Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
PLGA-PLL-PEG-Tf-based targeted nanoparticles drug delivery system enhance antitumor efficacy via intrinsic apoptosis pathway
Authors Bao W, Liu R, Wang Y, Wang F, Xia G, Zhang H, Li X, Yin H, Chen B
Received 29 September 2014
Accepted for publication 18 November 2014
Published 12 January 2015 Volume 2015:10(1) Pages 557—566
DOI https://doi.org/10.2147/IJN.S75090
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Wen Bao,1 Ran Liu,1 Yonglu Wang,1,2 Fei Wang,1 Guohua Xia,1 Haijun Zhang,1 Xueming Li,2 Haixiang Yin,2 Baoan Chen1
1Department of Hematology and Oncology, Key Medical Disciplines of Jiangsu Province, Zhongda Hospital, Medical School of Southeast University, 2College of Pharmacy, Nanjing University of Technology, Nanjing, People’s Republic of China
Abstract: Chemotherapy offers a systemic cancer treatment; however, it is limited in clinical administration due to its serious side effects. In cancer medicine, the use of nanoparticles (NPs) drug delivery system (DDS) can sustainedly release anticancer drug at the specific site and reduce the incidence of toxicity in normal tissues. In the present study, we aimed to evaluate the benefit of a novel chemotherapeutic DDS and its underlying mechanisms. Daunorubicin (DNR) was loaded into poly (lactic-co-glycolic acid) (PLGA)-poly-l-lysine (PLL)-polyethylene glycol (PEG)-transferrin (Tf) NPs to construct DNR-PLGA-PLL-PEG-Tf-NPs (DNR-loaded NPs) as a DDS. After incubating with PLGA-PLL-PEG-Tf-NPs, DNR, and DNR-loaded NPs, the leukemia K562 cells were collected and the intracellular concentration of DNR was detected by flow cytometry, respectively. Furthermore, the effect of drugs on the growth of tumors in K562 xenografts was observed and the relevant toxicity of therapeutic drugs on organs was investigated in vivo. Meanwhile, cell apoptosis in the excised xenografts was measured by transferase-mediated dUTP nick-end labeling assay, and the expression of apoptosis-related proteins, including Bcl-2, Bax, Caspase-9, Caspase-3, and cleaved-PARP, was determined by Western blotting analysis. Results showed that DNR-loaded NPs increased intracellular concentration of DNR in K562 cells in vitro and induced a remarkable improvement in anticancer activity in the xenografts in vivo. The expression of Bcl-2 protein was downregulated and that of Bax, Caspase-9, Caspase-3, and cleaved-PARP proteins were obviously upregulated in the DNR-loaded NPs group than that in other ones. Interestingly, pathological assessment showed no apparent damage to the main organs. In summary, the results obtained from this study showed that the novel NPs DDS could improve the efficacy of DNR in the treatment of leukemia and induce apoptosis via intrinsic pathway. Thus, it can be inferred that the new drug delivery may be a useful clinical tool.
Keywords: PLGA-PLL-PEG, daunorubicin, transferrin, K562 cells, apoptosis
Materials and methods
Main materials
DNR was pursed from Main Luck Pharmaceuticals Inc (Shenzhen, People’s Republic of China), RPMI-1640 from Gibco Chemical Co. (Carlsbad, CA, USA), 10% heat-inactivated fetal bovine serum from Sigma-Aldrich Co (St Louis, MO, USA), Hematoxylin–Eosin Staining Kit from Beyotime Institute of Biotechnology (Shanghai, People’s Republic of China), In Situ Cell Death Detection Kit from Nanjing KeyGen Biotech Co Ltd (Nanjing, People’s Republic of China), and monoclonal for Western blotting from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). All other reagents were of analytical grade.
Preparation and characterization of DNR-loaded NPs
PLGA-PLL-PEG-NPs18 were provided by College of Pharmacy, Nanjing University of Technology. The conjugation was analyzed with 1H NMR, and then the PLGA-PLL-PEG-NPs-loaded DNR was prepared using a modified double-emulsion solvent evaporation/diffusion method19 and separated using ultracentrifuge (GL-20G-H; Shanghai Anting Scientific Instrument Factory, Shanghai, People’s Republic of China) for 30 minutes (4°C, 1,500 rpm). Then, PLGA-PLL-PEG-NPs-loaded DNR was well dissolved in phosphate buffered saline (PBS) using ultrasound treatment for 30 seconds and then Tf was added (Tf/PLGA-PLL-PEG molar ratio 8:1) with stirring at room temperature for 3 hours to conjugate with the surface of DNR-PLGA-PLL-PEG via CDI (N,N-carbonyldiimidazole) of PEG. Thereafter, DNR-PLGA-PLL-PEG-Tf-NPs (DNR-loaded NPs) were extracted using ultracentrifuge for 20 minutes (4°C, 1,500 rpm).
Mean particle size and ZP value of DNR-loaded NPs were determined by a Zetasizer 3000HS system (Malvern Instruments, Malvern, UK), the diameters and width of particle distribution were calculated using a volume distribution from the Gaussian distribution, and morphological evaluation was observed under transmission electron microscope (Hitachi Ltd, Tokyo, Japan). Meanwhile, drug loading efficiency was calculated as the percent ratio of actual amount of DNR encapsulated into PLGA-PLL-PEG-Tf-NPs to total amounts of DNR-PLGA-PLL-PEG-Tf-NPs. The drug entrapping efficiency of DNR was calculated as the actual amount of DNR encapsulated into PLGA-PLL-PEG-Tf-NPs vs the theoretical of DNR in DNR-PLGA-PLL-PEG-Tf-NPs, respectively.19
Cell line and cell culture
Leukemia K562 cells were obtained from Institute of Hematology, Chinese Academy of Medical Sciences (Beijing, People’s Republic of China). The cells were cultured in RPMI-1640 media supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified atmosphere containing 5% carbon dioxide. The cells used in experiments were in logarithmic growth phase.
Cellular accumulation of DNR
Briefly, K562 cells were incubated with PLGA-PLL-PEG-Tf-NPs, DNR, and DNR-loaded NPs with equivalent concentration of DNR for 48 hours. Meanwhile, those were exposed to RPMI-1640 medium as control. Then cells were harvested and washed three times with phosphate buffered saline (PBS). Thereafter, cells were suspended in 500 μL PBS, and the cellular accumulation of DNR in each sample was directly detected by FACSCalibur flow cytometry (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) at excitation/emission wavelengths of 488/575 nm. The relative fluorescence intensity (RFI) of DNR was calculated as FItreated group/FIcontrol group.
Animals
Six-week age BALB/c nude mice, weighing 18–22 g, from the Shanghai National Center for Laboratory Animals were housed in cages under specific pathogen-free conditions where temperature was maintained at 22°C and humidity was in the range of 40%–50%, and sterile water and food were fed. All experiments involving the mice were carried out following the Guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health.
Experimental design and animal treatments
Mice were subcutaneously inoculated into the left thigh with an injection of 1×107 K562 cells suspended in 0.2 mL of RPMI-1640 medium. Tumor sizes were measured with a digital caliper, and the tumor volumes were calculated according to the following formula: tumor volume V (mm3) =(1/2)×a×b2, where a is the largest diameter (length) and b is the smallest diameter (width) of the tumor. When tumors reached an average volume of approximately 50 mm3, animals were divided into four groups with six animals each for treatment as follows: Group 1, saline water (control); Group 2, PLGA-PLL-PEG-Tf-NPs; Group 3, free DNR (1 mg/kg for the single injection); and Group 4, DNR-loaded NPs (containing 1 mg/kg DNR for the single injection).
The intravenous treatments were done once every other day for 9 times for 18 days, and antitumor activity was determined by calculating the tumor volume as described earlier twice a week before sacrifice. After intravenous treatment for 18 days, mice were sacrificed and their xenograft tumors were harvested and processed for histopathologic analysis and determination of protein expression. During the whole experiment, the trial animals were monitored for signs of toxicity.
Histopathologic examination
After the mice were sacrificed, the lung, heart, liver, kidney, and spleen were quickly removed and immediately fixed in 4% paraformaldehyde, dehydrated in a graded series of alcohol, and then embedded in paraffin. Tissue sections (4 μm) were prepared and stained with hematoxylin and eosin. Thereafter, the sections were examined and microphotographed by using Olympus IX51 microscope (400×; Olympus Corporation, Tokyo, Japan).
Transferase-mediated dUTP nick-end labeling
Based on the detection of single- and double-stranded DNA breaks by the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay, analysis of apoptosis was performed using an In Situ Cell Death Detection Kit according to the manufacturer’s instructions. Tissue sections were analyzed to detect the localized green fluorescence of apoptotic cells and blue fluorescence of cell nuclei by fluorescence microscope; fluorescent images were acquired and photographed under the Olympus IX51 microscope (400×).
Western blot assays
In brief, the tissues were extracted with Triton X-100, and then the proteins were subjected to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride membrane. The membrane was blocked with buffer containing 5% skim milk. Thereafter, Western blotting was performed with the primary antibodies for Bcl-2, Bax, c-PARP, Caspase-3, Caspase-9, and GAPDH overnight at 4°C; and then with horseradish peroxidase-conjugated secondary antibody for 2 hours at room temperature. The protein bands were detected by enhanced chemiluminescence (ECL system; Amersham, UK) and analyzed by Gel-Pro32 software.
Statistical analysis
All experiments were performed three times. The results were presented as mean ± standard deviation, and data were analyzed with SPSS software (version 13.0; SPSS Inc, Chicago, IL, USA). The statistical significance of differences was determined by one-way analysis of variance among multiple groups, and differences with P<0.05 were considered statistically significant.
Results
Characterization of DNR-loaded NPs
DNR-loaded NPs had a spherical shape and dispersed uniformly under transmission electron microscope (Figure 1A); the mean size and ZP of DNR-loaded NPs were 176.4±11.0 nm (Figure 1B Size distribution by intensity) and −19.24±0.12 mV, respectively. Figure 2 shows the schematic representation of DNR loading into NPs. The drug entrapping efficiency was 75.12%±1.68%, and the loading efficiency was 5.32%±0.15%. All these results demonstrated that DNR-loaded NPs were relatively homogenous in size and stably stored.
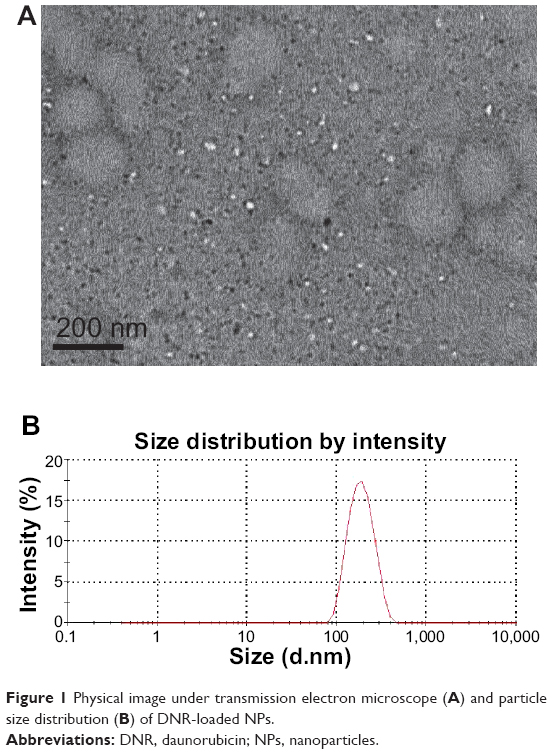 | Figure 1 Physical image under transmission electron microscope (A) and particle size distribution (B) of DNR-loaded NPs. |
 | Figure 2 Schematic representation of DNR loaded nanoparticles. |
Intracellular DNR
The relative fluorescence intensity of intracellular DNR (FItreated group/FIcontrol group) in K562 cells was lower in DNR group than that in DNR-loaded NPs (equivalent DNR concentration) group (8.14±0.29 vs 14.26±0.39) (Figure 3A), and there was a significant difference between them (P<0.05, Figure 3B). Otherwise, there was no significant difference between the PLGA-PLL-PEG-Tf-NPs group and the control groups (P>0.05, Figure 3B). These results suggested that DNR modified by PLGA-PLL-PEG-Tf-NPs could be targeted successfully to leukemia cells.
 | Figure 3 Intracellular accumulation of DNR in K562 cells incubated for 48 hours by FCM assay (A). Intracellular relative fluorescence intensity associated with DNR (B). |
Drug inhibits the growth of tumor tissue
After intravenous treatment for 18 days, the mean tumor volume was 1,099±238 mm3 for the saline water group, 1,034±178 mm3 for the PLGA-PLL-PEG-Tf-NPs group, 536±137 mm3 for the DNR group, and 237±85 mm3 for the DNR-loaded NPs group (Figure 4); and the inhibition of tumor growth in the DNR or DNR-loaded NPs groups was significantly higher than that in the PLGA-PLL-PEG-Tf-NPs group and the control group (P<0.05). Interestingly, the mean tumor volume in the DNR-loaded NPs group was smaller than that in the DNR group (P<0.05), while there was no significant difference between the PLGA-PLL-PEG-Tf-NPs group and control group (P>0.05). These results were further demonstrated by gross-observation (Figure 5A). Moreover, the weight of the tumors from the animals treated with either DNR or DNR-loaded NPs decreased significantly when compared with the PLGA-PLL-PEG-Tf-NPs group and control groups (P<0.05; Figure 5B). Thus, we inferred that the DNR-loaded NPs had a greater antitumor efficacy than DNR (P<0.05).
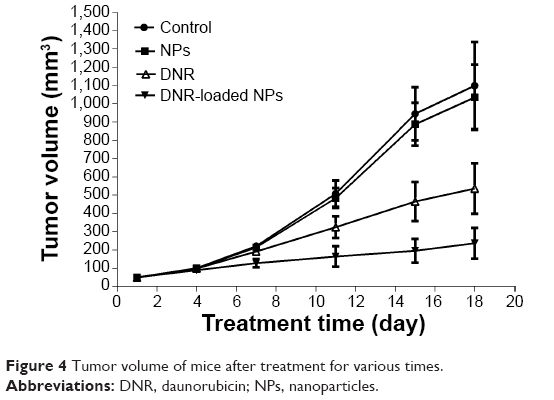 | Figure 4 Tumor volume of mice after treatment for various times. |
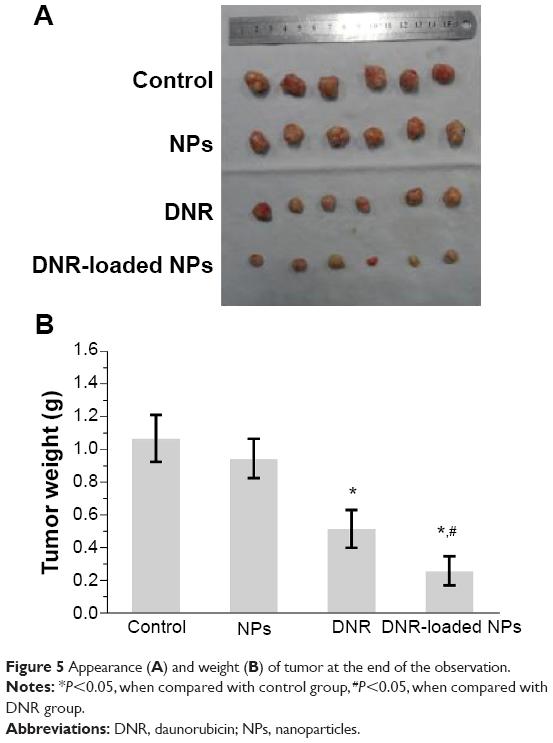 | Figure 5 Appearance (A) and weight (B) of tumor at the end of the observation. |
Toxicity in vivo
All the mice were alive, and they were weighed during the experiment period of 18 days. It is noteworthy that no significant differences in body weight (P>0.05; Figure 6) and no abnormal findings in any group were observed.
 | Figure 6 The body weight of mice pre- and post-treatment for various times. |
Next, we carried out histological bioanalysis of organs to evaluate the potential side effects of NPs on the main organs of mice in vivo. There were no apparent histopathologic changes in the tissues, including lung, heart, liver, kidney, and spleen (Figure 7). These results indicate that DNR-loaded NPs are safe and have no obvious toxic effects on the main organs of mice.
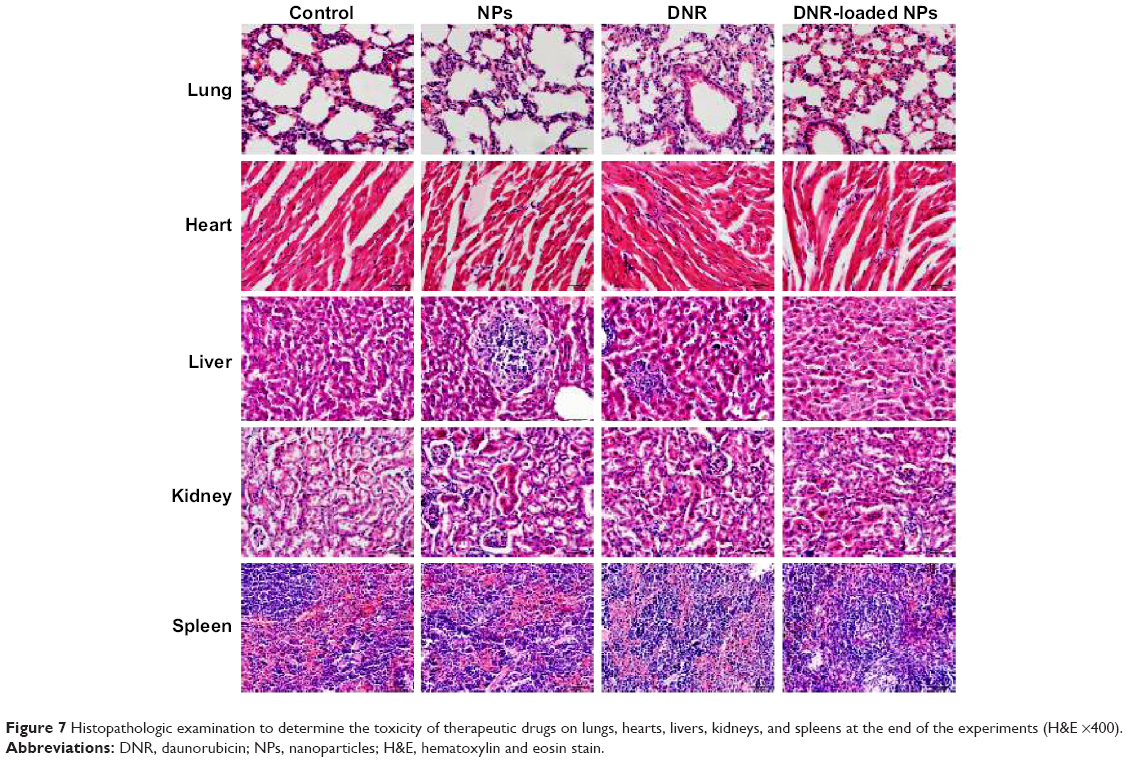 | Figure 7 Histopathologic examination to determine the toxicity of therapeutic drugs on lungs, hearts, livers, kidneys, and spleens at the end of the experiments (H&E ×400). |
Analysis of cell apoptosis in K562 xenograft tumors
Samples were analyzed to detect the localized green fluorescence of apoptotic cells and blue fluorescence of cell nuclei using fluorescence microscope (Figure 8A). The percentage of apoptotic cells compared to the average number of cells per field was determined and graphically represented (Figure 8B). The apoptotic rate in DNR-loaded NPs group increased significantly compared with that in DNR group (P<0.05). However, there was no significant difference between the PLGA-PLL-PEG-Tf-NPs group and the control group (P>0.05), indicating that DNR-loaded NPs are a more effective drug treatment to achieve apoptosis than DNR (P<0.05).
 | Figure 8 Apoptotic cells in K562 xenograft tumors. |
Expression of Bcl-2, Bax, cleaved-PARP, Caspase-3, and Caspase-9
To explore the possible signaling pathways through which DNR-loaded NPs induced more anticancer activity, we examined the changes in the expression levels of apoptosis-related proteins by Western blotting. In our study, the levels of apoptosis regulating proteins Bcl-2 in both DNR group and DNR-loaded NPs group were downregulated while that of Bax was upregulated compared with the control group (P<0.05). In addition, the expression of Bcl-2 and Bax proteins in DNR-loaded NPs group was more obviously regulated than that in DNR group (P<0.05). The expression of Caspase-9 was upregulated in the DNR or DNR-loaded NPs group compared with the control group (P<0.05), and the increase was much more dramatic in the DNR-loaded NPs group than that in the DNR group (P<0.05; Figure 9). Similar results for Caspase-3 and cleaved-PARP were detected. Notably, the protein expression in the PLGA-PLL-PEG-Tf-NPs group was not obviously changed when compared with the control group (P>0.05).
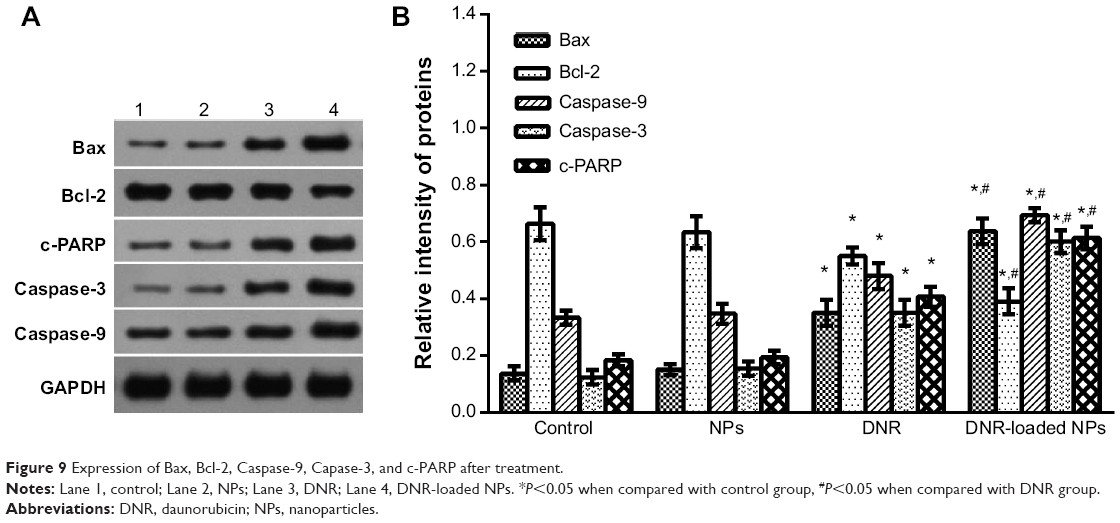 | Figure 9 Expression of Bax, Bcl-2, Caspase-9, Capase-3, and c-PARP after treatment. |
Discussion
Current cancer treatments are restricted to chemotherapy, radiation, and surgery. Radiotherapy and surgery are largely localized in nature and fail to remove occult metastatic disease that can lead to eventual recurrence and poor patient survival. Nowadays, chemotherapy, particularly with respect to drug delivery, has evolved from traditional methodology.20 DDS is an intriguing field of research that has captured the interest of researchers. Many studies focus on the safe and effective chemotherapeutic DDS, which can directly target tumor cells while sparing normal cells.1 Targeted DDS can convey drugs more effectively and conveniently than chemotherapy in the past.21
As we know, nanotechnology has provided a wide field of research among chemistry, biology, medicine, material science, engineering, and mathematics. These innovations are referred to as nanomedicines by the National Institutes of Health and have a significant impact on drug delivery. NPs as carrier can reduce the cytotoxic side effects and achieve targeting chemotherapy.22,23 We had previously evaluated the application of NPs and found that it would provide a promising strategy for chemotherapy.24
Tfs are a family of homologous iron-binding glycoproteins. A growing number of studies have demonstrated that use of the Tf uptake pathway is highly effective in cancer therapy.25 The TfR is a cell membrane-associated glycoprotein involved in the cellular uptake of iron and in the regulation of cell growth.26 Many studies have shown that the level of TfR expressed in malignant cells is many times higher than their normal counterparts. Its expression can be correlated with tumor stage or cancer progression.26,27 Because of the elevated levels of TfR in malignancies, it can be targeted by directly conjugating with its ligand Tf or by monoclonal antibodies specific for the extracellular domain. Compared with free drugs, the combination of Tf targeting was highly effective in increasing levels of DNA double-stranded breaks and apoptosis in leukemia cells without accumulation of conjugate in normal cells, retaining the conjugate for longer periods of time, and declining the rate of chemotherapeutic agent efflux.28 In an in vivo experiment, artemisinin-Tf conjugate demonstrated its ability to significantly retard the growth of breast cancer tumors in a rat model.29 Tf-based DDS for cancer therapy has been tested to produce tumor responses without systemic toxicity in patients.15 On the other hand, a possible limitation of Tf-based therapeutics is the short period that Tf remains within the cells to deliver its payload. Recently, nano-based drug delivery has been demonstrated to enhance the circulation time of targeted therapeutic drug.15 In accordance with these findings, we aim to explore whether the targeted delivery of a therapeutic drug DNR-loaded NPs can improve therapeutic benefit in leukemia. Currently, 100–200 nm is accepted as the optimal size for DDS because nanocarriers take advantage of the EPR effect in tumors and avoid filtration in the spleen.3 In the present study, spherical DNR-loaded NPs with diameters of 176.4±11.0 nm could be produced with a remarkably high DNR entrapping efficiency (75.12%±1.68%) and loading efficiency (5.32%±0.15%), which is suitable for drug delivery.
Drug targeting can be classified into two types: passive and active targeting. PEG-modification of NPs is useful to passively deliver the NPs to cancer tissues via an enhanced permeability and retention, avoiding the reticuloendothelial system.30,31 Active targeting allows the increased accumulation of drug in cancer tissue through specific interactions, such as antigen–antibody and ligand–receptor binding. Endocytosis plays a major role in active targeting.15 The formulation of DNR-PEG-PLGA-PLL-Tf-NPs in our study was based on either passive or active processes. In vitro, we found that DNR-loaded NPs showed higher DNR accumulation than the DNR alone (Figure 3). In vivo, the tumor volume of the mice injected with DNR-loaded NPs was smaller than that of the mice treated with DNR, and PLGA-PLL-PEG-Tf-NPs without DNR did not obviously affect the tumor volume in mice (Figures 4 and 5). These results indicated that DNR-loaded NPs showed more significant antitumor activity, and the intravenous delivery of DNR using NPs amplified the DNR antitumor that possibly elicited systemic targeting in leukemia. We infer that DNR-loaded NPs can be directly delivered into tumor cells by receptor-mediated endocytosis and drug-sustained release from NPs surface with the NPs skeleton degradation. Thereby, DNR-loaded NPs may render longer half-life of DNR in blood, and DNR concentration must be increased in the tumor tissue. Besides, there were no apparent histopathologic damages to the main organs of mice due to the size of the NPs.32 Based on mice body weights, the tolerability of the NPs formulation was acceptable. These results may advocate the feasibility of reducing the dose and intensifying the tumor-tissue targeting ability of DNR.
Apoptosis is a physiological process of programmed cell death that is essential for normal tissue development, and resistance to apoptosis is a hallmark feature of cancer; therefore, apoptosis induction of cancer cells is one of the most important and direct ways to control the development of cancer and eliminate tumors. In our present study, extensive cell apoptosis by TUNEL assay was observed in tumor tissues of xenografts treated with DNR-loaded NPs compared to the control one. Studies have shown that apoptosis can be triggered via two distinct pathways: the extrinsic and the intrinsic or mitochondria pathway.33 Once cellular stress signals arise, the intrinsic pathway is activated and then causes permeabilization of mitochondrial outer membrane, which allows Caspase-9-mediated mitochondrial damage, thereby activating the downstream member Caspase-3.34 To further explore the possible apoptosis pathways through which DNR-loaded NPs induced targeted chemotherapy, we assayed the expressions of some key apoptosis-associated proteins, including Caspase-3, Caspase-9, and c-PARP. In our study, the level of Caspase-9, Caspase-3, and c-PARP proteins was remarkably elevated in DNR-loaded NPs group compared to other groups. As we know, one of the most important functions of PARP is to help repair single-stranded DNA nicks, and the cleaved-PARP is critically involved in the intrinsic apoptosis pathway and considered to be a marker of apoptosis.35,36 In the present study, we observed the expression of proapoptotic and antiapoptotic members of the Bcl-2 family, which is the mitochondrial-related death switch. The members of Bcl-2 homology are divided into three main groups based on function and regions of the domains: multi-domain antiapoptotic proteins (Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and A1), multi-domain proapoptotic proteins (Bax and Bak), and BH3-only proapoptotic proteins (Bid, PUMA, Bim, and NOXA), trigger the activation of Bak and Bax.37 Our present results showed that the level of Bcl-2 protein was downregulated by the treatment of DNR-loaded NPs, compared to DNR group, whereas that of Bax was upregulated, and the ratio of Bax to Bcl-2 was increased. Taken together, these data indicate that DNR-loaded NPs can activate the intrinsic signaling pathway to trigger leukemia cells apoptosis.
Conclusion
In summary, the results obtained from the current study demonstrate that the novel NPs DDS enhances the activity of traditional anticancer drug DNR against leukemia cells through activation of the intrinsic apoptosis by both active and passive targeting pathways. This would ensure improved efficacy and minimize toxicity. Thus, novel NPs DDS may be a useful clinical tool.
Acknowledgments
This work was supported by the Jiangsu Province Health Department Medical Science and Technology Development Fund Project (No H201229), the National Natural Science Foundation of China (Nos 81170492, 81370673), the National High-Tech Research and Development Program 863 (No 2012AA022703), the National Key Basic Research Program 973 (No 2010CB732404), the National Innovative Research Program for College Students (No 1210286098), and Key Department of Jiangsu Medicine (No 2011-12).
Disclosure
The authors report no conflicts of interest in this work.
References
Sakhrani NM, Padh H. Organelle targeting: third level of drug targeting. Drug Des Devel Ther. 2013;7:585–599. | ||
Parveen S, Misra R, Sahoo SK. Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine. 2012;8:147–166. | ||
Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615–627. | ||
Davis ME, Zuckerman JE, Choi CH, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. | ||
Woodrow KA, Cu Y, Booth CJ, Saucier-Sawyer JK, Wood MJ, Saltzman WM. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat Mater. 2009;8:526–533. | ||
Zhang J, Sun H, Ma PX. Host-guest interaction mediated polymeric assemblies: multifunctional nanoparticles for drug and gene delivery. ACS Nano. 2010;4:1049–1059. | ||
Geels P, Eisenhauer E, Bezjak A, et al. Palliative effect of chemotherapy: objective tumor response is associated with symptom improvement in patients with metastatic breast cancer. J Clin Oncol. 2000; 18:2395–2405. | ||
Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63:170–183. | ||
Umano M, Uechi K, Uriuda T, et al. Tumor accumulation of epsilon-poly-lysines-based polyamines conjugated with boron clusters. Appl Radiat Isot. 2011;69:1765–1767. | ||
Tahara K, Furukawa S, Yamamoto H, Kawashima Y. Hybrid-modified poly(d,l-lactide-co-glycolide) nanospheres for a novel cellular drug delivery system. Int J Pharm. 2010;392:311–313. | ||
Duncanson WJ, Figa MA, Hallock K, Zalipsky S, Hamilton JA, Wong JY. Targeted binding of PLA microparticles with lipid-PEG-tethered ligands. Biomaterials. 2007;28:4991–4999. | ||
Gref R, Minamitake Y, Peracchia MT, et al. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263:1600–1603. | ||
Ryschich E, Huszty G, Knaebel HP, Hartel M, Büchler MW, Schmidt J. Transferrin receptor is a marker of malignant phenotype in human pancreatic cancer and in neuroendocrine carcinoma of the pancreas. Eur J Cancer. 2004;40:1418–1422. | ||
Sinha R, Kim GJ, Nie S, Shin DM. Nanotechnology in cancer therapeutics: bioconjugated nanoparticles for drug delivery. Mol Cancer Ther. 2006;5:1909–1917. | ||
Tortorella S, Karagiannis TC. Transferrin receptor-mediated endocytosis: a useful target for cancer therapy. J Membr Biol. 2014;247:291–307. | ||
Tortorella S, Karagiannis TC. The significance of transferrin receptors in oncology: the development of functional nano-based drug delivery systems. Curr Drug Deliv. 2014;11:427–443. | ||
Petre CE, Dittmer DP. Liposomal daunorubicin as treatment for Kaposi’s sarcoma. Int J Nanomedicine. 2007;2:277–288. | ||
Guo LT, Chen BA, Liu R, et al. Biocompatibility assessment of polyethylene glycol-poly l-lysine-poly lactic-co-glycolic acid (PEG-PLL-PLGA) nanoparticles in vitro and in vivo. J Nanosci Nanotechnol. 2014;14:1–10. | ||
Liu J, Qiu Z, Wang S, Zhou L, Zhang S. A modified double-emulsion method for the preparation of daunorubicin-loaded polymeric nanoparticle with enhanced in vitro anti-tumor activity. Biomed Mater. 2010; 5:065002. | ||
Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol. 2008;26:57–64. | ||
Sanna V, Pala N, Sechi M. Targeted therapy using nanotechnology: focus on cancer. Int J Nanomedicine. 2014;9:467–483. | ||
Sharma A, Jain N, Sareen R. Nanocarriers for diagnosis and targeting of breast cancer. Biomed Res Int. 2013;2013:960821. | ||
Kim BY, Rutka JT, Chan WC. Nanomedicine. N Engl J Med. 2010;363: 2434–2443. | ||
Zhang H, Wang C, Chen B, Wang X. Daunorubicin-TiO2 nanocomposites as a “smart” pH-responsive drug delivery system. Int J Nanomedicine. 2012;7:235–242. | ||
Brandsma ME, Jevnikar AM, Ma S. Recombinant human transferrin: beyond iron binding and transport. Biotechnol Adv. 2011; 29:230–238. | ||
Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121:144–158. | ||
Singh M, Mugler K, Hailoo DW, et al. Differential expression of transferrin receptor (TfR) in a spectrum of normal to malignant breast tissues: implications for in situ and invasive carcinoma. Appl Immunohistochem Mol Morphol. 2011;19:417–423. | ||
Szwed M, Matusiak A, Laroche-Clary A, Robert J, Marszalek I, Jozwiak Z. Transferrin as a drug carrier: cytotoxicity, cellular uptake and transport kinetics of doxorubicin transferrin conjugate in the human leukemia cells. Toxicol In Vitro. 2014;28:187–197. | ||
Lai H, Nakase I, Lacoste E, Singh NP, Sasaki T. Artemisinin-transferrin conjugate retards growth of breast tumors in the rat. Anticancer Res. 2009;29:3807–3810. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136–151. | ||
Fumoto S, Kawakami S. Combination of nanoparticles with physical stimuli toward cancer therapy. Biol Pharm Bull. 2014;37:212–216. | ||
Kettiger H, Schipanski A, Wick P, Huwyler J. Engineered nanomaterial uptake and tissue distribution: from cell to organism. Int J Nanomedicine. 2013;8:3255–3269. | ||
Packham G, Stevenson FK. Bodyguards and assassins: Bcl-2 family proteins and apoptosis control in chronic lymphocytic leukaemia. Immunology. 2005;114:441–449. | ||
Elkholi R, Floros KV, Chipuk JE. The role of BH3-only proteins in tumor cell development, signaling, and treatment. Genes Cancer. 2011;2: 523–537. | ||
Agarwal A, Mahfouz RZ, Sharma RK, Sarkar O, Mangrola D, Mathur PP. Potential biological role of poly (ADP-ribose) polymerase (PARP) in male gametes. Reprod Biol Endocrinol. 2009;7:143. | ||
Danial NN. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin Cancer Res. 2007;13:7254–7263. | ||
Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435: 677–681. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.