Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
PM2.5 Induces the Expression of Inflammatory Cytokines via the Wnt5a/Ror2 Pathway in Human Bronchial Epithelial Cells
Authors Zou W ![]() , Wang X, Hong W, He F, Hu J, Sheng Q, Zhu T, Ran P
, Wang X, Hong W, He F, Hu J, Sheng Q, Zhu T, Ran P
Received 20 July 2020
Accepted for publication 23 September 2020
Published 23 October 2020 Volume 2020:15 Pages 2653—2662
DOI https://doi.org/10.2147/COPD.S270762
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Weifeng Zou,1 Xiaoqian Wang,2 Wei Hong,3 Fang He,4 Jinxing Hu,1 Qing Sheng,1 Tao Zhu,1 Pixin Ran5
1State Key Laboratory of Respiratory Disease, Guangzhou Chest Hospital, Guangzhou, Guangdong, People’s Republic of China; 2State Key Laboratory of Respiratory Disease, Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China; 3GMU-GIBH Joint School of Life Sciences, Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China; 4The Research Center of Experiment Medicine, Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China; 5State Key Laboratory of Respiratory Diseases, National Clinical Research Center for Respiratory Diseases, Guangzhou Institute of Respiratory Health, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China
Correspondence: Pixin Ran
State Key Laboratory of Respiratory Diseases, National Clinical Research Center for Respiratory Diseases, Guangzhou Institute of Respiratory Health, The First Affiliated Hospital of Guangzhou Medical University, 151 Yanjiang Road, Yuexiu District, Guangzhou, Guangdong 510120, People’s Republic of China
, Tel +86 13922765811
Email [email protected]
Background and Purpose: Recently, fine particulate matter (PM2.5) was identified as the main exposure risk for COPD, and inflammation is central to the development of COPD. In this study, we investigated whether PM2.5 can induce the secretion of interleukin-6 (IL-6), IL-8 and IL-1β in human bronchial epithelial cells (HBECs) in vitro via the wingless-related integration site 5A (Wnt5a)/receptor tyrosine kinase-like orphan receptor 2 (Ror2) signaling.
Methods: The expression of Wnt5a and Ror2 was assessed by immunohistochemistry in motor vehicle exhaust (MVE)-induced Sprague-Dawley rats. HBECs were transfected with small interfering RNA (siRNA) targeting Wnt5a or Ror2 and subsequently stimulated with PM2.5.The secretion of IL-6, IL-8 and IL-1β was assessed by ELISAs, and the expression of Wnt5a/Ror2 signaling were assessed by RT-PCR and Western blotting.
Results: Both Wnt5a and Ror2 protein were increased in the lung of MVE-induced rats. HBECs exposed to PM2.5 for 24 h significantly upregulated Wnt5a and Ror2 expression and subsequently promoted the nuclear translocation of NF-κB, which increased the production of IL-1β, IL-6 and IL-8. Wnt5a siRNA prevented these outcomes. Wnt5a antagonist (BOX5) also prevented inflammatory effects. Furthermore, Ror2 siRNA blocked the NF-κB activity and inhibited the release of IL-6, IL-8 and IL-1β from PM2.5-exposed HBECs.
Conclusion: PM2.5 induces the secretion of IL-6, IL-8 and IL-1β in HBECs via the Wnt5a/Ror2 signaling, demonstrating a novel mechanism for PM2.5-associated airway inflammation.
Key words: COPD, PM2.5, Wnt5a/ROR2, inflammatory cytokines, HBECs
Introduction
Chronic obstructive pulmonary disease (COPD) is one of the leading causes of morbidity and mortality worldwide, resulting in a growing social and economic burden. COPD is a chronic disease characterized by the blocking of lung airways because of degeneration of lung parenchyma and inflammation. Recently, PM2.5 exposure was identified as a major risk factor for COPD,1 and substantial epidemiological studies revealed that PM2.5 is associated not only with an enhanced risk of COPD hospitalization, morbidity and mortality,2,3 but also with the exacerbated and aggravated respiratory dysfunction and symptoms experienced by COPD patients, such as shortness of breath, coughing and wheezing.4,5 Air pollution amelioration would substantially benefit COPD patients.6 However, the mechanisms by which PM2.5 induced the airway inflammation of COPD remained to be fully elucidated.
Recently, several studies have demonstrated that increased noncanonical Wnt signaling activated by Wnt5a contributes to COPD pathogenesis.7 Increased expression of Wnt5a was detected in two distinct animal models of emphysema/COPD.Wnt5a has positive or negative regulatory effects on canonical Wnt signaling according to the kind of receptor involved.8 Moreover, further investigation demonstrated that CSE exposure increased Wnt5a expression in conjunction with inflammatory cytokines production in the model of COPD.9
Inflammation is central to the development of COPD.10 However, the research on Wnt5a participating in airway inflammation of COPD have been actually unsubstantial, therefore, we designed the present study to explore the effects of Wnt5a in the airway inflammation of COPD induced by PM2.5.
Materials and Methods
Cell Culture and Treatment
Human bronchial epithelial cell line 16HBE was provided by Guangzhou Medical University (Guangzhou, China), an agent of ATCC. 16HBEC were cultured in DMEM (Sigma, Louis, USA) containing 10% FBS (Sigma, Louis, USA) and 1% penicillin (Sigma, Louis, USA). The cells were serum starved for 24 h, subsequently transfected with BOX5 (Sigma, Louis, USA), siRNA duplexes (either control siRNA, Wnt5a siRNA, or Ror2 siRNA; Santa Cruz, CA, USA) for 48 h and treat with 5–20 μg/mL PM2.5 in medium for 3–24 h.
Cell Counting Kit-8 (CCK8) Cell Viability Assay
Cells were exposed to PM2.5 (5, 10, 20, or 40μg/mL) in medium for 24 hours. Then CCK8 (Dojindo, Kumamoto, Japan) assays were used to detect the viability of 16HBE cells according to the manufacturer’s instructions, and the optical density (OD) of each well was read at 490 nm.
Animals and Sample Lung Tissues
Eighteen female Sprague-Dawley rats (body weight of 180–200 g, 6–8 weeks old) were socially housed in the Laboratory Animal Center of the First Affiliated Hospital of Guangzhou Medical University. All in vivo manipulations were approved by the Institutional Animal Care and Use Committee of Guangzhou Medical University, and performed following the regulations designated by the Chinese Association for Laboratory Animal Science Policy. The experimental environment is carried out according to our previous study.11 The rats were randomly allocated to an MVE group and a clean air control group. The rats in the MVE group were exposed to MVE (1.5 mg/m3) for 2 h periods, 5 days per week for 1 month. In the MVE exposure room, the concentrations of PM2.5 were 1.46± 0.034mg/m3.
Rats were sacrificed by intraperitoneal injection of sodium pentobarbital (100 mg/kg) for the duration of the testing. The lung tissues were then embedded in paraffin, then cut into 4 μm thick section, which were stained for hematoxylin and eosin (H&E), immunohistochemistry.
Collection and Extraction of PM2.5
The aerodynamic impactors were used for the collection of traffic ambient PM2.5, Soluble parts of PM2.5 were extracted by DMSO. Sufficient specific samples for biological experiments were collected by high volume impactors assembled with glass fiber filter from arterial traffic road. The specific physicochemical characterizations of PM2.5 were analyzed by gravimetric analysis. The details of PM2.5 samples and analysis methods can be found in our previous research.12 The PM2.5 sample was stored at −80°C until use.
siRNA Transfection
HBECs were cultured to reach 60% density prior to transfection. The cells transiently transfected for 48h with targeting siRNA or control siRNA. Transfection was performed with the ratio of the plasmids, and the transfection reagent consisted of 1 mg:2 mL. Western blot analysis were performed to examine the efficiency.
RNA Isolation and Quantitative Real-Time PCR
Total RNA was extracted from cultured HBECs using the RNeasy plus mini kit (Qiagen), which was performed as manufacturer’s described. The primer sequences used was: GAPDH, 5′-ATCACTGCCACCCAGAAG-3′ and 5′-TCCACGACGGA CACATTG-3′. Wnt5a, 5′-GCCAGTATCAATTCCGACATCG-3′ and 5′-TCACCGCGTATGTGAAGGC-3′; Ror2, 5ʹ-GGCAGAACCCATCCTCGTG-3ʹ and 5ʹ-CGACTGCGAATCCAGGACC-3ʹ. GAPDH served as the housekeeping control. The data were expressed relative to the control (control, set as 1.0).
Histology and Immunohistochemistry
The samples of lung tissue were stained with H&E for morphometric measurement. The sections were deparaffinized and incubated with Wnt5a antibodies (1:200, Santa Cruz, CA, USA) and Ror2 antibodies (1:400, Santa Cruz, CA, USA), respectively. These sections were then incubated with the peroxidase (HRP)-conjugated anti-rabbit or anti-mouse IgG and diaminobenzidine, which was performed following manufacturer’s protocols. Immunochemistry staining of the sections was evaluated with the confocal microscope at 200× magnification. We quantified the optical density (OD) of Wnt5a- and Ror2-positive cells using a semiquantitative scoring system.
Protein Extraction and Western Blot Analysis
Protein extraction and Western blot were performed according to our previous description12 and the manufacturer’s instructions. The concentrations of the antibodies were as follows: Wnt5a antibody (1:1000), Ror2 antibody (1:1000), and NF-κB p65 antibody (1:1000, Santa Cruz, CA, USA). GAPDH (1:5000), β-tubulin (1:2000), and lamin B (1:500) antibodies were used as loading controls. Results were expressed relative to the control (control, set as 1.0).
ELISAs
Cell supernatants were collected, and IL-6, IL-8 and IL-1β (R&D systems, Minneapolis, MN, USA) levels were measured using IL-6, IL-8 and IL-1β ELISA kits, following protocols provided by the ELISA kit manufacturer. Standard curve was prepared using standard solution, and the concentrations of samples were calculated according to the standard curve.
Statistical Analysis
All statistical analyses were performed using SPSS 25.0 statistical software, the results are presented as the means±standard deviation (SD) of three independent experiments. Student’s t-test was used to evaluate statistical significance, and that among multiple groups was determined by ANOVAs. p<0.05 was considered statistically significant.
Results
MVE-Induced Inflammatory Changes and the Upregulation of Wnt5a and Ror2 in the Lungs of Rats
H&E staining was utilized to observe the changes of lung histology, and lung histologic analysis revealed that MVE exposure led to the increase of inflammatory cells and displayed enlarged alveolar space (Figure 1A). Furthermore, immunostaining results revealed that the expression of Wnt5a and Ror2 in the lungs tissues was increased in the MVE-exposed rats compared to those exposed to air as controls (Figure 1B and 1C).
 |
Figure 1 MVE-induced inflammatory changes and the upregulation of Wnt5a and Ror2 in the lungs of rats. |
Effect of PM2.5 on Viability of the HBECs
To assess the cytotoxicity of PM2.5 on HBECs, cells were exposed to various concentrations of PM2.5 (0, 5, 10, 20, 40μg/mL). After exposure for 24h, the cell viability was measured by CCK8 assay. The results showed that the cell viability did not significantly change after 24 h of PM2.5 (5–20μg/mL) exposure, but the cell viability were significantly declined at PM2.5 treatment of 40 μg/mL in contrast to the control group (Figure 2). So concentrations of PM2.5 (5–20μg/mL) were selected for the following experiments.
 |
Figure 2 Effect of PM2.5 on viability of the HBECs. |
Wnt5a and Ror2 Expression is Induced by PM2.5 Stimulation in HBECs
Since PM2.5 is the major ingredients of an MVE exposure space in our previous study, we examined whether PM2.5 stimuli enhance the expression of Wnt5a and Ror2 in the HBECs. As expected, Western blotting data revealed Wnt5a production did not significantly change after 24 h of PM2.5 (5–10μg/mL) exposure, but the production were significantly increased at PM2.5 treatment of 20 μg/mL in contrast to the control group (Figure 3A), RT-PCR revealed a dramatic upregulation of Wnt5a mRNA and Ror2 mRNA expression after 24 h of PM2.5 (20 μg/mL) stimulation (Figure 3B), which suggested that Wnt5a and Ror2 upregulation occurred at the transcriptional level. Moreover, Western blotting data also revealed that Wnt5a and Ror2 expression did not significantly change after 3 h and 12 h of PM2.5 (20 μg/mL) stimulation, but the expression were significantly increased after 24 h of PM2.5 exposure compared with the control group (Figure 3C).
 |
Figure 3 Wnt5a and Ror2 expression is induced by PM2.5 stimulation in HBECs. |
Wnt5a Knockdown Reduced Ror2 Expression in PM2.5-Exposed HBECs
Next, we sought to examine the effect of Wnt5a in PM2.5-modulated the production of Wnt5a receptors. We specifically knocked down Wnt5a expression with Wnt5a siRNA. Wnt5a siRNA completely blocked the production of Wnt5a and Ror2 in PM2.5-exposed HBECs (Figure 4). On the other side, control siRNA did not prevent the elevated Wnt5a and Ror2 expression (Figure 4), demonstrating that the elevation of Ror2 was predominantly modulated by Wnt5a in PM2.5-exposed HBECs.
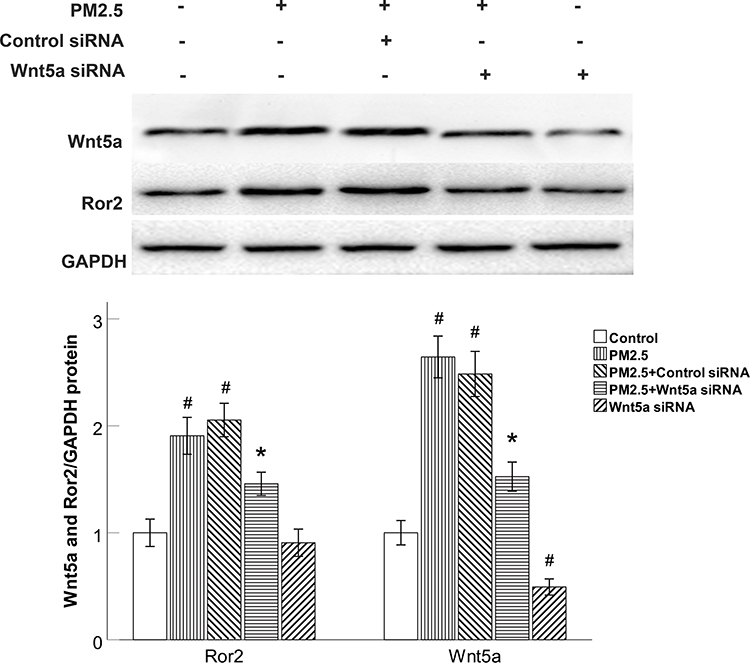 |
Figure 4 Wnt5a knockdown reduced Ror2 expression in PM2.5-exposed HBECs. |
The NF-κB Activity Was Regulated by Wnt5a/Ror2 Signaling in PM2.5-Exposed HBECs
We previously demonstrated that HBECs treated with PM2.5 exhibited remarkably reduced cytoplasmic extracts and elevated nuclear extracts of NF-κB. Since Wnt5a has been demonstrated to activate NF-κB pathway dependent survival signalling in vitro. We examined whether the expression of NF-κB is modulated by Wnt5a/Ror2 signaling in PM2.5-exposed HBECs, siRNAs were used to deplete Wnt5a or Ror2. In comparison to the control siRNA groups, Wnt5a siRNA led to increased cytoplasmic accumulation and inhibited nuclear translocation of NF-κB in PM2.5-exposed HBECs (Figure 5A). Ror2 siRNA suppressed the PM2.5-induced expression of Ror2 (Figure 5B). Consistently, Ror2 siRNA led to increased cytoplasmic accumulation and inhibited nuclear translocation of NF-κB in PM2.5-exposed HBECs (Figure 5C), indicating that PM2.5-upregulated the NF-κB activation is dependent on Wnt5a/Ror2 signaling in HBECs.
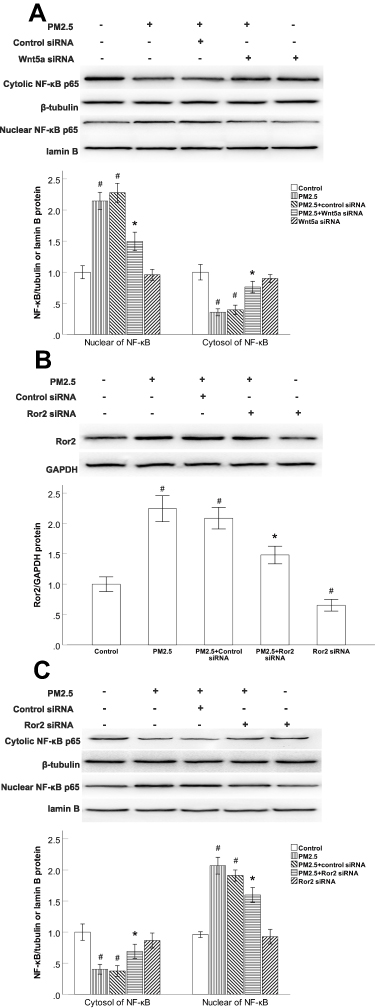 |
Figure 5 The NF-κB activity was regulated by Wnt5a/Ror2 signaling in PM2.5-exposed HBECs. |
PM2.5 Increased the Production of IL-6, IL-8 and IL-1β Through Wnt5a/Ror2 Signaling in HBECs
To gain further insight into the mechanism underlying PM2.5-modulated the production of IL-6, IL-8 and IL-1β in HBECs, we examined Wnt5a/Ror2 signaling and the production of IL-6, IL-8 and IL-1β.ELISA results showed that the levels of IL-6, IL-8 and IL-1β protein were increased in these cells after 24 h of exposure to PM2.5 (Figure 6A and B). Not surprisingly, Cells transfected with Wnt5a or Ror2 siRNA showed decreased secretion of the inflammatory cytokines IL-6, IL-8 and IL-1β compared with cells stimulated with PM2.5, the cells transfected with the control siRNA did not show attenuation of the enhanced production of IL-6, IL-8 and IL-1β after PM2.5 treatment (Figure 6A and B).To further validate the specificity of Wnt5a siRNA in airway inflammation, we also used BOX5 (200μM), the Wnt5a-derived antagonistic peptide, to prevent the production of cytokines. As expected, BOX5 prevented the upregulation of IL-6, IL-8 and IL-1β induced by PM2.5 (Figure 6C). To sum up, these data suggest that inhibition of the Wnt5a/Ror2 signaling attenuated the production of IL-6, IL-8 and IL-1β in PM2.5-exposed HBECs.
 |
Figure 6 PM2.5 increased IL-6, IL-8 and IL-1β production through Wnt5a/Ror2 signaling in HBECs. |
Discussion
Recent epidemiologic reports have revealed the relationship between PM2.5 exposure and the exacerbation of preexisting COPD.13,14 In the current study, we demonstrate that exposure to MVE causes aberrant upregulation of Wnt5a and Ror2 in vivo in rats and PM2.5 also induces the expression of Wnt5a in vitro in HBEC models. Upregulated Wnt5a leads to upregulated Ror2 expression, facilitates the nuclear translocation of NF-kB, and induces the production of the inflammatory cytokines IL-6, IL-8 and IL-1β in HBECs, targeting the Wnt5a/Ror2 signaling may present as an effective therapeutic strategy for PM2.5- associated airway inflammation in COPD.
HBECs in the airway form the first barrier against environmental insults, such as inhaled PM2.5, and they can also initiate the pathogenesis of COPD. Chronic inflammation is a critical characteristic in the pathogenesis of COPD.15 Consistently, a variety of proinflammatory cytokines produced by AECs are increased in COPD, including IL-1β, IL-6, IL-8 and TNF-a.16 In accordance with previous studies, our present results revealed that the production of IL-1β, IL-6 and IL-8 were increased in PM2.5-exposed HBECs. In addition, our previous study also showed that the profibrotic cytokines is induced by PM2.5 stimulation in HBECs. These results suggest that PM2.5 exposure is involved in the formation of airway inflammation in COPD.
Canonical and noncanonical Wnt signaling are both indispensable for proper lung development. It has been reported that Wnt5a increases the production of inflammatory cytokines such as IL-6 and TNF-α and is increased in experimental and human COPD.17 Lung-specific overexpression of Wnt5a during development has been demonstrated to negatively affect epithelial branching in vivo.18 Furthermore, 100 µg/mL of PM2.5 exposure promotes Wnt5a production and airway inflammation in COPD,19 which is differences from the dose of PM2.5 (20 ug/mL) in our study. Some studies showed that compositions of ambient PM2.5 are not completely similar to those of extracted PM2.5 for the methods and marker analyzed.20 Therefore, we analyze the inconsistency of dose is due to different extraction methods lead to the compositional alterations of PM2.5. Combined with previous reports, the relationship between Wnt5a and inflammatory cytokines due to PM2.5 exposure was also established in our study.
Ror2 is a key recipient of Wnt5a-induced effects realized during development, as demonstrated by the remarkable phenotypic resemblance between Ror2- and Wnt5a-knockout mice.21 A study indicated that higher levels of Wnt5a and ROR2 expression were found in human bronchial epithelial cells.22 It has previously been indicated that Wnt5a interacts with Ror2, which participates in the regulation of inflammatory signaling pathways.23 Wnt5a activates the translocation of NF-κB to the nucleus to promote the secretion of inflammatory cytokines.24,25 Some studies have shown that NF-κB is an important transcription factor, as it activates several inflammatory genes after PM stimulus.26 In our study, we also found that PM2.5 induced HBECs to upregulate Wnt5a, elevated Ror2 expression, and increased the amount of NF-κB in the nuclei. Reports have previously indicated that inhibition of Wnt5a reduced the increase of inflammatory cytokines produced in COPD.19 Our findings demonstrated that Wnt5a siRNA or BOX5 (a specific Wnt5a antagonist) alone significantly inhibited the PM2.5-induced release of IL-1β, IL-6 and IL-8. Wnt5a siRNA decreased Ror2 production, and suppressed the NF-κB activity, demonstrating that Wnt5a is predominantly participated in the elevated inflammatory cytokines IL-1β, IL-6 and IL-8 caused by PM2.5 in COPD.
In addition, to further test whether Ror2 is the important receptor involved in the Wnt5a-mediated inflammatory response in PM2.5-exposed HBECs, Knockdown of Ror2 to prevent the production of cytokines and activate NF-κB. Our current data demonstrated that Ror2 siRNA influenced NF-κB activation and the production of the inflammatory cytokines IL-1β, IL-6 and IL-8 after PM2.5 exposure, demonstrating that Ror2 plays an important role during the process of PM2.5-mediated airway inflammation. Recent studies also show that Bufei Huoxue Capsule reduces expression of inflammatory factors in PM2.5-induced mouse lungs and may via regulation of related signaling pathways (BCL2, STAT3, andP38).27 Based on these findings, we speculate that although the combined inhibition of Wnt5a and Ror2 will not lead to complete suppression of PM2.5 induced inflammatory responses, but Wnt5a/Ror2 pathway plays a critical role in PM2.5-induced airway inflammation.
Conclusion
In summary, our report demonstrates that PM2.5 induces the upregulation of Wnt5a that in turn elevates Ror2 expression, accompanies with positively regulating NF-κB activity, which promotes the production of IL-1β, IL-6 and IL-8 in HBECs. The present study provides evidence that Wnt5a/Ror2 signaling may be used as an effective therapeutic target for PM2.5-associated airway inflammation in COPD. Further in vivo manipulation of Wnt5a/Ror2 signaling is needed to characterize its role during airway inflammation.
Funding
This study was supported by the Natural Science Foundation of Guangdong, China (2020A1515010264 and 2017A030310419), Science and Technology Program of Guangzhou, China (202002030080), National Natural Science Foundation of China (81900044).Characteristic Innovation Projects of Universities in Guangdong Province (2019KTSCX139).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lancet T. Air pollution: a major threat to lung health. Lancet. 2019;393(10183):1774. doi:10.1016/S0140-6736(19)30992-4
2. Atkinson RW, Carey IM, Kent AJ, van Staa TP, Anderson HR, Cook DG. Long-term exposure to outdoor air pollution and the incidence of chronic obstructive pulmonary disease in a national English cohort. Occup Environ Med. 2015;72(1):42–48. doi:10.1136/oemed-2014-102266
3. Zhao B, Zheng H, Wang S, et al. Change in household fuels dominates the decrease in PM2.5 exposure and premature mortality in China in 2005–2015. Proc Natl Acad Sci U S A. 2018;115(49):12401–12406. doi:10.1073/pnas.1812955115
4. Schultz ES, Litonjua AA, Melen E. Effects of long-term exposure to traffic-related air pollution on lung function in children. Curr Allergy Asthma Rep. 2017;17(6):41. doi:10.1007/s11882-017-0709-y
5. Huang W, Wang G, Lu SE, et al. Inflammatory and oxidative stress responses of healthy young adults to changes in air quality during the Beijing olympics. Am J Respir Crit Care Med. 2012;186(11):1150–1159. doi:10.1164/rccm.201205-0850OC
6. Kopp J, Flessa S, Lieb W, et al. Association of PNPLA3 rs738409 and TM6SF2 rs58542926 with health services utilization in a population-based study. BMC Health Serv Res. 2016;16:41. doi:10.1186/s12913-016-1289-6
7. Baarsma HA, Skronska-Wasek W, Mutze K, et al. Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. J Exp Med. 2017;214(1):143–163. doi:10.1084/jem.20160675
8. Liao HD, Mao Y, Ying YG. The involvement of the laminin-integrin α7β1 signaling pathway in mechanical ventilation-induced pulmonary fibrosis. J Thorac Dis. 2017;9(10):3961–3972. doi:10.21037/jtd.2017.09.60
9. Feller D, Kun J, Ruzsics I, et al. Cigarette smoke-induced pulmonary inflammation becomes systemic by circulating extracellular vesicles containing Wnt5a and inflammatory cytokines. Front Immunol. 2018;9:1724. doi:10.3389/fimmu.2018.01724
10. Wang Y, Xu J, Meng Y, Adcock IM, Yao X. Role of inflammatory cells in airway remodeling in COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:3341–3348. doi:10.2147/COPD.S176122
11. He F, Liao B, Pu J, et al. Exposure to ambient particulate matter induced COPD in a rat model and a description of the underlying mechanism. Sci Rep. 2017;7:45666. doi:10.1038/srep45666
12. Zou W, He F, Liu S, et al. PM2.5 induced the expression of fibrogenic mediators via HMGB1-RAGE signaling in human airway epithelial cells. Can Respir J. 2018;2018:1817398. doi:10.1155/2018/1817398
13. Liu S, Zhou Y, Liu S, et al. Association between exposure to ambient particulate matter and chronic obstructive pulmonary disease: results from a cross-sectional study in China. Thorax. 2017;72(9):788–795. doi:10.1136/thoraxjnl-2016-208910
14. Zhao J, Li M, Wang Z, et al. Role of PM2.5 in the development and progression of COPD and its mechanisms. Respir Res. 2019;20(1):120. doi:10.1186/s12931-019-1081-3
15. Ezegbunam W, Foronjy R. Posttranscriptional control of airway inflammation. Wiley Interdiscip Rev RNA. 2018;9(1):e1455. doi:10.1002/wrna.1455
16. Chen Y, Thomas PS, Kumar RK, Herbert C. The role of noncoding RNAs in regulating epithelial responses in COPD. Am J Physiol Lung Cell Mol Physiol. 2018;315(2):L184–L92. doi:10.1152/ajplung.00063.2018
17. Catalan V, Gomez-Ambrosi J, Rodriguez A, et al. Activation of noncanonical Wnt signaling through WNT5A in visceral adipose tissue of obese subjects is related to inflammation. J Clin Endocrinol Metab. 2014;99(8):E1407–17. doi:10.1210/jc.2014-1191
18. Li C, Hu L, Xiao J, et al. Wnt5a regulates Shh and Fgf10 signaling during lung development. Dev Biol. 2005;287(1):86–97. doi:10.1016/j.ydbio.2005.08.035
19. Wang Z, Zhao J, Wang T, Du X, Xie J. Fine-particulate matter aggravates cigarette smoke extract-induced airway inflammation via Wnt5a-ERK pathway in COPD. Int J Chron Obstruct Pulmon Dis. 2019;14:979–994. doi:10.2147/COPD.S195794
20. Roper C, Chubb LG, Cambal L, et al. Association of IL-6 with PM2.5 components: importance of characterizing filter-based PM2.5 following extraction. Water Air Soil Pollut. 2017;228. doi:10.1007/s11270-016-3219-y
21. Ho HY, Susman MW, Bikoff JB, et al. Wnt5a-Ror-Dishevelled signaling constitutes a core developmental pathway that controls tissue morphogenesis. Proc Natl Acad Sci U S A. 2012;109(11):4044–4051. doi:10.1073/pnas.1200421109
22. Wang B, Tang Z, Gong H, Zhu L, Liu X. Wnt5a promotes epithelial-to-mesenchymal transition and metastasis in non-small-cell lung cancer. Biosci Rep. 2017;37(6). doi:10.1042/BSR20171092
23. Sato A, Kayama H, Shojima K, et al. The Wnt5a-Ror2 axis promotes the signaling circuit between interleukin-12 and interferon-gamma in colitis. Sci Rep. 2015;5:10536.
24. Barbero G, Castro MV, Villanueva MB, et al. An autocrine Wnt5a loop promotes NF-kappaB pathway activation and cytokine/chemokine secretion in melanoma. Cells. 2019;8(9):1060. doi:10.3390/cells8091060
25. Yu T, Dong D, Guan J, Sun J, Guo M, Wang Q. Alprostadil attenuates LPS-induced cardiomyocyte injury by inhibiting the Wnt5a/JNK/NF-kappaB pathway. Herz. 2019. doi:10.1007/s00059-019-4837-0
26. Kafoury RM, Kelley J. Ozone enhances diesel exhaust particles (DEP)-induced interleukin-8 (IL-8) gene expression in human airway epithelial cells through activation of nuclear factors- kappaB (NF-kappaB) and IL-6 (NF-IL6). Int J Environ Res Public Health. 2005;2(3–4):403–410. doi:10.3390/ijerph2005030004
27. Jing Y, Zhang H, Cai Z, et al. Bufei Huo Xue capsule attenuates PM2.5-induced pulmonary inflammation in mice. Evid Based Complement Alternat Med. 2017;2017:1575793. doi:10.1155/2017/1575793
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.