominant genetic diseases, it appears that it is the recessive genotype that causes significant alterations in the epithelial cells of the biliary ducts, thus triggering PLD.14,29,33
 |
Table 1 Main Genes Involved in PLD Development |
Ductal Plate Malformation
Hepatic organogenesis begins in the 4th week of embryonic development from the endoderm of the foregut. The hepatic diverticulum is formed, which later divides into a cranial portion (precursor to the liver) and a caudal portion (precursor to the gallbladder). The biliary tree begins to form between the 7th-8th week of development from the migration of precursor cells from the liver (hepatoblasts) towards the portal vein. Elevated levels of transforming growth factor beta (TGF-β) are expressed near the portal vein, stimulating the migration of hepatoblasts positive for cytokeratin (CK) 8, CK18 and CK19 and expressing TGF-β receptor II (TGF-βRII). Hepatoblasts surround the portal mesenchyme forming a primary layer of cells (pre-biliary cells) called the ductal plate. Subsequently, a second layer of hepatoblasts expressing TGF-βRII is formed, thus allowing the formation of a lumen between the two layers.34–37
The differentiation of hepatoblasts into biliary tree cells (cholangiocytes) is regulated by the interaction between the Notch, TGF-β and Wnt signaling pathways, components of the extracellular matrix and intracellular transcription factors.34,37–40 The main transcription factors involved in this process are: hematopoietically expressed homeobox protein (HHEX), onecut domain family member 2 (OC2), hepatocyte nuclear factor 6 (HNF-6), SRY-box transcription factor 9 (SOX9) and hepatocyte nuclear factor 1β (HNF-1β). The increase in Notch pathway activity stimulates the expression of cholangiocyte transcription factors, mainly HNF-6 and HNF-1β, and inhibits the expression of hepatocyte transcription factors.34,35,37,40,41
Ductal plaque remodeling consists of three main events: formation of the primary layer of pre-biliary cells; maturation of the primary layer of pre-biliary cells into cholangiocytes and formation of the second layer of pre-biliary cells, with consequent production of a lumen between the two layers; maturation of the second layer of pre-biliary cells into cholangiocytes, with consequent formation of bile ducts. This process is dependent on three main signaling gradients, expressed at higher levels in the vicinity of the portal vein: Notch, TGF-β and Wnt.34,35,37
Hepatic cystogenesis appears to be related to an abnormal ductal plaque remodeling process, which occurs from at least three distinct origins of ductal plaque malformations (DPMs): DPM 1, impaired differentiation of bile precursor cells; DPM 2, impaired maturation of ductular structures; and DPM 3, abnormal expansion of bile ducts.37,40 The formation of bile ducts requires a network of epithelial-mesenchymal interactions and the presence of growth and transcription factors to control migration, adhesion and differentiation of cholangiocytes. Aberrant expression and signaling profiles result in impaired remodeling and subsequently dilated or disconnected ductal plate cells, favoring the development of cystic biliary structures.42–47
Ciliary Dysfunction
The primary cilium is a non-motile sensory structure composed of the basal body, axoneme, cilioplasm, and ciliary membrane. Cholangiocytes have a single primary cilium that protrudes from the apical membrane into the lumen of the bile duct. The primary cilium detects changes in the composition and osmolarity of the bile stream.48–50 Hepatic cystogenesis is believed to be related to disturbances in the sensitivity of the primary cilium, which are caused by structural and functional changes resulting from aberrant expression of ciliary proteins.1
Polycystin 1 (PC1) is an extremely important protein in PLD. Alterations in its expression or synthesis are directly related to the presentation, progression, and severity of PLD because PC1 reduction is inversely proportional to cyst size.18,51 PC1 interacts with polycystin 2 (PC2), forming a functional complex that acts on the surface of the ciliary membrane to regulate intracellular calcium levels. PC1 activation stimulates calcium uptake by PC2.13 The two proteins, after being synthesized inside cholangiocytes, form the PC1-PC2 complex. This association promotes the migration of the complex toward the primary cilium. Isolated molecules may reach the cellular surface but in a lower degree than in the form of a PC1-PC2 complex. Consequently, alterations in PC2 synthesis that compromise the formation of the PC1-PC2 complex can contribute to the reduction of PC1 expression in the primary cilium.20,52
Most proteins involved in hepatic cystogenesis are located in the endoplasmic reticulum. Dysfunction of these proteins interferes with the synthesis, processing, translocation, traffic or expression of other proteins, including PC1. Mutations in the PRKSCH, SEC63, SEC61B, GANAB, ALG8, ALG9, and DNAJB11 genes are associated with reduced PC1 levels in the primary cilium, which favors PLD development. It appears that cholangiocytes are less tolerant to reduced PC1 levels than renal epithelial cells. In some cases, this could explain why cysts are formed in the liver but not in the kidneys.12,18,26,53,54
Within the primary cilium, the bidirectional transport of substances along the axoneme is guaranteed by intraflagellar transport (IFT). There are two main modalities of IFT: the intraflagellar transport complex B (IFT-B), which interacts with kinesin to mediate anterograde intraflagellar transport, and the intraflagellar transport complex A (IFT-A), which interacts with dynein to mediate retrograde intraflagellar transport.55,56 Experimental studies in mice with ADPKD revealed that deletion of IFT-B genes attenuates hepatic and renal cystogenesis, thus reducing the severity of PLD, but deletion of IFT-A genes appears not to present the same results, being associated with inadequate development of the bile duct, biliary fibrosis, shortening of the primary cilium and increase in the Notch signaling pathway. These findings reveal that the increase in Notch signaling in cystic cholangiocytes may represent an important factor in the progression of liver cystogenesis, and may be seen as a potential therapeutic target in the future.55,57,58
Action of Hormones and Growth Factors
Liver cysts are not connected to the biliary tree and their growth depends on the autocrine and/or paracrine effect of cytokines and growth factors produced by the cystic epithelium. The cholangiocytes of this epithelium exhibit phenotypic and functional specificities, such as overexpression of insulin-like growth factor 1 (IGF1), vascular endothelium growth factor (VEGF), angiopoietin 1, and their respective receptors, that is, IGF1 receptor (IGF1R), VEGF receptor-2 (VEGFR2), and the receptor tyrosine kinase with immunoglobulin-like and EGF-like domains 2 (TIE2).59,60 The mechanism that allows cystic cholangiocytes to produce growth factors abnormally is unclear, but it may be a consequence of a relative lack of cell differentiation or a necessary response to cellular stress.61 Some hormones also contribute to the hyperproliferative phenotype observed in PLD, including estrogen. Healthy cholangiocytes normally lack estrogen receptors. Conversely, cells of the cystic epithelium express estrogen receptors alpha (ERα) and beta (ERβ) and are more sensitive to the proliferative effects of estrogens. These hormones exert a mitogenic effect on cystic cholangiocytes directly or by stimulating IGF1 synthesis and secretion.59,62
Cell Signaling Abnormalities
The hyperproliferative phenotype of cholangiocytes in PLD is directly related to changes in the concentration of intracellular mediators and dysfunction of signaling pathways. The process of cystic growth progresses with increased cyclic adenosine 3’,5’-monophosphate (cAMP) and reduced intracellular calcium levels. The main signaling pathways implicated are the mitogen-activated protein kinase (MAPK) and mammalian rapamycin target protein (mTOR) signaling pathways.61,63–68
In normal cells, mitogenic activity is controlled by the binding of growth factors to tyrosine kinase receptors with sequential activation of the MAPK pathway. Substances such as hormones (vasopressin, secretin, parathyroid hormone, and vasoactive intestinal peptide) and autacoids (prostaglandins and adenosine) and intracellular concentration of some ions stimulate the enzyme adenylate cyclase (AC) to convert adenosine triphosphate (ATP) into cAMP. The increase in cAMP activates two intracellular mediators, protein kinase A (PKA) and exchange protein directly activated by cAMP (EPAC). Under physiological conditions, PKA acts by inhibiting cell proliferation and blocking the activity of rapidly accelerated fibrosarcoma protein 1 (RAF1), thereby preventing receptor-mediated MAPK activation (Figure 1A). EPAC activity is proportional to cAMP levels. EPAC stimulates the cell proliferation pathway that, under normal conditions, ensures adequate cell renewal. The intracellular calcium level is also regulated by a series of mechanisms, among them the PC1-PC2 complex. Adequate intracellular calcium levels ensure the activity of phosphatidylinositol 3-kinase protein (PI3K), which acts by stimulating the protein kinase B (AKT). The AKT protein phosphorylates the rapidly accelerated fibrosarcoma B-type (BRAF), rendering it inactive.13,63,64,69–72
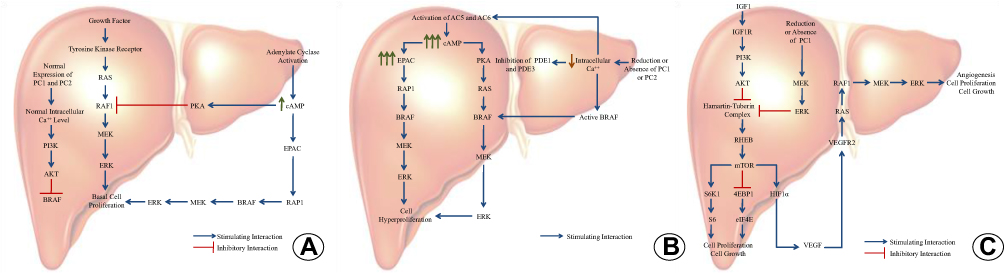 |
Figure 1 Main cell signaling pathways involved in PLD. (A) Normal functioning of the mitogen-activated protein kinase (MAPK) pathway in the liver. (B) Dysfunction of the mitogen-activated protein kinase (MAPK) pathway and influence of intracellular calcium concentration on the pathogenesis of polycystic liver disease. (C) Mammalian target of rapamycin (mTOR) dysfunction and influence of growth factors on the pathogenesis of polycystic liver disease. Abbreviations: PC1, polycystin1; PC2, polycystin2; Ca++, calcium; BRAF, rapidly accelerated fibrosarcoma B-type; MEK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; PKA, protein kinase A; RAS, rat sarcoma protein; RAF1, rapidly accelerated fibrosarcoma protein 1; cAMP, cyclic adenosine 3’-5’-monophosphate; EPAC, exchange protein directly activated by cAMP; RAP1, Ras-associated protein 1; PI3K, phosphatidylinositol-3-kinase; AKT, protein kinase B; AC5, adenylate cyclase 5; AC6, adenylate cyclase 6; PDE1, phosphodisterase1; PDE3, phosphodisterase3; IGF1, insulin-like growth factor 1; IGF1R, insulin-like growth factor 1 receptor; VEGF, vascular endothelial growth factor; VEGFR2, vascular endothelial growth factor receptor 2; RHEB, Ras homolog enriched in the brain; S6K1, ribosomal protein S6 kinase beta-1; 4EBP1, eukaryotic initiation factor 4E-binding protein 1; HIF1α, hypoxia-inducible factor 1-alpha; S6, ribosomal protein S6; eIF4E, eukaryotic translation initiation factor 4E. |
In pathological conditions caused by reduced expression of PC1 or PC2, there is a decrease in the intracellular calcium level. This reduction prevents the activity of PI3K and AKT and induces BRAF activation; induces the activity of adenylate cyclase 5 (AC5) and adenylate cyclase 6 (AC6), which are inactive in adequate calcium concentrations; inhibits the activity of phosphodiesterase 1 and phosphodiesterase 3, which are responsible for cAMP intracellular degradation; and stimulates the mitogenic activity of PKA. These events lead to an increase in cAMP levels and promote cell hyperproliferation. The increase in cAMP levels activates PKA, which, in the presence of reduced calcium levels, promotes an alternative cellular proliferation pathway that uses BRAF as an intermediate (Figure 1B). As a result of the significant increase in cAMP, the cAMP pathway becomes hyperactivated, which contributes considerably to the hyperproliferative state.63–65,72–76
mTOR is a signaling molecule involved in cell growth and proliferation processes. Under normal conditions, most cytoplasmic mTORs are controlled by the action of a protein complex composed of two proteins: tuberous sclerosis complex protein 1 (TSC1 or hamartin) and tuberous sclerosis complex protein 2 (TSC2 or tuberin). This complex inhibits the activity of the protein RAS homolog enriched in the brain (Rheb), thus preventing it from activating mTOR. Overexpression of growth factors, including IGF1, contributes to increased levels of active mTOR inside the cells of the cystic epithelium. IGF1 stimulates the activation of the AKT protein, which promotes tuberin phosphorylation and consequent dissociation of the hamartin/tuberin complex (Figure 1C). The loss of the inhibitory complex activates Rheb, which then stimulates and activates mTOR. Activated mTOR promotes cell growth and proliferation by stimulating ribosomal protein S6 kinase beta 1 (S6K1), inhibiting eukaryotic initiation factor 4E-binding protein 1 (4EBP1), and promoting VEGF synthesis via activation of hypoxia-inducible factor 1-alpha (HIF1α).61,77–79 Some studies have demonstrated that reduction of PC1 expression also contributes to the activation of the mTOR pathway by directly interfering with the functional activity of the hamartin/tuberin complex and/or allowing the activation of the MEK/ERK pathway, normally inhibited by PC1, resulting in increased tuberin phosphorylation by the ERK protein.80,81
Change in Bile Acid Metabolism
Changes in bile acid metabolism may also be involved in PLD development. Bile acids are known to regulate cholangiocyte functions, including cell proliferation. A study conducted by researchers in Spain showed that rats with PLD had increased intrahepatic concentration of bile acids, especially cytotoxic species, and reduced concentration of acids in the bile secreted by the liver. The researchers also found that patients with PLD had elevated bile acid levels in the cystic fluid and that some bile acids, such as glycodeoxycholic acid, had the ability to stimulate the proliferation of cystic cholangiocytes in vitro and promote hepatic cystogenesis.82 Lithocholic acid, taurocholic acid, and taurolithocholic acid are also known to stimulate cholangiocyte proliferation.83
Modifications in Cell-Matrix Interaction
Cell-matrix interactions are also involved in the progression and development of PLD. The extracellular matrix (ECM) is composed mainly of proteins and amorphous fundamental substance. The ECM remodeling process is regulated by the action of matrix metalloproteinases (MMPs), inhibitors of metalloproteinases, and hormones. Cystic epithelial cells show increased secretion of interleukin 6 (IL-6) and interleukin 8 (IL-8). These cytokines, which act in an autocrine/paracrine manner, interact with the IL-6 receptor and CXC chemokine receptor type 1 (CXCR1), present on the plasma membrane of cystic cholangiocytes, and induce MMP overexpression. These enzymes remodel the ECM around the liver cysts, thus allowing their growth and expansion. Estrogens also act on cystic cholangiocytes by stimulating the expression and secretion of MMPs.9,84,85
Fibrosis
Fibrosis is characterized by the accumulation of ECM proteins, mainly collagen, in response to a process of chronic cell injury.86,87 It is a common alteration in patients with PLD.11 PLD can promote liver damage and of the bile ducts in different ways, but the main mechanisms involved are: biliary stasis, generated by abnormal dilation of the bile ducts, mechanical compression of the hepatic parenchyma around the hepatic cysts and complications related to hepatic cysts (rupture, hemorrhage and infection).29,86–88 Hepatic stellate cells (HSCs), under physiological conditions, are quiescent and are mainly related to the storage and metabolism of vitamin A, showing a low proliferative rate, modest expression of fibrogenic genes, low secretion of cytokines and absence of contractile properties, but in situations of cell injury these cells become activated, assuming a pro-inflammatory profile active, fibrogenic, contractile and with a high rate of proliferation.86,87
Chronic inflammation, with production of inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), lymphotoxin, interleukin 1β (IL-1β) and lipid peroxidation products and the synthesis and release of cytokines and growth factors by damaged neighboring cells, such as transforming growth factor β1 (TGF-β1), platelet-derived growth factor (PDGF) and endothelin 1 (ET-1), stimulate the activation of HSCs. Activated HSCs (myofibroblasts) sustain their own activation from autocrine loops, including TGF-β1 secretion, and upregulation of their receptors, especially platelet-derived growth factor receptor β (PDGFR-β). The proliferation and fibrogenic activity of HSCs are mainly stimulated by PDGF and TGF-β1, respectively. Another characteristic of activated HSCs is their ability to resist apoptosis. During liver injury, hepatocytes and HSCs express IGF1, an important cell survival factor. TGF-β1 itself is also a factor that contributes to the survival of these cells.86,87
Activated HSCs synthesize fibrillar collagen and other extracellular matrix proteins that are eventually deposited in the space of Disse. This process can further enhance the fibrogenic activity of HSCs through 3 mechanisms. First, fibrillar collagens can bind to discoid domain receptors (DDRs) and activate cell signaling pathways that favor fibrogenesis. Second, the altered ECM can serve as a reservoir for several growth factors (PDGF, TGF-β), which are released and reach neighboring cells, including HSCs. Third, HSCs express several integrins on their surface whose ligands are ECM proteins. The activation of these integrins modulates a series of extremely important functions for HSCs, such as cell proliferation, contraction, migration and collagen synthesis. Activated HSCs also play an important role in hepatic inflammation, as they have the ability to secrete several pro-inflammatory cytokines and chemokines. Several vasoconstrictor agents (ET-1, norepinephrine, angiotensin II, and thrombin) are also produced locally in the injured liver and stimulate the fibrogenic activity of activated HSCs. Adipokines can also modulate fibrogenesis. Elevated levels of leptin and/or reduced levels of adiponectin favor the process of hepatic fibrosis.86,87
PLD is part of the spectrum of fibrocystic liver diseases. This group of pathologies is characterized by the presence of malformations of the ductal plate and varying degrees of cysts and fibrosis in the liver and includes, in addition to polycystic liver disease, congenital hepatic fibrosis, Caroli’s disease and Von Meyenburg complexes or biliary hamartomas.29
Increased Fluid Secretion
Another factor that plays a key role in hepatic cystogenesis is increased secretion of fluid in the cystic lumen. Under physiological conditions, cholangiocytes regulatebile fluidity and alkalinity. The main hormone that controls this function is secretin.89,90 Secretin is a hormone produced by the S cells of the intestinal mucosa in response to a decrease in pH in the duodenum. Cholangiocytes have secretin receptors in the basolateral region of the membrane. Binding of secretin to its receptor increases intracellular cAMP levels and activates PKA, which in turn stimulates the insertion of multiple transporters and ion channels into the apical membrane of cholangiocytes, including the cystic fibrosis transmembrane conductance regulator (CFTR), which acts as a chloride channel, the anion exchange protein 2 (AE2), which acts as a chloride/bicarbonate exchanger, and aquaporin water channel 1 (AQP1).89–92 When activated, CFTR releases chloride into the lumen of the bile ducts, which returns to the interior of the cholangiocytes when bicarbonate exits via AE2. The ionic flow involving CFTR and AE2 stimulates the displacement of water from the interior of the cholangiocytes to the lumen of the bile ducts via AQP1, thus increasing bile fluidity.89 In PLD, these proteins are overexpressed and mislocalized in cholangiocytes, which results in increased fluid secretion, dilation of bile ducts, and consequent expansion of cysts.93
Epigenetic Changes
Some studies have shown that epigenetic mechanisms may also be involved in hepatic cystogenesis. The abnormal expression of some groups of microRNAs (miRNAs) has been associated with the hyperproliferative phenotype of cystic cholangiocytes. Particularly, miRNA-15a appears to be underexpressed in some cases of PLD. Negative regulation of miRNA-15a promotes overexpression of cell division cycle 25A (CDC 25A) protein, thereby stimulating cell proliferation and accelerated growth of liver cysts.94 miRNA-30a, which is important for ductal plaque formation, is also underexpressed in cystic cholangiocytes.95 Histone deacetylase 6 (HDAC6), an enzyme predominantly located in the cytoplasm and involved in the regulation of various cellular processes, including the cell cycle, has also been associated with PLD pathogenesis. Some carriers of the disease show overexpression of HDAC6 in cystic cholangiocytes. High HDAC6 levels promote deacetylation of α-tubulin, which is present in the primary cilium, an event that may lead to ciliary malformation and dysfunction and also stimulate cell proliferation by regulating Wnt signaling through β-catenin deacetylation.96–98
Increased Autophagy
Autophagy, a process related to the degradation and recycling of cytoplasmic constituents, is also altered in PLD. Some organelles involved in the autophagic machinery show changes in cystic cholangiocytes. Autophagosomes, lysosomes and autolysosomes show an increase in number and size in patients with polycystic liver disease, indicating an important dysfunction in the lysosomal autophagic pathway. Cystic cholangiocytes express aberrant proteins related to the modulation and control of autophagy, thus promoting increased autophagic flux.9,99
The increase in autophagy is directly related to impaired ciliogenesis, increased cell proliferation and progression of hepatic cystogenesis. PLD carriers show overexpression of HDAC6 in the primary cilium region. The deacetylase 1 domain (DAC1) of HDAC6 also functions as a ubiquitin ligase and acts by ubiquitinating structural proteins of the primary cilium, specifically ARL3 and ARL13B, which are forwarded to autophagosomes for further degradation. Depletion of ARL3 and ARL13B not only alters the morphology of the primary cilium but also affects the ciliary location of the G protein-coupled bile acid receptor 1 (TGR5). TGR5 is a receptor related to the cAMP signaling pathway and one of the main sensory/signaling proteins of the primary cilium.100–103
In PLD, due to the reduced levels of ARL3 and ARL13B, TGR5 is abnormally expressed in the apical region of the primary cilium and its activation promotes an increase in intracellular levels of cAMP, an event that favors the cell proliferation of cystic cholangiocytes and, consequently, the hepatic cystogenesis. Pharmacological inhibition of autophagy by mefloquine and verteporfin restores levels of ARL3 and ARL13B and restores proper localization of TGR5, thus reducing hepatic cystogenesis. Thus, the inhibition of the autophagic pathway becomes an interesting therapeutic strategy for the control of PLD.100,104
Abnormal Proteostasis
Most genes involved in PLD encode proteins residing in the endoplasmic reticulum (ER). These proteins play an important role in the regulation of protein synthesis, folding, trafficking and degradation in the ER. Patients with PLD have altered proteomic profiles, mainly related to proteostasis. Proteostasis disorders cause accumulation of aberrant proteins and consequent ER stress, triggering 3 adaptive mechanisms: activation of the unfolded protein response (UPR), ER lumen dilation and hyperactivation of the ubiquitin-proteasome system (UPS). When ER stress levels are low, adaptive mechanisms are able to restore proteostasis and reduce stress, reinforcing the survival and proliferation of cystic cholangiocytes. However, under conditions of chronic and high stress, adaptive mechanisms fail to restore proteostasis and cystic cholangiocytes are induced to cell death. The use of ER stress inhibitors, specifically 4-phenylbutyric acid (4-PBA), contributes to the restoration of proteostasis, thus delaying the progression of PLD.10 Therefore, the resumption of proteostasis, with a consequent reduction in the stress of the RE, represents a promising strategy in the treatment of PLD.
Other Mechanisms Involved in Hepatic Cystogenesis
It was recently discovered that several proteins of the hemostatic system are upregulated in PLD. In the presence of tissue and vascular injury, the coagulation cascade is activated, culminating in the enzymatic conversion of fibrinogen to fibrin, the latter being further degraded by plasmin during the fibrinolysis process. A recent study based on quantitative proteomics revealed overexpression of several proteins of the fibrinogen complex in animals with PLD, including the serum amyloid P component (SAMP), the fibrinogen beta chain (FIBB), the fibrinogen alpha chain (FIBA) and fibrinogen gamma chain (FGG).105 It is known that fibrinogen is related to the distribution of interstitial collagen and formation of fibrotic lesions in the liver.106 Elevated levels of annexin A2 (ANXA2), a protein related to several cellular functions, have also been observed, including fibrinolysis, inflammation regulation and tissue repair. Dysfunctions in ANXA2 have been related to changes in ECM remodeling and hepatic fibrosis.105,107–109 In view of the above, the fibrinogen complex is identified as a possible factor involved in the progression of PLD and as a possible therapeutic target in the future.
Clinical Presentation
Approximately 95–98% of PLD cases are clinically asymptomatic, but a small proportion develop disabling symptoms and a significant reduction in quality of life.7,110–112 The number, size, location, and distribution of the cysts determine the spectrum of symptoms, with most being associated with mass effect or compression imposed by hepatomegaly.113 Abdominal distension usually occurs in patients with significant hepatomegaly. Increased tension in Glisson’s capsule can cause abdominal pain. Symptoms of early satiety, postprandial fullness, gastroesophageal reflux, and digestive intolerance can be observed in cases of gastric and/or duodenal compression. These symptoms may lead the patient to restrict the number of meals, which can lead to weight loss and severe malnutrition. Malnutrition is generally diagnosed late because the reduction in body weight is partially masked by abdominal distension and increased liver weight. The presence of cysts in the region of the hepatic dome that promotes diaphragm elevation and, therefore, restriction of thoracic expansion is directly related to dyspnea symptoms.16,112,114–116 Moreover, some patients report lumbar pain, which is possibly associated with compression of neighboring structures.110 Patients with expressive symptoms have significant impairment in the quality of life. The progressive increase in abdominal volume affects body image and limits the performance of simple tasks, such as activities involving trunk flexion, which significantly affects the self-esteem of these individuals.4,117
Compression of important vascular structures by hepatic cysts can occur but rarely leads to significant clinical and hemodynamic changes.115,118–120 The growth of cysts in the region of the hepatic hilum may compress the portal vein, resulting in portal hypertension (PH). Secondary complications of PH result from severe hepatic fibrosis, including splenomegaly, ascites, variceal hemorrhage, and encephalopathy, but the development of liver cirrhosis is a rare event.121–125 Compression of the inferior vena cava frequently occurs, but in most cases, there are no hemodynamic repercussions.115,119,120 Conversely, compression of hepatic veins is much more common, leading to the formation of intraparenchymal and subcapsular veno-venous collaterals, a situation that may increase the risks of hemorrhagic events during surgical interventions.115,126,127 Intrahepatic bile ducts are often compressed by hepatic cysts, but hilar compression of extrahepatic bile ducts is rare. Thus, compression of the biliary tree may be associated with the development of jaundice in these patients.124,128–130 Liver failure is rarely observed and usually associated with a very advanced disease stage.110,116
PLD can develop in both sexes, but women usually have a more severe hepatic phenotype, especially those with a history of multiple pregnancies and prolonged exposure to exogenous estrogen.131,132 Women have a higher mean liver volume, present the condition earlier, and are more susceptible to progressive PLD.110,116,133 Liver volume increases during the reproductive period and stabilizes in postmenopause, secondary to reduced endogenous estrogen synthesis.7,134 There is no consensus on the effects of pregnancy on liver volume in women with DHP. Pregnancy is not contraindicated in these women, but they should receive preconception counseling about the risk of transmitting the disease to the newborn.7
The main risk factors involved in the progression of PLD are as follows: advanced age, female sex, exposure to estrogen (use of oral contraceptives, estrogen replacement therapy), severity of renal dysfunction, and volume of renal cysts.112,135
Measuring the severity of the disease can be difficult, since the liver volume identified in the imaging tests may not be consistent with the symptoms expressed by the patient. Therefore, the assessment of the severity of symptoms can be obtained more reliably from the application of specific questionnaires for patients with PLD. Currently, two validated instruments have been used: the polycystic liver disease questionnaire (PLD-Q) and polycystic liver disease complaint-especific assessment (POLCA).7
Complications
Patients with PLD may also present with acute complications associated with hepatic cysts. Approximately half of patients with advanced PLD had cyst hemorrhage, rupture, or infection at some point. These complications appear to be more frequent in individuals presenting with ADPKD than in those with ADPLD.114,116,124
Intracystic hemorrhage occurs more commonly in large cysts. It usually manifests as sudden, non-radiating pain in the right hypochondrium. In some cases, abdominal pain is reported as colicky and accompanied by vomiting. Usually, the pain spontaneously disappears after a few days without fever or significant changes in laboratory test results. Elevated intracystic pressure, rapid cyst growth, and direct trauma are presumed to be triggering factors for this complication. The diagnosis of intracystic hemorrhage is dependent on imaging. Ultrasonography (US) or magnetic resonance imaging (MRI) show internal septations and/or deposits of fibrin aggregates inside the cyst. Conservative treatment is recommended for patients with mild symptoms. In the case of patients with significant symptoms and unremitting pain, fenestration or enucleation of the hemorrhagic cyst may be considered.136–142
Hepatic cyst infection is a serious complication due to the high risk of progression to sepsis. It usually has an indolent course and high risk of recurrence. The identification of inflammatory cells and bacteria in the cystic aspirate is considered the gold standard for diagnosis. If cystic aspirate is not available, clinical, biochemical, and radiological parameters should be considered to establish the diagnosis. Clinically, cystic infection is characterized by right upper quadrant pain, malaise, and fever. Elevated C-reactive protein and carbohydrate antigen 19–9 (CA 19–9) levels may be detected in patients presenting with hepatic cyst infection. Imaging studies showing cyst wall thickening and heterogeneous fluid (cell debris) are suggestive of infection but are inaccurate. It is believed that hepatic cyst infection occurs due to translocation of intestinal bacteria. Escherichia coli and Klebsiella are the main bacteria isolated in aspirates from infected cysts. The initial treatment is usually based on broad-spectrum antibiotic therapy. However, complete remission of the infectious presentation is not achieved in 64% of patients treated with antibiotics, and in these cases, drainage of the infected cyst is recommended.5,143–147
Hepatic cyst rupture is an extremely rare complication. Rupture is associated with a substantial increase in cystic volume that may spontaneously occur or may be secondary to hemorrhage. The increased volume reduces the stability of the cyst and increases the risk of rupture. The most common symptom is acute, severe abdominal pain. Imaging examinations show fluid around the liver and often a residual cyst. In most cases, treatment is conservative. Some patients may present hemodynamic instability, requiring percutaneous drainage of ascites and liver cysts or surgical intervention.148–150
Diagnosis
Asymptomatic individuals usually do not show changes in laboratory test results. Liver function is not affected in most patients, even in the presence of numerous cysts. Symptomatic patients or those with advanced disease may have elevated serum gamma glutamyl transferase, alkaline phosphatase, aspartate amino transferase, and total bilirubin levels.136,151–154
Elevated levels of CA 19–9 can also be observed in patients with PLD and are directly related to polycystic liver volume. Some studies suggest using CA 19–9 as a biomarker for PLD, which can be used to monitor disease progression and treatment efficiency, but its sensitivity and specificity have not yet been characterized, thus making its routine use in clinical practice difficult.155–157 It is important to point out that the levels of CA 19–9, whether in the blood or in the cystic fluid, do not allow the differentiation between hepatic cysts (solitary or associated with PLD) and mucinous cystic neoplasms.7
The levels of other tumor markers, such as carbohydrate antigen 125, carcinoembryonic antigen, and alpha-fetoprotein, may also be elevated in patients with PLD but less frequently than the CA19-9 level. The sensitivity and specificity of these other markers is also not established, and studies with higher quality and sample size are needed for better conclusions of their indications and applicability in the management and monitoring of the progression of PLD.155,158–161
The diagnosis of PLD is based on the family history of polycystic liver and/or kidney disease, clinical manifestations, and radiological evidence of the presence of a cystic phenotype in the kidneys and/or liver. Imaging examinations are also an important tool to exclude other causes of hepatomegaly. US and computed tomography (CT) are the most widely used modalities because of image quality, availability, and reasonable cost. US is a fast and easily accessible method that does not use ionizing radiation, and should be the initial imaging test to diagnose PLD.7 CT is highly sensitive in the characterization of the distribution, location, and size of hepatic cysts. It is an excellent tool for the evaluation of PLD complications and identification of cysts in other abdominal sites (Figure 2). MRI is not routinely used but is highly sensitive in the detection of cysts and provides valuable information for surgical planning (Figure 3). There is no indication for radiological monitoring of patients with asymptomatic PLD.4,7,151,162,163
 |
Figure 2 Contrast-enhanced computed tomography image, portal phase. (A) Axial section. (B) Sagittal section, showing liver with increased size and a sequence of images of multiple of liver cysts of varying sizes, with fluid density and thin and regular walls, and without enhancement after contrast injection. |
 |
Figure 3 Magnetic resonance image. (A) Axial section showing multiple liver cysts with low signal on T1-weighted sequence. (B) Axial section showing multiple liver cysts with low signal on the T1-weighted sequence, without enhancement after infusion of paramagnetic contrast in the portal phase. (C) Axial section showing multiple liver cysts with high signal on T2-weighted sequence. (D) Coronal section, 2D multiplanar reconstruction, in T2 sequence. |
Nuclear medicine tests can also be used in the management of PLD. Although imaging methods have made great advances in recent decades, the diagnosis of liver cyst infection remains a challenge, largely due to nonspecific signs and symptoms and the reduced accuracy of conventional imaging techniques. Recent studies have suggested positron emission computed tomography with 18 - fluorodeoxyglucose (18FDG PET/CT) as the test of choice in the diagnosis of cystic infection.164–16718FDG is a glucose analogue that accumulates inside inflammatory cells. In the presence of inflammatory processes, these cells, aiming to increase their metabolic rate, increase the expression of glucose receptors (GLUT-1 and GLUT-3) and the synthesis of enzymes involved in the glycolytic pathway, consequently increasing the uptake of 18FDG.164,166,168
The sensitivity and specificity of 18FDG PET/CT in the diagnosis of cystic infections are still unclear. The main advantages of 18FDG PET/CT are speed in obtaining images, high target/background ratio, higher spatial resolution and low radiation dose, while its disadvantages include high cost and restricted availability. In addition, 18FDG uptake is not exclusive to infectious processes, but may occur in other clinical conditions, such as neoplasms.165,166,169 Scintigraphic techniques using gallium citrate 67 (67Ga) and leukocytes labeled with indium 111 (111In) have also been described in the literature, but more consistent studies are needed to determine their real benefits and indications in the diagnosis of cystic infections.170–173
Genetic tests are mainly used to differentiate between polycystic disorders in cases of atypical clinical presentation and to confirm the diagnosis of ADPKD in the following situations: presence of imaging tests with atypical kidney changes or not consistent with the clinical presentation and sporadic polycystic kidney disease without history familial.7,174,175 Analysis of the PKD1 and PKD2 genes can help to distinguish between ADPLD and ADPKD, especially in young patients, with few renal cysts and several liver cysts and who do not meet Ravine’s diagnostic criteria.174,176 Unfortunately, only 30–45% of patients diagnosed with ADPLD have some known mutation identified in genetic tests.7,14,25 Currently, genetic tests are not routinely used in the diagnosis and management of PLD, since the genotype-phenotype correlation is not completely established and also because the results obtained do not change the therapeutic management of the disease.7
The great advancement of genetic sequencing technologies in recent years has enabled the development of faster, more comprehensive and affordable genetic tests. Next-generation sequencing techniques, including whole exome sequencing, have been used in the diagnosis of PLD, mainly due to the high number of disease-related genes. Some institutions have developed specific gene panels to investigate liver diseases, including PLD, thus optimizing the diagnostic process. Open exome sequencing can be used in the absence of a pathogenic variant identified in the gene panels. The results of genetic tests can cause additional problems for people with PLD, mainly related to life insurance, health insurance, pension fund, mortgage or employment. Thus, every patient must be previously instructed before performing these exams.174
Currently, the presence of more than 10 hepatic cysts confirms the diagnosis of PLD.7 However, distinguishing between the different diseases included in the spectrum of PLD can be difficult because patients with ADPLD may present with renal cysts and patients with ADPKD and ARPKD may present with hepatic cysts.116,177–179 Some factors, such as family history, age, liver phenotype, and radiological features, may help in this differentiation. Figure 4 show the diagnostic criteria for ADPLD.
 |
Figure 4 Diagnostic criteria for ADPLD. Notes: Data from Reynolds et al,180 Cnossen and Drenth,181 and Patel et al.182 |
Classification
Most patients with PLD remain asymptomatic over time; however, the presence of symptoms requires imaging examinations to assess the disease severity. To date, four clinical classifications have been proposed to assess the severity of PLD: Gigot, Schnelldorfer, Qian, and Mayo classifications.6,116,183
The Gigot classification (Table 2) is most commonly used in clinical practice. It classifies PLD into three types based on CT images and the number and size of liver cysts and amount of preserved liver parenchyma. Patients with a limited number of large cysts are classified as Gigot type I. Gigot type II classification includes patients with diffuse distribution of multiple medium-sized cysts in the liver parenchyma and large areas of non-cystic liver parenchyma. In Gigot type III, the most severe form of PLD, there is massive and diffuse involvement of the hepatic parenchyma by small- and medium-sized hepatic cysts, with small areas of normal hepatic parenchyma. Patients classified as Gigot type I are not technically classified as patients with PLD because of the low number of hepatic cysts. This is a classification that does not consider the degree of vascular involvement of the hepatic parenchyma or the symptoms presented by the patient for the purpose of categorization. The Gigot classification aims to identify patients who would benefit from fenestration of hepatic cysts and is inadequate to evaluate disease progression.154
 |
Table 2 Gigot Classification for Polycystic Liver Disease |
The Schnelldorfer classification (Table 3) is also based on CT imaging. It classifies PLD severity into four types and, in addition to the characteristics of the cysts and area of preserved liver parenchyma, considers the venous involvement and presence or absence of symptoms for categorization purposes. This classification aims to identify patients who would benefit from partial hepatectomy or liver transplantation (LT).184
 |
Table 3 Schnelldorfer Classification for Polycystic Liver Disease |
The Qian classification (Table 4) is based on US findings and the number of cysts and presence of symptomatic hepatomegaly. It classifies the PLD severity into five grades. This classification does not aid in the therapeutic decision and is highly simplified. Therefore, it is rarely used in clinical practice, being mainly used in the context of familial screening.110
 |
Table 4 Qian Classification for Polycystic Liver Disease |
Recently, researchers from the Mayo Clinic in the United States have proposed a new classification for PLD (Table 5). This classification is based on CT or MRI findings and categorizes the disease into four types according to severity. The Mayo classification also aims to identify patients who would benefit from partial hepatectomy with fenestration of remaining cysts or LT.183
 |
Table 5 Mayo Classification for Polycystic Liver Disease |
Despite the various proposed classifications, there is no consensus or guideline that determines the most appropriate option for use in clinical practice, but the Gigot and Schnelldorfer classifications seem to be the most used in most clinical and observational studies of DHP.177,182,185,186
Treatment
PLD treatment is indicated only for symptomatic patients, and its main objective is to reduce the symptoms caused by increased liver volume.7,187 Recently, the European Association for the Study of the Liver (EASL) released guidelines for the management of cystic liver diseases, including PLD.7
The use of oral contraceptives and estrogen-based hormone replacement therapy worsens the severity of PLD and should be discouraged in women with the disease.6,7,62,188 A recent study revealed that the use of estrogen-containing oral contraceptives in premenopausal women promoted a 1.45% increase in height-adjusted total liver volume (hTLV) for each year of exposure, compared to women not exposed to estrogen.188 Women with PLD of childbearing age should be instructed to use non-hormonal contraceptive methods and their daughters should avoid exposure to exogenous estrogen until disease involvement is ruled out.188
The treatment of PLD can be divided into three modalities: pharmacological, percutaneous and surgical.177 The algorithm for the treatment of PLD can be seen in Figure 5. Post-treatment imaging tests are not indicated, as the effectiveness of the treatment is defined for the relief of symptoms and not for the reduction of the volume of the hepatic cysts. Thus, it is recommended the use of specific instruments to assess the severity of symptoms (PLD-Q or POLCA) and consequent more reliable analysis of the effectiveness of the proposed treatment.7,189
 |
Figure 5 Algorithm for the treatment of polycystic liver disease. Note: Data from van Aerts RMM.6 |
Pharmacological Treatment
Somatostatin Analogues
Somatostatin receptors (SSTRs) belong to the family of G protein-coupled receptors and are directly related to the Giα subunit. They are classified into five different subtypes: SSTR1, SSTR2, SSTR3, SSTR4, and SSTR5. The binding of somatostatin or its analogues to these receptors promotes Giα subunit activation and consequent inhibition of the enzyme adenylate cyclase. Consequently, intracellular cAMP levels decrease, thereby reducing fluid secretion within hepatic cysts and activation of cell proliferation pathways.83,190–194 Cholangiocytes express all subtypes of somatostatin receptors.9
Octreotide and lanreotide were the first somatostatin analogues introduced in clinical practice. These drugs have higher affinity for SSTR2, lower affinity for SSTR5 and SSTR3, and almost no affinity for the SSTR1 and SSTR4. Pasireotide is a second-generation somatostatin analogue that has a broader binding spectrum, interacting with SSTR1, SSTR2, SSTR3, and SSTR5, and has higher binding affinity, with the exception of the SSTR2 receptor than octreotide and lanreotide. Currently, these drugs are the main pharmacological treatment option for patients with PLD, being prescribed off-label in most cases.9,194–197
The reduction in liver volume promoted by somatostatin analogues occurs independently of the type of inherited polycystic disorder, but efficacy appears to be directly related to the administered dose.198–200
There is some controversy regarding the clinical indication of somatostatin analogues and characteristics of patients who would benefit the most from this treatment. Some authors advocate the use of these drugs only in patients with significant symptoms (Schnelldorfer types C and D) and who have contraindications for surgical intervention or unsatisfactory results with conventional treatment.6,112,131,182,201–203 Other authors argue that somatostatin analogues should be used at an early stage, specifically in asymptomatic patients or those with mild symptoms (Schnelldorfer type A) to avoid disease progression and the need for invasive procedures in the future.177,204,205 The latest EASL guideline recommends the use of somatostatin analogues in patients who have numerous small to medium sized cysts distributed throughout the liver.7 Young (age ≤ 48 years) and female patients with PLD seem to benefit the most from the use of these drugs.206
In recent years, several clinical studies have sought to demonstrate the effectiveness of somatostatin analogues in reducing liver volume and controlling the progression of PLD. Pisani et al found a 130 ± 133.3 mL (7.8% ± 7.4%) reduction in total liver volume (TLV) in patients who received extended-release ocreotide and an increase of 144.3 ± 316.8 mL (6, 1% ± 14.1%) in the control group after a period of 3 years of treatment. The authors also observed that the reductions in TLV were maintained for 2 years after the end of treatment.205 Another clinical study by van Aerts et al found a reduction in liver volume of 5.9% in patients who used lanreotide for 120 weeks when compared to the control group. The authors also noted that the reduction was still present 4 months after stopping lanreotide treatment.207 Hogan et al observed a reduction in height-adjusted total liver volume (hTLV) from 2582 ± 1381 mL/m to 2479 ± 1317 in patients who received extended-release pasireotide and an increase in TLV from 2387 ± 759 mL/m to 2533 ± 770 mL/m in the control group within 12 months of treatment.208 The main adverse events observed in these studies and related to use of these drugs were: cholelithiasis, acute cholecystitis, cystic infection, hyperglycemia, diarrhea, dizziness, abdominal pain, nausea, fatigue, alopecia, bradycardia and diabetes.205,207,208
Recently, some meta-analyses sought to provide evidence to justify the use of somatostatin analogues in clinical practice. Griffiths et al performed an analysis involving 311 patients with PLD. The authors observed a significant reduction in TLV when compared to the control group (mean difference - 0.15 L, [95% CI - 0.26 to - 0.03], 5 studies, p = 0.01, I2 = 0%), but the dosage of the somatostin analogues used in these studies was very varied. The authors also noted that lanreotide appears to have better results in reducing TLV and total renal volume (TKV) when compared to ocreotide, but to date there is no consensus on the most suitable agent for use.209 Another meta-analysis performed by Suwabe et al involving 363 patients with PLD showed that the use of these drugs was associated with a lower rate of increase in TLV when compared to the control group (mean difference - 6.37%, [CI 95% −7.90 to −4.84], 6 studies, p < 0.00001, I2 = 14%).210 Garofalo et al performed an analysis involving 332 patients with PLD. The results revealed a greater reduction in liver volume in patients using somatostatin analogues when compared to the placebo group (mean difference - 176 mL, [CI 95% - 406 to 54], 6 studies, p < 0.133, I2 = 0%), but this difference was not statistically significant.211 Despite the results presented, more studies are needed to define the cost-effectiveness of the treatment in the long-term.
mTOR Inhibitors
These agents bind to FK506-binding protein 1A to form a drug-protein complex that inhibits cytoplasmic mTOR activation. mTOR inhibition decreases hepatic cystogenesis by preventing the activation of signaling pathways related to cell growth and proliferation.212
There are few studies evaluating the effectiveness of mTOR inhibitors in the treatment of PLD. A clinical trial involving 44 patients with PLD compared the efficacy of everolimus combined therapy plus octreotide to octreotide monotherapy in reducing liver volume. The results showed no significant difference between the two treatment regimens. Thus, there was no additive effect of everolimus in reducing liver volume.213 A systematic review that evaluated the effectiveness of these drugs in reducing liver volume revealed that there are no significant benefits from the use of everolimus or sirolimus in the treatment of PLD.204
Adverse events associated with the use of these drugs are directly related to their immunosuppressive and antiproliferative properties.79,214,215 The main adverse events described in the literature include: microangiopathic thrombosis, thrombocytopenia, lymphopenia, anemia, interstitial pneumonitis, skin rashes, hyperglycemia, dyslipidemia, reduced fertility male, delayed healing and lymphocele.8,177,215 Due to their toxicity and lack of evidence, these drugs should not be used in the treatment of patients with PLD.7,8,112,177,204,213
Vasopressin V2 Receptor Antagonists
Vasopressin is a peptide hormone that has three different receptors: V1a, V1b, and V2.216 The V2 receptor (V2R) is expressed mainly in the renal tubular epithelium, but cholangiocytes also express it.217 V2R is a G protein-coupled receptor, specifically related to the Gsα subunit, that promotes the activation of the enzyme adenylate cyclase when stimulated. Activated AC increases intracellular cAMP levels and promotes subsequent activation of PKA. In cystic cholangiocytes, these events increase the rate of cell proliferation and secretory activity, thereby potentiating hepatic cystogenesis.216,217
The efficacy of V2R antagonists in PLD treatment has been rarely investigated. Isolated case studies have shown that Tolvaptan promotes liver volume reduction and improvement of abdominal symptoms in patients with PLD.218,219 The main adverse events related to the use of these drugs are thirst, polyuria, nocturia, polydipsia, and increased liver enzyme and sodium and uric acid levels.220 Studies with better methodological quality and larger sample sizes are necessary to confirm these drugs as potential therapeutic options in PLD treatment.
Ursodeoxycholic Acid
Ursodeoxycholic acid (UDCA) is a hydrophilic bile acid that acts by reducing the rate of cholangiocyte cell proliferation, stimulating hepatobiliary secretion, and protecting cholangiocytes from the cytotoxic effects of hydrophobic bile acids. UDCA acts on cholangiocytes and hepatocytes by increasing intracellular calcium levels. This increase is mediated by the recruitment of intracellular calcium stores sensitive to inositol 1,4,5-triphosphate (IP3), independently of IP3 synthesis, and influx of extracellular calcium through unknown mechanisms. Restoring physiological calcium concentrations in cystic cholangiocytes restores the activity of PI3K enzyme, thereby inhibiting BRAF-dependent cell proliferation pathways.82,221–227
In hepatocytes, increased intracellular calcium promotes the activation of protein kinase C alpha, which promotes vesicular exocytosis and subsequent insertion of multidrug resistance-associated protein 2 (MRP2) (related to secretion of organic anions) and bile salt export pump (BSEP) (related to bile acid secretion) in the canalicular membrane. UDCA also increases the expression of these proteins by positively regulating gene transcription. Moreover, the drug increases bicarbonate secretion in the bile ducts. It is speculated that this process is associated with calcium-dependent mechanisms through the activation of calcium-dependent chloride channels and concomitant stimulation of chloride/bicarbonate exchange via AE2. Increased expression of MRP2 and BSEP and increased bicarbonate secretion favor the synthesis and secretion of bile, thereby reducing the retention of bile acids and other potentially toxic compounds in the liver.225–227
Few studies evaluated the efficacy of UDCA in PLD treatment. D’Agnolo et al conducted a multicenter, randomized, controlled clinical trial involving 34 patients with PLD, with both ADPKD and ADPLD, to assess the effects of UDCA on total liver volume. The results showed that UDCA was unable to promote total liver volume reduction; however, it decreased cystic liver volume in patients with ADPKD.228 Diarrhea is the main adverse event related to the use of this drug.228 Currently, treatment with UDCA is not indicated for patients with HPD.7
Percutaneous Treatment
Aspiration with Sclerotherapy
Aspiration with sclerotherapy is a minimally invasive procedure that is considered safe and effective. It is guided by imaging and consists of puncture and aspiration of the cystic content, followed by injection of a sclerosing agent that remains in contact with the inner wall of the cyst for a pre-established period and is subsequently aspirated. The sclerosing substance promotes the destruction of the epithelial lining of the cyst, thus inhibiting the production and accumulation of fluid inside it.229–231 The main sclerosing agents used are ethanol, ethanolamine oleate, minocycline, and tetracycline.187,232–234 The procedure is indicated for patients classified as Gigot type I or Schnelldorfer B and who present symptoms directly related to the presence of a dominant cyst (diameter > 5 cm).5,8,177,232 In most cases, a single session is sufficient to abolish the activity of the dominant cyst although some patients may require more sessions to achieve a satisfactory therapeutic response.235
Wijnands et al performed a systematic review evaluating the efficacy of aspiration with sclerotherapy. The review involved 16 studies, which included 526 patients with a total of 588 treated cysts. The authors reported a rate of cystic volume reduction between 76% and 100%. With regard to improvement of the clinical presentation, 72–100% of patients reported improvement of symptoms and 56–100% reported disappearance of symptoms.236 Abdominal pain, mainly related to peritoneal irritation caused by the sclerosing agent, and bleeding of the liver cyst are the main complications associated with the procedure.236,237 It is important to emphasize that, despite its benefits, aspiration with sclerotherapy is a procedure rarely used in clinical practice because most patients diagnosed with PLD have multiple cysts or cysts whose size does not justify this approach.8,177
Transcatheter Arterial Embolization
Transcatheter arterial embolization (TAE) is a treatment modality that aims to selectively embolize the hepatic artery branches that supply the liver regions affected by cysts. Blood supply restriction reduces the production of fluid inside the cysts, leading to a decrease in the volume of the cysts and, consequently, in the total volume of the liver, thus improving the symptoms caused by hepatomegaly. The main embolizing agents used are microcoils and polyvinyl alcohol particles.238–241
A recent study conducted by Yan et al involving 14 patients with PLD showed improvement of symptoms in 93% of patients after TAE. There was also a reduction in total liver volume, from 9,776 ± 2,219 cm3 in the pre-treatment to 8,303 ± 2,009 cm3 6–12 months after the procedure.242 Despite the expressive results, a retrospective study involving 18 patients undergoing TAE in PLD treatment showed a significant rate of treatment failure. Of the 23 procedures performed during the study, only seven showed satisfactory results; that is, the treatment failure rate was 69.6%.243 The main complications related to TAE are abdominal pain, low fever, reduced appetite, nausea, and leukocytosis.239,244 In most cases, these symptoms are self-limited and reversible. Severe complications, such as infection of hepatic cysts and liver failure, are also described in the literature.245 More robust, preferably controlled studies are needed to confirm whether TAE is safe and effective in PLD treatment.4,8,115,177
Surgical Treatment
Fenestration
Fenestration was for a long time considered the main treatment option in PLD management.246 It is a technique that combines aspiration and disruption of the cyst wall. The main advantage of fenestration is that it addresses multiple cysts in a single intervention.237,247 It can be performed by laparotomy or laparoscopy.248,249 Laparoscopic fenestration is the procedure of choice in most cases, but in situations of difficult access to hepatic cysts, such as cysts located in the right posterior segments (VI and VII) or liver dome (segment VIII), open fenestration is the best option.247,248,250 The comparison of the recurrence rate of cysts between laparoscopic and open fenestration shows no significant difference between the techniques.249
A systematic review by Bernts et al evaluating 1314 patients submitted to laparoscopic fenestration showed improvement of symptoms in 90.2% of cases. The rate of symptom recurrence and reintervention was 9.6% and 7.1%, respectively. Postoperative complications developed in 10.8% of cases, and the mortality rate related to the procedure was 1.0%. This study included symptomatic patients with non-parasitic liver cysts. After a subgroup analysis, the authors detected significantly higher rates of symptom recurrence (33.7%), reintervention (26.4%), and complications (29.3%) among patients with PLD.251 The main predictors of procedure failure areas follows: history of abdominal surgical procedures, presence of deep cysts, incomplete deroofing, cysts located in segments VII and VIII, and presence of diffuse PLD.237
Fenestration is indicated mainly for patients with multiple large cysts classified as Schnelldorfer B or Gigot type I. The procedure can also be used in cases of unsatisfactory results of aspiration with sclerotherapy.154,177,248 The main complications related to fenestration include ascites, pleural effusion, hemorrhage, and bile leakage.252 Laparoscopic fenestration is associated with shorter hospital stay and lower complication rates than open fenestration and therefore considered the treatment of choice for symptomatic patients with a limited number of large cysts. Despite the small number of quality studies, fenestration appears to be an effective option for the treatment of patients with PLD.237,251,253,254
Hepatic Resection
Liver resection is a therapeutic option aimed at patients with symptomatic PLD with localized involvement of some segments of the hepatic parenchyma and significant hepatomegaly, but who have normal or little affected liver segments.7 The procedure is indicated mainly for patients with Gigot type II or Schnelldorfer C classifications.177,184 Hepatic resection can also be performed in patients in whom hepatic fenestration does not significantly reduce liver volume or in cases in which LT is contraindicated or has a high probability of failure.237
It is recommended that the remaining normal hepatic parenchyma corresponds to at least 25–30% of the total liver volume and that the preserved area does not present alterations in venous drainage.126,127,183,255,256 Distortion of intrahepatic vascularization, biliary tree, and fibrous capsule of liver (Glisson’s capsule) caused by the compression of cysts, and limited mobility of the liver related to the rigidity of the liver parenchyma are some factors that make liver resection difficult.8,237 The main complications related to the procedure are ascites, hemorrhage, pleural effusion, and bile leakage.237
In a review of the literature, 26 studies involving 337 patients who underwent liver resection were analyzed. Symptom relief was achieved in 86% of patients. The authors observed morbidity and mortality rates of 51% and 3%, respectively. The mean hospital stay ranged from 10 to 15 days, and cyst recurrence was observed in 34% of patients.237 Liver resection promotes liver volume reduction and symptom relief but should not be considered as the treatment of choice because it is associated with significant morbidity and mortality rates. Another disadvantage is the high occurrence of post-procedure abdominal adhesions that may hinder LT in the future.8,237 Therefore, careful analysis to determine which patients would benefit from hepatectomy and the appropriate time to perform the procedure and presence of a team with experience in the procedure are decisive factors for treatment success.6,177
Liver Transplantation
LT is the only curative treatment; however, it is recommended for a select group of patients, including those with severe PLD and refractory to other treatment modalities.6,7,177,182 LT is mainly indicated for patients with Gigot type III or Schnelldorfer D classifications.154,177,184,257 Patients presenting with massive hepatomegaly associated with severe malnutrition, ascites, and sarcopenia, recurrent or intractable complications, such as cyst infections and portal hypertension, failure of conventional therapy, or extremely disabling symptoms leading to significant impairment of quality of life should be considered for LT.115,203,258–261
The model for end-stage liver disease (MELD), used as parameter for the indication of liver transplantation, does not adequately represent advanced-stage PLD because most patients have normal liver function. In these cases, the exception criteria are used to calculate MELD.259,262 According to the Organ Procurement and Transplantation Network (OPTN), the exception criteria should be applied to patients with severe symptoms (classified as Mayo type C or D) and who present with at least one of the following characteristics: hepatic decompensation, concomitant hemodialysis, glomerular filtration rate < 20 mL/min, previous kidney transplantation, and moderate to severe protein-calorie malnutrition.263 The OPTN recommends LT for patients with diffuse liver disease, classified as Mayo type C (with prior resection/fenestration or alternative therapy excluded) or D with at least two of the following: hepatic decompensation, concomitant renal failure (on dialysis therapy), and presence of compensated comorbidities.263
A recent study conducted in China evaluated 11 patients with severe PLD who underwent LT in a 10-year period. Most patients in the study (n = 9) underwent isolated liver transplantation. The authors reported morbidity and mortality rates of 54.5% and 18.2%, respectively. The 1-, 5-, and 10-year survival rates were 81.8%, 81.8% and 65.5%, respectively.264 A retrospective study by Baber et al evaluated the influence of previous interventions used in the management of PLD on post-liver transplantation morbidity and mortality. The study included 28 participants who underwent isolated LT or combined liver and kidney transplantation, who were divided into two groups. The first group (n = 12) was composed of patients who had a history of open surgical intervention (liver resection, fenestration by laparotomy, or nephrectomy), and the second group (n = 16) was composed of patients who had no or minimal previous intervention (aspiration with percutaneous sclerotherapy or laparoscopic fenestration). All participants who died during the follow-up period had a history of open surgical intervention. The 30-day, 1-year, and 5-year patient survival rates were 83%, 83%, and 48% in the first group versus 100%, 100%, and 100% in the second group, respectively. Moreover, the authors observed that operative time, length of hospital stay, and complication rates were longer and higher in the group of patients who had undergone previous abdominal Surgery.265 Although LT is effective in the management of severe PLD, its performance should be evaluated with caution considering the complexity of the procedure, risks of morbidity and mortality, limited availability of organs for transplantation, and long-term effects of immunosuppressive therapy.6,115,177
Conclusion
Although PLD is a progressive disease, only a small number of patients develop symptoms and require treatment. The diagnosis is based on family history of polycystic disease, age, clinical presentation, and presence of multiple hepatic and/or renal cysts on imaging tests. Treatment is aimed at reducing liver volume. Currently, the main therapeutic options are aspiration with sclerotherapy, fenestration, liver resection, and liver transplantation, the latter being the only curative option. Drug treatment, mainly based on somatostatin analogues, is not yet a reality in clinical practice and needs to be better studied. Therefore, understanding the mechanisms that modulate hepatic cystogenesis is extremely important to identify potential therapeutic targets in the future.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors have no conflicts of interest to declare regarding the present work.
References
1. Perugorria MJ, Masyuk TV, Marin JJ, et al. Polycystic liver diseases: advanced insights into the molecular mechanisms. Nat Rev Gastroenterol Hepatol. 2014;11(12):750–761.
2. Harris PC, Torres VE. Polycystic kidney disease, autossomal dominant. In: Adam MP, Ardinger HH, Pagon RA, et al, editors. GeneReviews [Internet]. Seattle: University of Washington; 2002:1–44.
3. Sweeney WE, Avner ED. Polycystic kidney disease, autossomal recessive. In: Adam MP, Ardinger HH, Pagon RA, et al, editors. GeneReviews [Internet]. Seattle: University of Washington; 2001:1–26.
4. Chandok N. Polycystic liver disease: a clinical review. Ann Hepatol. 2012;11(6):819–826.
5. Gevers TJG, Drenth JPH. Diagnosis and management of polycystic liver disease. Nat Rev Gastroenterol Hepatol. 2013;10(2):101–108.
6. van Aerts RMM, Van de Laarschot LFM, Banales JM, Drenth JPH. Clinical management of polycystic liver disease. J Hepatol. 2018;68(4):827–837.
7. European Association for the Study of the Liver. EASL clinical practice guidelines on management of cystic liver diseases. J Hepatol. 2022;77(4):1083–1108.
8. Wong MYW, McCaughan GW, Strasser SI. An update on the pathophysiology and management of polycystic liver disease. Expert Rev Gastroenterol Hepatol. 2017;11(6):569–581.
9. Masyuk T, Masyuk A, LaRusso N. Polycystic Liver Diseases: genetics, Mechanisms, and Therapies. In: Arias IM, Alter HJ, Boyer JL, et al, editors. The Liver: Biology and Pathobiology.
10. Santos-Laso A, Izquierdo-Sanchez L, Rodrigues PM, et al. Proteostasis disturbances and endoplasmic reticulum stress contribute to polycystic liver disease: new therapeutic targets. Liver Int. 2020;40(7):1670–1685.
11. Fabris L, Fiorotto R, Spirli C, et al. Pathobiology of inherited biliary diseases: a roadmap to understand acquired liver diseases. Nat Rev Gastroenterol Hepatol. 2019;16(8):497–511.
12. Masyuk TV, Masyuk AI, LaRusso NF. Polycystic liver disease: the interplay of genes causative for hepatic and renal cystogenesis. Hepatology. 2018;67(6):2462–2464.
13. Ghata J, Cowley BD. Polycystic kidney disease. Compr Physiol. 2017;7(3):945–975.
14. Lee-Law PY, Van de Laarschot LFM, Banales JM, Drenth JPH. Genetics of polycystic liver diseases. Curr Opin Gastroenterol. 2019;35(2):65–72.
15. Ong AC, Harris PC. Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int. 2005;67(4):1234–1247.
16. Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–168.
17. Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers. 2018;4(1):50.
18. Fedeles SV, Gallagher AR, Somlo S. Polycystin-1: a master regulator of intersecting cystic pathways. Trends Mol Med. 2014;20(5):251–260.
19. Bergmann C, Senderek J, Kupper F, et al. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat. 2004;23(5):453–463.
20. Besse W, Dong K, Choi J, et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest. 2017;127(5):1772–1785.
21. Lu H, Galeano MCR, Ott E, et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat Genet. 2017;49(7):1025–1034.
22. Drenth JPH, teMorsche RHM, Smink R, Bonifacino JS, Jansen JBMJ. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet. 2003;33(3):345–347.
23. Li A, Davila S, Furu L, et al. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003;72(3):691–703.
24. Davila S, Furu L, Gharavi AG, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36(6):575–577.
25. Cornec-le Gall E, Torres VE, Harris PC. Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J Am Soc Nephrol. 2018;29(1):13–23.
26. Besse W, Chang AR, Luo JZ, et al. ALG9 mutation carriers develop kidney and liver cysts. J Am Soc Nephrol. 2019;30(11):2091–2102.
27. Cnossen WR, teMorsche RHM, Hoischen A, et al. Whole-exome sequencing reveals LRP5 mutations and canonical Wnt signaling associated with hepatic cystogenesis. Proc Natl Acad Sci USA. 2014;111(14):5343–5348.
28. Besse W, Choi J, Ahram D, et al. A noncoding variant in GANAB explains isolated polycystic liver disease (PCLD) in a large family. Hum Mutat. 2018;39(3):378–382.
29. Mirza H, Besse W, Somlo S, Weinreb J, Kenney B, Jain D. An update on ductal plate malformations and fibropolycystic diseases of the liver. Hum Pathol. 2022. doi:10.1016/j.humpath.2022.06.022
30. Devuyst O, Olinger E, Weber S, et al. Autosomal dominant tubulointerstitial kidney disease. Nat Rev Dis Primers. 2019;5(1):60.
31. Mabillard H, Sayer JA, Olinger E. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease. Nephrol Dial Transplant. 2021;gfab268. doi:10.1093/ndt/gfab268
32. Knudson AG. Antioncogenes and human cancer. Proc Nat Acad Sci USA. 1993;90(23):10914–10921.
33. Watnick TJ, Torres VE, Gandolph MA, et al. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol Cell. 1998;2(2):247–251.
34. Antoniou A, Raynaud P, Cordi S, et al. Intrahepatic bile ducts develop according to a new mode of tubulogenesis regulated by the transcription factor SOX9. Gastroenterology. 2009;136(7):2325–2333.
35. Zaret KS, Bort R, Duncan SA. Embryonic Development of the Liver. In: Arias IM, Alter HJ, Boyer JL, et al, editors. The Liver: Biology and Pathobiology.
36. Moore KL, Persaud TVN, Torchia MG. Embriologia Clínica.
37. Wills ES, Roepman R, Drenth JP. Polycystic liver disease: ductal plate malformation and the primary cilium. Trends Mol Med. 2014;20(5):261–270.
38. Yanai M, Tatsumi N, Hasunuma N, Katsu K, Endo F, Yokouchi Y. FGF signaling segregates biliary cell-lineage from chick hepatoblasts cooperatively with BMP4 and ECM components in vitro. Dev Dyn. 2008;237(5):1268–1283.
39. Bogue CW. Embryology and development of the ductal plate. In: Murray K, Larson A, editors. Fibrocystic Diseases of the Liver. Clinical Gastroenterology. Totowa (NJ): Humana Press; 2010:3–21.
40. Raynaud P, Tate J, Callens C, et al. A classification of ductal plate malformations based on distinct pathogenic mechanisms of biliary dysmorphogenesis. Hepatology. 2011;53(6):1959–1966.
41. Tanimizu N, Miyajima A. Notch signaling controls hepatoblast differentiation by altering the expression of liver-enriched transcription factors. J Cell Sci. 2004;117(Pt 15):3165–3174.
42. Roskams T, Desmet V. Embryology of extra- and intrahepatic bile ducts, the ductal plate. Anat Rec. 2008;291(6):628–635.
43. Raynaud P, Carpentier R, Antoniou A, Lemaigre FP. Biliary differentiation and bile duct morphogenesis in development and disease. Int J Biochem Cell Biol. 2011;43(2):245–256.
44. Strazzabosco M, Fabris L. Development of the bile ducts: essentials for the clinical hepatologist. J Hepatol. 2012;56(5):1159–1170.
45. Lopes SS, Lourenço R, Pacheco L, Moreno N, Kreiling J, Saúde L. Notch signalling regulates left-right asymmetry through ciliary length control. Development. 2010;137(21):3625–3632.
46. Hassane S, Leonhard WN, Van der Wal A, et al. Elevated TGFβ-Smadsignalling in experimental Pkd1 models and human patients with polycystic kidney disease. J Pathol. 2010;222(1):21–31.
47. May-Simera HL, Kelley MW. Cilia, Wnt signaling, and the cytoskeleton. Cilia. 2012;1:7.
48. Pala R, Alomari R, Nauli SM. Primary cilium-dependent signaling mechanisms. Int J Mol Sci. 2017;18(11):2272.
49. Masyuk AI, Masyuk TV, LaRusso NF. Cholangiocyte primary cilia in liver health and disease. Dev Dyn. 2008;237(8):2007–2012.
50. Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRusso NF. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131(3):911–920.
51. Fedeles SV, Tian X, Gallagher AR, et al. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat Genet. 2011;43(7):639–647.
52. Su X, Wu M, Yao G, et al. Regulation of polycystin-1 ciliary trafficking by motifs at its C-terminus and polycystin-2 but not by cleavage at the GPS site. J Cell Sci. 2015;128(22):4063–4073.
53. Van der Laarschot LFM, Drenth JPH. Genetics and mechanisms of hepatic cystogenesis. BiochimBiophys Acta Mol Basis Dis. 2018;1864(4 pt B):1491–1497.
54. Cornec-le Gall E, Olson RJ, Besse W, et al. Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet. 2018;102(5):832–844.
55. Wang W, Pottorf TS, Wang HH, et al. IFT-A deficiency in juvenile mice impairs biliary development and exacerbates ADPKD liver disease. J Pathol. 2021;254(3):289–302.
56. Fu W, Wang L, Kim S, Li J, Dynlacht BD. Role for the IFT-A complex in selective transport to the primary cilium. Cell Rep. 2016;17(6):1505–1517.
57. Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet. 2013;45(9):1004–1012.
58. Shao L, El-Jouni W, Kong F, et al. Genetic reduction of cilium length by targeting intraflagellar transport 88 protein impedes kidney and liver cyst formation in mouse models of autosomal polycystic kidney disease. Kidney Int. 2020;98(5):1225–1241.
59. Alvaro D, Onori P, Alpini G, et al. Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease. Am J Pathol. 2008;172(2):321–332.
60. Fabris L, Cadamuro M, Fiorotto R, et al. Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology. 2006;43(5):1001–1012.
61. Spirli C, Okolicsanyi S, Fiorotto R, et al. Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin 2 defective mice. Hepatology. 2010;51(5):1778–1788.
62. Alvaro D, Mancino MG, Onori P, et al. Estrogens and the pathophysiology of the biliary tree. World J Gastroenterol. 2006;12(22):3537–3545.
63. Yamaguchi T, Nagao S, Wallace DP, et al. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003;63(6):1983–1994.
64. Banales JM, Masyuk TV, Grandilone SA, Masyuk AI, Medina JF, LaRusso NF. The cAMP effectors Epac and protein kinase A (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD). Hepatology. 2009;49(1):160–174.
65. Spirli C, Locatelli L, Fiorotto R, et al. Altered store operated calcium entry increases cAMP production and ERK1/2 phosphorylation in polycystin-2 defective cholangiocytes. Hepatology. 2012;55(3):856–868.
66. Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol. 2006;17(1):178–187.
67. Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMPdependent growth-stimulated phenotype. J Biol Chem. 2004;279(39):40419–40430.
68. Fischer DC, Jacoby U, Pape L, et al. Activation of the AKT/mTOR pathway in autosomal recessive polycystic kidney disease (ARPKD). Nephrol Dial Transplant. 2009;24(6):1819–1827.
69. Calvet JP. Strategies to inhibit cyst formation in ADPKD. Clin J Am Soc Nephrol. 2008;3(4):1205–1211.
70. Peyssonnaux C, Eychène A. The RAF/MERK/ERK pathway: new concepts of activation. Biol Cell. 2001;93(1–2):53–62.
71. Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG. Cell physiology of cAMP sensor EPAC. J Physiol. 2006;577(Pt 1):5–15.
72. Breckler M, Berthouze M, Laurent AC, Crozatier B, Morel E, Lezoualc’h F. Rap-linked cAMP signaling Epac proteins: compartmentation, functioning and disease implications. Cell Signal. 2011;23(8):1257–1266.
73. Halls ML, Cooper DMF. Regulation by Ca2+ -Signaling Pathways of Adenylyl Cyclases. Cold Spring HarbPerspect Biol. 2011;3(1):a004143.
74. Strazzabosco M, Fiorotto R, Melero S, et al. Differentially expressed adenylyl cyclase isoforms mediate secretory functions in cholangiocyte subpopulation. Hepatology. 2009;50(1):244–252.
75. Ye H, Wang X, Sussman CR, et al. Modulation of Polycystic kidney disease severity by phosphodiesterase 1 and 3 subfamilies. J Am Soc Nephrol. 2016;27(5):1312–1320.
76. Boswell-Smith V, Spina D, Page CP. Phosphodiesterase Inhibitors. Br J Pharmacol. 2006;147(Suppl 1):S252–S257.
77. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293.
78. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976.
79. Qian Q, Du H, King BF, et al. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol. 2008;19(3):631–638.
80. Distefano G, Boca M, Rowe I, et al. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol Cell Biol. 2009;29(9):2359–2371.
81. Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. ProcNatlAcadSci USA. 2006;103(14):5466–5471.
82. Munoz-Garrido P, Marin JJG, Perugorria MJ, et al. Ursodeoxycholic acid inhibits hepatic cystogenesis in experimental models of polycystic liver disease. J Hepatol. 2015;63(4):942–961.
83. Alvaro D, Gigliozzi A, Attili AF. Regulation and deregulation of cholangiocyte proliferation. J Hepatol. 2000;33(2):333–340.
84. Urribarri AD, Munoz-Garrido P, Perugorria MJ, et al. Inhibition of metalloprotease hyperactivity in cystic cholangiocytes halts the development of polycystic liver diseases. Gut. 2014;63(10):1658–1667.
85. Murray SL, Grubman SA, Perrone RD, et al. Matrix metalloproteinase activity in human intrahepatic biliary epithelial cell lines from patients with autosomal dominant polycystic kidney disease. Connect Tissue Res. 1996;33(4):249–256.
86. Bataller R, Brenner DA. Hepatic Fibrosis. In: Arias IM, Alter HJ, Boyer JL, et al, editors. The Liver: Biology and Pathobiology.
87. Kumar V, Abbas AK, Fausto N, Aster JC. Robbins & Cotran - Patologia: Bases Patológicas das Doenças.
88. Patil A, Sweeney WE Jr, Avner ED, Pan C. Childhood polycystic kidney disease. In: Li X, editor. Polycystic Kidney Disease [Internet]. Brisbane (AU): Codon Publications; 2015. doi:10.15586/codon.pkd.2015.ch2
89. Banales JM, Prieto J, Medina JF. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J Gastroenterol. 2006;12(22):3496–3511.
90. Banales JM, Arenas F, Rodríguez-Ortigosa CM, et al. Bicarbonate-rich choleresis induced by secretin in normal rat is taurocholate-dependent and involves AE2 anion exchanger. Hepatology. 2006;43(2):266–275.
91. Tietz PS, Marinelli RA, Chen XM, et al. Agonist-induced coordinated trafficking of functionally related transport proteins for water and ions in cholangiocytes. J Biol Chem. 2003;278(22):20413–20419.
92. Afroze S, Meng F, Jensen K, et al. The physiological roles of secretin and its receptor. Ann Transl Med. 2013;1(3):29.
93. Banales JM, Masyuk TV, Bogert PS, et al. Hepatic cystogenesis is associated with abnormal expression and location of ion transporters and water channels in an animal model of autosomal recessive polycystic kidney disease. Am J Pathol. 2008;173(6):1637–1646.
94. Lee SO, Masyuk T, Splinter P, et al. MicroRNA15a modulates expression of the cell-cycle regulator Cdc25A and affects hepatic cystogenesis in a rat model of polycystic kidney disease. J Clin Invest. 2008;118(11):3714–3724.
95. Hand NJ, Master ZR, EauClaire SF, Weinblatt DE, Matthews RP, Friedman JR. The microRNA-30 family is required for vertebrate hepatobiliary development. Gastroenterology. 2009;136(3):1081–1090.
96. Gradilone SA, Habringer S, Masyuk TV, Howard BN, Masyuk AI, Larusso NF. HDAC6 is overexpressed in cystic cholangiocytes and its inhibition reduces cystogenesis. Am J Pathol. 2014;184(3):600–608.
97. Gradilone SA, Radtke BN, Bogert PS, Huang BQ, Gajdos GB, LaRusso NF. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res. 2013;73(7):2259–2270.
98. Li Y, Zhang X, Polakiewicz RD, Yao TP, Comb MJ. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J Biol Chem. 2008;283(19):12686–12690.
99. Masyuk AI, Masyuk TV, Lorenzo Pisarello MJ, et al. Cholangiocyte autophagy contributes to hepatic cystogenesis in polycystic liver disease and represents a potential therapeutic target. Hepatology. 2018;67(3):1088–1108.
100. Masyuk AI, Masyuk TV, Trussoni CE, Pirius NE, LaRusso NF. Autophagy promotes hepatic cystogenesis in polycystic liver disease by depletion of cholangiocyte ciliogenic proteins. Hepatology. 2022;75(5):1110–1122.
101. Zhang M, Xiang S, Joo HY, et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSα. Mol Cell. 2014;55(1):31–46.
102. Masyuk AI, Huang BQ, Radtke BN, et al. Ciliary subcellular localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. Am J Physiol Gastrointest Liver Physiol. 2013;304(11):G1013–G1024.
103. Keitel V, Ullmer C, Häussinger D. The membrane-bound bile acid receptor TGR5 (Gpbar-1) is localized in the primary cilium of cholangiocytes. Biol Chem. 2010;391(7):785–789.
104. Masyuk TV, Masyuk AI, Lorenzo Pisarello M, et al. TGR5 contributes to hepatic cystogenesis in rodents with polycystic liver diseases through cyclic adenosine monophosphate/Gαs signaling. Hepatology. 2017;66(4):1197–1218.
105. Cordido A, Vizoso-Gonzalez M, Nuñez-Gonzalez L, et al. Quantitative proteomic study unmasks fibrinogen pathway in polycystic liver disease. Biomedicines. 2022;10(2):290.
106. Kopec AK, Luyendyk JP. Role of Fibrin(ogen) in Progression of liver disease: guilt by association? Semin Thromb Hemost. 2016;42(4):397–407.
107. Mayer G, Poirier S, Seidah NG. Annexin A2 is a C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. J Biol Chem. 2008;283(46):31791–31801.
108. Zhang L, Peng X, Zhang Z, et al. Subcellular proteome analysis unraveled annexin A2 related to immune liver fibrosis. J Cell Biochem. 2010;110(1):219–228.
109. Yang M, Wang C, Li S, et al. Annexin A2 promotes liver fibrosis by mediating von Willebrand factor secretion. Dig Liver Dis. 2017;49(7):780–788.
110. Qian Q, Li A, King BF, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37(1):164–171.
111. Bistritz L, Tamboli C, Bigam D, Bain VG. Polycystic liver disease: experience at a teaching hospital. Am J Gastroenterol. 2005;100(10):2212–2217.
112. Abu-Wasel B, Walsh C, Keough V, Molinari M. Pathophysiology, epidemiology, classification and treatment options for polycystic liver diseases. World J Gastroenterol. 2013;19(35):5775–5786.
113. Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17(2):173–180.
114. Hoevenaren IA, Wester R, Schrier RW, et al. Polycystic liver: clinical characteristics of patients with isolated polycystic liver disease compared with patients with polycystic liver and autosomal dominant polycystic kidney disease. Liver Int. 2008;28(2):264–270.
115. Aussilhou B, Dokmak S, Dondero F, et al. Treatment of polycystic liver disease. Update on the management. J Visc Surg. 2018;155(6):471–481.
116. van Keimpema L, De Koning DB, Van Hoek B, et al. Patients with isolated polycystic liver disease referred to liver centres: clinical characterization of 137 cases. Liver Int. 2011;31(1):92–98.
117. Kirchner GI, Rifai K, Cantz T, et al. Outcome and quality of life in patients with polycystic liver disease after liver or combined liver-kidney transplantation. Liver Transpl. 2006;12(8):1268–1277.
118. Chauveau D, Fakhouri F, Grünfeld JP. Liver involvement in autosomal-dominant polycystic kidney disease: therapeutic dilemma. J Am Soc Nephrol. 2000;11(9):1767–1775.
119. Johnstone AJ, Turnbull LW, Allan PL, Garden OJ. Cholangitis and Budd Chiari syndrome as complications of simple cystic liver disease. HPB Surg. 1993;6(3):223–228.
120. Grams J, Teh SH, Torres VE, Andrews JC, Nagorney DM. Inferior vena cava stenting: a safe and effective treatment for intractable ascites in patients with polycystic liver disease. J Gastrointest Surg. 2007;11(8):985–990.
121. Bernts LHP, Drenth JPH, Tjwa ETTL. Management of portal hypertension and ascites in polycystic liver disease. Liver Int. 2019;39(11):2024–2033.
122. Bernts LHP, Tjwa ETTL, D’Agnolo HMA, Jenniskens SFM, Drenth JPH. Venous stent placement for refractory ascites due to hepatic venous outflow obstruction in polycystic liver disease. J VascIntervRadiol. 2019;30(10):1617–1619.
123. Everson GT. Polycystic liver disease. Gastroenterol Hepatol. 2008;4(3):179–181.
124. Macutkiewicz C, Plastow R, Chrispijn M, et al. Complications arising in simple and polycystic liver cysts. World J Hepatol. 2012;4(12):406–411.
125. Madigan CG, Wang JY. Cirrhosis in an active duty soldier with concomitant isolated polycystic disease and H63D homozygosity. Mil Med. 2016;181(8):965–967.
126. Barbier L, Ronot M, Aussilhou B, et al. Polycystic liver disease: hepatic venous outflow obstruction lesions of the noncystic parenchyma have major consequences. Hepatology. 2017;68(2):652–662.
127. Aussilhou B, Douflé G, Hubert C, et al. Extended liver resection for polycystic liver disease can challenge liver transplantation. Ann Surg. 2010;252(5):735–743.
128. Ishikawa H, Uchida S, Yokokura Y, et al. Nonparasitic solitary huge liver cysts causing intracystic hemorrhage or obstructive jaundice. J Hepatobiliary Pancreat Surg. 2002;9(6):764–768.
129. Rosenfeld L, Abergel A, Bonny C, et al. Complicated polycystic liver disease with intracystic hemorrhage and obstructive jaundice. Gastroenterol Clin Biol. 2001;25(8–9):818–822.
130. Lerner ME, Roshkow JE, Smithline A, Ng C. Polycystic liver disease with obstructive jaundice: treatment with ultrasound-guided cyst aspiration. GastrointestRadiol. 1992;17(1):46–48.
131. Chapman AB. Cystic disease in women: clinical characteristics and medical management. Adv Ren Replace Ther. 2003;10(1):24–30.
132. Sherstha R, McKinley C, Russ P, et al. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology. 1997;26(5):1282–1286.
133. Bae KT, Zhu F, Chapman AB, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the consortium for radiologic imaging studies of polycystic kidney disease cohort. Clin J Am Soc Nephrol. 2006;1(1):64–69.
134. Chebib FT, Jung Y, Heyer CM, et al. Effect of genotype on the severity and volume progression of polycystic liver disease in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2016;31(6):952–960.
135. Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, Everson GT. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11(6):1033–1037.
136. Everson GT, Scherzinger A, Berger-Leff N, et al. Polycystic liver disease: quantitation of parenchymal and cyst volumes from computed tomography images and clinical correlates of hepatic cysts. Hepatology. 1988;8(6):1627–1634.
137. Wilcox DM, Weinreb JC, Lesh P. MR imaging of a hemorrhagic hepatic cyst in a patient with polycystic liver disease. J Comput Assist Tomogr. 1985;9(1):183–185.
138. Mortele KJ, Ros PR. Cystic focal liver lesions in the adult: differential CTand MR imaging features. Radiographics. 2001;21(4):895–910.
139. Fong ZV, Wolf AM, Doria C, Berger AC, Rosato EL, Palazzo F. Hemorrhagic hepatic cyst: report of a case and review of the literature with emphasis on clinical approach and management. J Gastroint Surg. 2012;16(9):1782–1789.
140. Tonolini M, Rigiroli F, Bianco R. Symptomatic and complicated non hereditary developmental liver cysts: cross-sectional imaging findings. Emerg Radiol. 2014;21(3):301–308.
141. Murphy BJ, Casillas J, Ros PR, Morillo G, Albores-Saavedra J, Rolfes DB. The CT appearance of cystic masses of the liver. Radiographics. 1989;9(2):307–322.
142. Vachha B, Sun MR, Siewert B, Eisenberg RL. Cystic lesions of the liver. AJR Am J Roentgenol. 2011;196:W355–W366.
143. Jouret F, Lhommel R, Devuyst O, et al. Diagnosis of cyst infection in patients with autosomal dominant polycystic kidney disease: attributes and limitations of the current modalities. Nephrol Dial Transplant. 2012;27(10):3746–3751.
144. Lantinga MA, Drenth JP, Gevers TJ. Diagnostic criteria in renal and hepatic cyst infection. Nephrol Dialysis Transplant. 2015;30(5):744–751.
145. Kanaan N, Goffin E, Pirson Y, Devuyst O, Hassoun Z. Carbohydrate antigen 19-9 as a diagnostic marker for hepatic cyst infection in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2010;55(5):916–922.
146. Lantinga MA, de Sevaux RG, Drenth JPH. 18F-FDG PET/CT during diagnosis and follow-up of recurrent hepatic cyst infection in autosomal dominant polycystic kidney disease. Clin Nephrol. 2015;84(1):61–64.
147. Suwabe T, Araoka H, Ubara Y, et al. Cyst infection in autosomal dominant polycystic kidney disease: causative microorganisms and susceptibility to lipid-soluble antibiotics. Eur J Clin Microbiol Infect Dis. 2015;34(7):1369–1379.
148. Shutsha E, Brenard R. Hepatic cyst rupture after a coughing fit. J Hepatol. 2003;38(6):870.
149. Miliadis L, Giannakopoulos T, Boutsikos G, Terzis I, Kyriazanos ID. Spontaneous rupture of a large non-parasitic liver cyst: a case report. J Med Case Rep. 2010;4:2.
150. Marion Y, Brevartt C, Plard L, Chiche L. Hemorrhagic liver cyst rupture: an unusual life-threatening complication of hepatic cyst and literature review. Ann Hepatol. 2013;12(2):336–339.
151. Arnold HL, Harrison SA. New advances in evaluation and management of patients with polycystic liver disease. Am J Gastroenterol. 2005;100(11):2569–2582.
152. Que F, Nagorney DM, Gross JB, Torres VE. Liver resection and cyst fenestration in the treatment of severe polycystic liver disease. Gastroenterology. 1995;108:487–494.
153. Kabbej M, Sauvanet A, Chauveau D, Farges O, Belghiti J. Laparoscopic fenestration in polycystic liver disease. Br J Surg. 1996;83(12):1697–1701.
154. Gigot JF, Jadoul P, Que F, et al. Adult polycystic liver disease: is fenestration the most adequate operation for long-term management? Ann Surg. 1997;225(3):286–294.
155. Waanders E, van Keimpema L, Brouwer JT, et al. Carbohydrate antigen 19–9 is extremely elevated in polycystic liver disease. Liver Int. 2009;29(9):1389–1395.
156. Fukasawa H, Kaneko M, Niwa H, Yasuda H, Kumagai H, Furuya R. Carbohydrate antigen 19-9 is significantly elevated in autosomal dominant polycystic kidney disease. Nephrology. 2018;23(3):210–216.
157. Fliszkiewicz M, Niemczyk M, Kulesza A, Pączek L. Carbohydrate antigen 19-9 level in patients with autosomal dominant polycystic kidney disease. Transplant Proc. 2018;50(6):1631–1633.
158. Deschênes M, Michel RP, Alpert E, Barkun JS, Metrakos P, Tchervenkov J. Elevation of CA-125 level is due to abdominal distension in liver transplantation candidates. Transplantation. 2001;72(9):1519–1522.
159. Iwase K, Takenaka H, Oshima S, et al. Determination of tumor marker levels in cystic fluid of benign liver cysts. Dig Dis Sci. 1992;37(11):1648–1654.
160. McCormick SE, Sjogren MH, Goodman ZD. A 22-year-old man with a liver mass and markedly elevated serum alpha fetoprotein. Semin Liver Dis. 1994;14(4):395–403.
161. Waanders E, Van Krieken JH, Lameris AL, Drenth JP. Disrupted cell adhesion but not proliferation mediates cyst formation in polycystic liver disease. Mod Pathol. 2008;21(11):1293–1302.
162. Levine E, Cook LT, Grantham JJ. Liver cysts in autosomal-dominant polycystic kidney disease: clinical and computed tomographic study. AJR Am J Roentgenol. 1985;145(2):229–233.
163. Nicolau C, Torra R, Bianchi L, et al. Abdominal sonographic study of autosomal dominant polycystic kidney disease. J Clin Ultrasound. 2000;28(6):277–282.
164. Banzo J, Ubieto MA, Gil D, et al. 18F-FDG PET/CT diagnosis of liver cyst infection in a patient with autosomal dominant polycystic kidney disease and fever of unknown origin. Rev Esp Med Nucl Imagen Mol. 2013;32(3):187–189.
165. Jouret F, Lhommel R, Beguin C, et al. Positron-emission computed tomography in cyst infection diagnosis in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2011;6(7):1644–1650.
166. Bleeker-Rovers CP, de Sévaux RG, van Hamersvelt HW, Corstens FH, Oyen WJ. Diagnosis of renal and hepatic cyst infections by 18-F-fluorodeoxyglucose positron emission tomography in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2003;41(6):E18–E21.
167. Sallée M, Rafat C, Zahar JR, et al. Cyst infections in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2009;4(7):1183–1189.
168. Peters AM. Nuclear medicine in vasculitis. Rheumatology. 2000;39(5):463–470.
169. Meller J, Sahlmann CO, Scheel AK. 18F-FDG PET and PET/CT in fever of unknown origin. J Nucl Med. 2007;48(1):35–45.
170. Jiménez-Bonilla JF, Quirce R, Calabia ER, Banzo I, Martínez-Rodríguez I, Carril JM. Hepatorenal polycystic disease and fever: diagnostic contribution of gallium citrate Ga 67 scan and fluorine F 18 FDG-PET/CT. Eur Urol. 2011;59(2):297–299.
171. Belair JA, Joyce JM, Kwak EJ. Detection of infected liver cysts by 111In WBC SPECT/CT in polycystic liver disease. Clin Nucl Med. 2014;39(1):59–61.
172. Even-Sapir E, Barnes DC, Iles SE. Remnants of normal tissue in polycystic disease of the liver. A cause for difficulty in the interpretation of indium-111 white blood cell study. Clin Nucl Med. 1993;18(11):967–969.
173. Schwab SJ, Bander SJ, Klahr S. Renal infection in autosomal dominant polycystic kidney disease. Am J Med. 1987;82(4):714–718.
174. Boerrigter MM, Bongers EMHF, Lugtenberg D, Nevens F, Drenth JPH. Polycystic liver disease genes: practical considerations for genetic testing. Eur J Med Genet. 2021;64(3):104160.
175. Hogan MC, Abebe K, Torres VE, et al. Liver involvement in early autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol. 2015;13(1):155–64.e6.
176. Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20(1):205–212.
177. Zhang ZY, Wang ZM, Huang Y. Polycystic liver disease: classification, diagnosis, treatment process, and clinical management. World J Hepatol. 2020;12(3):72–83.
178. Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343(8901):824–827.
179. Carrim ZI, Murchison JT. The prevalence of simple renal and hepatic cysts detected by spiral computed tomography. Clin Radiol. 2003;58(8):626–629.
180. Reynolds DM, Falk CT, Li A, et al. Identification of a locus for autosomal dominant polycystic liver disease, on chromosome 19p13.2-13.1. Am J Hum Genet. 2000;67(6):1598–1604.
181. Cnossen WR, Drenth JPH. Polycystic liver disease: an overview of pathogenesis, clinical manifestations and management. Orphanet J Rare Dis. 2014;9(1):69.
182. Patel A, Chapman AB, Mikolajczyk AE. A practical approach to polycystic liver disease. Clin Liver Dis. 2019;14(5):176–179.
183. Chebib FT, Harmon A, Irazabal Mira MV, et al. Outcomes and durability of hepatic reduction after combined partial hepatectomy and cyst fenestration for massive polycystic liver disease. J Am Coll Surg. 2016;223(1):118–126.
184. Schnelldorfer T, Torres VE, Zakaria S, Rosen CB, Nagorney DM. Polycystic liver disease: a critical appraisal of hepatic resection, cyst fenestration and liver transplantation. Ann Surg. 2009;250(1):112–118.
185. Tang H, Ding F, Yao J, et al. Liver transplantation for polycystic liver disease: 11 cases report and literature review. Zhonghua Yi Xue Za Zhi. 2019;99(10):767–770.
186. Kataoka H, Watanabe S, Sato M, et al. Predicting liver cyst severity by mutations in patients with autosomal-dominant polycystic kidney disease. Hepatol Int. 2021;15(3):791–803.
187. van Keimpema L, Höckerstedt K. Treatment of polycystic liver disease. Br J Surg. 2009;96(12):1379–1380.
188. van Aerts RMM, Bernts LHP, Gevers TJG, et al. Estrogen-containing oral contraceptives are associated with polycystic liver disease severity in premenopausal patients. Clin Pharmacol Ther. 2019;106(6):1338–1345.
189. Neijenhuis MK, Wijnands TFM, Kievit W, et al. Symptom relief and not cyst reduction determines treatment success in aspiration sclerotherapy of hepatic cysts. Eur Radiol. 2019;29(6):3062–3068.
190. Tietz PS, Aipini G, Pham LD, LaRusso NF. Somatostatin inhibits secretin-induced ductal choleresisin viva and exocytosis by cholangiocytes. Am J Physiol. 1995;269(1 Pt 1):G110–G118.
191. Tan CK, Podila PV, Taylor JE, et al. Human cholangiocarcinomas express somatostatin receptors and respond to somatostatin with growth inhibition. Gastroenterology. 1995;108(6):1908–1916.
192. Weckbecker G, Lewis I, Albert R, Schmid HA, Hoyer D, Bruns C. Opportunities in somatostatin research: biological, chemical and therapeutic aspects. Nat Rev Drug Discov. 2003;2(12):999–1017.
193. Florio T. Molecular mechanisms of the antiproliferative activity of somatostatin receptors (SSTRs) in neuroendocrine tumors. Front Biosci. 2008;13:822–840.
194. Bueno, CBF. Polimorfismos dos genes dos receptores de dopamina D2 e de somatostatina subtipos 2 e 5 e resposta ao tratamento medicamentoso de pacientes portadores de adenomas hipofisários [tese]. [The influence of dopamine receptor type 2 and somatostatin receptors type 2 and 5 polymorphisms in medical treatment of pituitary adenomas [Thesis]. São Paulo: Faculdade de Medicina, Universidade de São Paulo; 2016. Portuguese
195. Schmid HA, Schoeffter P. Functional activity of the multiligand analog SOM230 at human recombinant somatostatin receptor subtypes supports its usefulness in neuroendocrine tumors. Neuroendocrinology. 2004;80(Suppl 1):47–50.
196. Paragliola RM, Salvatori R. Novel somatostatin receptor ligands therapies for acromegaly. Front Endocrinol. 2018;9:78.
197. Bronstein MD. Acromegaly: molecular expression of somatostatin receptor subtypes and treatment outcome. Front Horm Res. 2006;35:129–134.
198. Hogan MC, Masyuk TV, Page LJ, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. Clin J Am Nephrol. 2010;21(6):1052–1061.
199. van Keimpema L, Nevens F, Vanslembrouck R, et al. Lanreotide reduces the volume of polycystic liver: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2009;137(5):1661–8.e1–2.
200. Temmerman F, Gevers T, Ho TA, et al. Safety and efficacy of different lanreotide doses in the treatment of polycystic liver disease: pooled analysis of individual patient data. Aliment PharmacolTher. 2013;38(4):397–406.
201. Hogan MC, Masyuk T, Bergstralh E, et al. Efficacy of 4 years of octreotide long-acting release therapy in patients with severe polycystic liver disease. Mayo Clin Proc. 2015;90(8):1030–1037.
202. Chrispijn M, Nevens F, Gevers TJG, et al. The long-term outcome of patients with polycystic liver disease treated with lanreotide. Aliment Pharmacol Ther. 2012;35(2):266–274.
203. Temmerman F, Missiaen L, Bammens B, et al. Systematic review: the pathophysiology and management of polycystic liver disease. Aliment Pharmacol Ther. 2011;34(7):702–713.
204. Khan S, Dennison A, Garcea G. Medical therapy for polycystic liver disease. Ann R Coll Surg Engl. 2016;98(1):18–23.
205. Pisani A, Sabbatini M, Imbriaco M, et al. Long-term effects of octreotide on liver volume in patients with polycystic kidney and liver disease. Clin Gastroenterol Hepatol. 2016;14(7):1022–1030.
206. Gevers TJG, Inthout J, Caroli A, et al. Young women with polycystic liver disease respond best to somatostatin analogues: a pooled analysis of individual patient data. Gastroenterology. 2013;145(2):357–65.e1–2.
207. van Aerts RMM, Kievit W, D’Agnolo HMA, et al. Lanreotide reduces liver growth in patients with autosomal dominant polycystic liver and kidney disease. Gastroenterology. 2019;157(2):481–491.
208. Hogan MC, Chamberlin JA, Vaughan LE, et al. Pansomatostatin agonist pasireotide long-acting release for patients with autosomal dominant polycystic kidney or liver disease with severe liver involvement: a randomized clinical trial. Clin J Am Soc Nephrol. 2020;15(9):1267–1278.
209. Griffiths J, Mills MT, Ong AC. Long-acting somatostatin analogue treatments in autosomal dominant polycystic kidney disease and polycystic liver disease: a systematic review and meta-analysis. BMJ Open. 2020;10(1):e032620.
210. Suwabe T, Barrera FJ, Rodriguez-Gutierrez R, Ubara Y, Hogan MC. Somatostatin analog therapy effectiveness on the progression of polycystic kidney and liver disease: a systematic review and meta-analysis of randomized clinical trials. PLoS One. 2021;16(9):e0257606.
211. Garofalo C, Capuano I, Pennino L, et al. The effects of somatostatin analogues on liver volume and quality of life in polycystic liver disease: a meta-analysis of randomized controlled trials. Sci Rep. 2021;11:23500.
212. Hartford CM, Ratain MJ. Something old, something new, sometimes borrowed and now renewed. Clin Pharmacol Ther. 2007;82:381–388.
213. Chrispijn M, Gevers TJG, Hol JC, Monshouwer R, Dekker HM, Drenth JPH. Everolimus does not further reduce polycystic liver volume when added to long acting octreotide: results from a randomized controlled trial. J Hepatol. 2013;59(1):153–159.
214. Walz G. Therapeutic approaches in autosomal dominant polycystic kidney disease (ADPKD): is there light at the end of the tunnel? Nephrol Dial Transplant. 2006;21(7):1752–1757.
215. Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf. 2013;12(2):177–186.
216. Birnbaumer M. Vasopressin receptors. Trends Endocrinol Metab. 2000;11(10):406–410.
217. Mancinelli R, Franchitto A, Glaser S, et al. Vasopressin regulates the growth of the biliary epithelium in polycystic liver disease. Lab Invest. 2016;96(11):1147–5.
218. Takenaka T, Miura S, Kitajima M. The management of polycystic liver disease by tolvaptan. Clin Mol Hepatol. 2020;26(1):70–73.
219. Mizuno H, Hoshino J, Suwabe T, et al. Tolvaptan for the Treatment of Enlarged Polycystic Liver Disease. Case Rep Nephrol Dial. 2017;7(2):108–111.
220. Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407–2418.
221. Beuers U, Nathanson MH, Boyer JL. Effects of tauroursodeoxycholic acid on cytosolic Ca2+ signals in isolated rat hepatocytes. Gastroenterology. 1993;104(2):604–612.
222. Bouscarel B, Fromm H, Nussbaum R. Ursodeoxycholate mobilizes intracellular Ca2+ and activates phosphorylase a in isolated hepatocytes. Am J Physiol. 1993;264(2 Pt 1):G243–G251.
223. Beuers U, Nathanson MH, Isales CM, Boyer JL. Tauroursodeoxycholic acid stimulates hepatocellular exocytosis and mobilizes extracellular Ca++ mechanisms defective in cholestasis. J Clin Invest. 1993;92(6):2984–2993.
224. Alpini G, Baiocchi L, Glaser S, et al. Ursodeoxycholate and tauroursodeoxycholate inhibit cholangiocyte growth and secretion of BDL rats through activation of PKC alpha. Hepatology. 2002;35(5):1041–1052.
225. Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology. 2002;36(3):525–531.
226. Poupon R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: an overview of their mechanisms of action. Clin Res Hepatol Gastroenterol. 2012;36(Suppl 1):S3–S12.
227. Lazaridis KN, Gores GJ, Lindor KD. Ursodeoxycholic acid ‘mechanisms of action and clinical use in hepatobiliary disorders’. J Hepatol. 2001;35(1):134–146.
228. D’Agnolo HMA, Kievit W, Takkenberg RB, et al. Ursodeoxycholic acid in advanced polycystic liver disease: a Phase 2 multicenter randomized controlled trial. J Hepatol. 2016;65(3):601–607.
229. Saini S, Mueller PR, Ferrucci JT, Simeone JF, Wittenberg J, Butch RJ. Percutaneous aspiration of hepatic cysts does not provide definitive therapy. AJR Am J Roentgenol. 1983;141(3):559–560.
230. Bean WJ, Rodan BA. Hepatic cysts: treatment with alcohol. AJR Am J Roentgenol. 1985;144(2):237–241.
231. van Keimpema L, de Koning DB, Strijk SP, Drenth JP. Aspiration-sclerotherapy results in effective control of liver volume in patients with liver cysts. Dig Dis Sci. 2008;53(8):2251–2257.
232. Yamada N, Shinzawa H, Ukai K, et al. Treatment of symptomatic hepatic cysts by percutaneous instillation of minocycline hydrochloride. DigDis Sci. 1994;39(11):2503–2509.
233. Nakaoka R, Das K, Kudo M, Chung H, Innoue T. Percutaneous aspiration and ethanolamine oleate sclerotherapy for sustained resolution of symptomatic polycystic liver disease: an initial experience. AJR Am J Roentgenol. 2009;193(6):1540–1545.
234. Benzimra J, Ronot M, Fuks D, et al. Hepatic cysts treated with percutaneous ethanol sclerotherapy: time to extend the indications to haemorrhagic cysts and polycystic liver disease. EurRadiol. 2014;24(5):1030–1038.
235. Kairaluoma MI, Leinonen A, Stahlberg M, Paivansalo M, Kiviniemi H, Siniluoto T. Percutaneous aspiration and alcohol sclerotherapy for symptomatic hepatic cysts. An alternative to surgical intervention. Ann Surg. 1989;210(2):208–215.
236. Wijnands TFM, Görtjes APM, Gevers TJG, et al. Efficacy and safety of aspiration sclerotherapy of simple hepatic cysts: a systematic review. AJR Am J Roentgenol. 2017;208(1):201–207.
237. Drenth JPH, Chrispijn M, Nagorney DM, Kamath PS, Torres VE. Medical and surgical treatment options for polycystic liver disease. Hepatology. 2010;52(6):2223–2230.
238. Wang MQ, Duan F, Liu FY, Wang ZJ, Song P. Treatment of symptomatic polycystic liver disease: transcatheter super-selective hepatic arterial embolization using a mixture of NBCA and iodized oil. Abdom Imaging. 2013;38(3):465–473.
239. Takei R, Ubara Y, Hoshino J, et al. Percutaneous transcatheter hepatic artery embolization for liver cysts in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2007;49(6):744–752.
240. Bello-Reuss E, Holubec K, Rajaraman S. Angiogenesis in autosomal-dominant polycystic kidney disease. Kidney Int. 2001;60(1):37–45.
241. Park HC, Kim CW, Ro H, et al. Transcatheter arterial embolization therapy for a massive polycystic liver in autosomal dominant polycystic kidney disease patients. J Korean Med Sci. 2009;24(1):57–61.
242. Yan JY, Zhang JL, Yuan K, et al. Transarterial embolisation with bleomycin and N-butyl-2-cyanoacrylate-Lipiodol mixture for symptomatic polycystic liver disease: preliminary experience. Clin Radiol. 2019;74(12):975.e11–975.e16.
243. Yang J, Ryu H, Han M, et al. Comparison of volume-reductive therapies for massive polycystic liver disease in autosomal dominant polycystic kidney disease. Hepatol Res. 2016;46(2):183–191.
244. Zhang JL, Yuan K, Wang MQ, et al. Transarterial embolization for treatment of symptomatic polycystic liver disease: more than 2-year follow-up. Chin Med J. 2017;130(16):1938–1944.
245. Hoshino J, Ubara Y, Suwabe T, et al. Intravascular embolization therapy in patients with enlarged polycystic liver. Am J Kidney Dis. 2014;63(6):937–944.
246. Lin TY, Chen CC, Wang SM. Treatment of non-parasitic cystic disease of the liver: a new approach to therapy with polycystic liver. Ann Surg. 1968;168(5):921–927.
247. van Keimpema L, Ruurda JP, Ernst MF, van Geffen HJ, Drenth JP. Laparoscopic fenestration of liver cysts in polycystic liver disease results in a median volume reduction of 12.5%. J Gastrointest Surg. 2008;12(3):477–482.
248. Russell RT, Pinson CW. Surgical management of polycystic liver disease. World J Gastroenterol. 2007;13(38):5052–5059.
249. Zhang JY, Liu Y, Liu HY, Chen L, Su DW, Wang YB. Comparison of the recurrence rates of nonparasitic hepatic cysts treated with laparoscopy or with open fenestration: a meta-analysis. Surg LaparoscEndoscPercutan Tech. 2018;28(2):67–72.
250. Gall TMH, Oniscu GC, Madhavan K, Parks RW, Garden OJ. Surgical management and long term follow-up of non-parasitic hepatic cysts. HPB. 2009;11(3):235–241.
251. Bernts LHP, Echternach SG, Kievit W, Rosman C, Drenth JPH. Clinical response after laparoscopic fenestration of symptomatic hepatic cysts: a systematic review and meta-analysis. Surg Endosc. 2019;33(3):691–704.
252. van Keimpema L, Drenth JP. Polycystic liver disease: a critical appraisal of hepatic resection, cyst fenestration, and liver transplantation. Ann Surg. 2011;253(2):419.
253. Martin IJ, McKinley AJ, Currie EJ, Holmes P, Garden OJ. Tailoring the management of nonparasitic liver cysts. Ann Surg. 1998;228(2):167–172.
254. Antonacci N, Ricci C, Taffurelli G, Casadei R, Minni F. Systematic review of laparoscopic versus open surgery in the treatment of non-parasitic liver cysts. Updates Surg. 2014;66(4):231–238.
255. Schindl MJ, Redhead DN, Fearon KC, Garden OJ, Wigmore SJ. The value of residual liver volume as a predictor of hepatic dysfunction and infection after major liver resection. Gut. 2005;54(2):289–296.
256. Newman KD, Torres VE, Rakela J, Nagorney DM. Treatment of highly symptomatic polycystic liver disease. Preliminary experience with a combined hepatic resection-fenestration procedure. Ann Surg. 1990;212(1):30–37.
257. Ogawa K, Fukunaga K, Takeuchi T, et al. Current treatment status of polycystic liver disease in Japan. Hepatol Res. 2014;44(11):1110–1118.
258. Everson GT, Taylor MRG, Doctor RB. Polycystic disease of the liver. Hepatology. 2004;40(4):774–782.
259. Arrazola L, Moonka D, Gish RG, Everson GT. Model for end-stage liver disease (MELD) exception for polycystic liver disease. Liver Transplant. 2006;12(12 Suppl 3):S110–S111.
260. Swenson K, Seu P, Kinkhabwala M, et al. Liver transplantation for adult polycystic liver disease. Hepatology. 1998;28(2):412–415.
261. Starzl TE, Reyes J, Tzakis A, et al. Liver transplantation for polycystic liver disease. Arch Surg. 1990;125:575–577.
262. Freeman RB, Gish RG, Harper A, et al. Model for end-stage liver disease (MELD) exception guidelines: results and recommendations from the MELD Exception Study Group and Conference (MESSAGE) for the approval of patients who need liver transplantation with diseases not considered by the standard MELD formula. Liver Transpl. 2006;12(12 Suppl 3):S128–S136.
263. Organ Procurement and Transplantation Network (OPTN). Guidance to liver transplant programs and the national liver review board for: adult meld exception review [internet]. Washington (DC): US Department of Health & Human Services; 2021. Available from: https://optn.transplant.hrsa.gov/resources/guidance/liver-review-board-guidance/.
264. Ding F, Tang H, Zhao H, et al. Long-term results of liver transplantation for polycystic liver disease: single-center experience in China. Exp Ther Med. 2019;17(5):4183–4189.
265. Baber JT, Hiatt JR, Busuttil RW, Agopian VG. A 20-year experience with liver transplantation for polycystic liver disease: does previous palliative surgical intervention affect outcomes? J Am Coll Surg. 2014;219(4):695–703.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.