")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
PLGA-encapsulated tea polyphenols enhance the chemotherapeutic efficacy of cisplatin against human cancer cells and mice bearing Ehrlich ascites carcinoma
Authors Singh M, Bhatnagar P, Mishra S, Kumar P , Shukla Y, Gupta KC
Received 18 December 2014
Accepted for publication 18 March 2015
Published 30 October 2015 Volume 2015:10(1) Pages 6789—6809
DOI https://doi.org/10.2147/IJN.S79489
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
This paper has been retracted
Madhulika Singh,1 Priyanka Bhatnagar,2 Sanjay Mishra,1 Pradeep Kumar,2 Yogeshwer Shukla,1 Kailash Chand Gupta1,2
1CSIR-Indian Institute of Toxicology Research, Lucknow, 2CSIR-Institute of Genomics and Integrative Biology, Delhi University Campus, Delhi, India
Abstract: The clinical success of the applicability of tea polyphenols awaits efficient systemic delivery and bioavailability. Herein, following the concept of nanochemoprevention, which uses nanotechnology for enhancing the efficacy of chemotherapeutic drugs, we employed tea polyphenols, namely theaflavin (TF) and epigallocatechin-3-gallate (EGCG) encapsulated in a biodegradable nanoparticulate formulation based on poly(lactide-co-glycolide) (PLGA) with approximately 26% and 18% encapsulation efficiency, respectively. It was observed that TF/EGCG encapsulated PLGA nanoparticles (NPs) offered an up to ~7-fold dose advantage when compared with bulk TF/EGCG in terms of exerting its antiproliferative effects and also enhanced the anticancer potential of cisplatin (CDDP) in A549 (lung carcinoma), HeLa (cervical carcinoma), and THP-1 (acute monocytic leukemia) cells. Cell cycle analysis revealed that TF/EGCG-NPs were more efficient than bulk TF/EGCG in sensitizing A549 cells to CDDP-induced apoptosis, with a dose advantage of up to 20-fold. Further, TF/EGCG-NPs, alone or in combination with CDDP, were more effective in inhibiting NF-κB activation and in suppressing the expression of cyclin D1, matrix metalloproteinase-9, and vascular endothelial growth factor, involved in cell proliferation, metastasis, and angiogenesis, respectively. EGCG and TF-NPs were also found to be more effective than bulk TF/EGCG in inducing the cleavage of caspase-3 and caspase-9 and Bax/Bcl2 ratio in favor of apoptosis. Further, in vivo evaluation of these NPs in combination with CDDP showed an increase in life span (P<0.05) in mice bearing Ehrlich’s ascites carcinoma cells, with apparent regression of tumor volume in comparison with mice treated with bulk doses with CDDP. These results indicate that EGCG and TF-NPs have superior cancer chemosensitization activity when compared with bulk TF/EGCG.
Keywords: anticancer, poly(lactide-co-glycolide), nanoparticles, cisplatin, tea polyphenols
Introduction
Cisplatin (cis-diamminedichloroplatinum (II), CDDP, molecular weight 300.04 Da), is one of the most commonly used anticancer drugs, and is used extensively in the treatment of many cancers. CDDP needs to be administered at high doses to obtain the required therapeutic response, thereby leading to severe adverse effects.1 Low aqueous solubility, high protein binding that reduces drug potency, systemic toxicity, and inherent/or acquired resistance have been reported as major disadvantages of CDDP in cancer therapy.2,3 Therefore, finding novel ways of lowering the dosage needed without loss of efficacy is very important. To this end, anticancer drugs are now frequently being combined with dietary phytochemicals in an effort to enhance their antitumor efficacy while lowering their toxic effects.4,5
Tea (Camellia sinensis) leaves contain polyphenolic compounds, in particular catechins, which are potent antioxidants and have biological activity relevant to the prevention and treatment of cancer. Catechins found in both black tea and green tea have been shown to suppress several key pathways linked to oncogenesis, including those involved in cell survival, proliferation, and invasion, along with angiogenesis.6–10 Earlier reports from our laboratory have established that tea polyphenols have excellent potential to chemosensitize cancer cells towards chemotherapeutic drugs like CDDP.4,11 Further, combined treatment with tea polyphenols and CDDP synergistically induced apoptosis by activation of caspase-8 and caspase-9 and overexpression of p53.4,12 In spite of promising preclinical findings, the applicability of tea in human cancer therapy is still not up to expectations, seemingly due to lack of efficient systemic delivery and bioavailability.13
Technological advancements have brought about innovative drug delivery systems, which are being assessed in the area of cancer therapeutics and management.14 Numerous nanomedical approaches to drug delivery are focused on developing nanoscale particles to improve drug bioavailability.15 The structure and tunable surface functionality of nanoparticles (NPs) allow them to encapsulate/conjugate entities either in their core or on their surface, rendering them ideal carriers for anticancer drugs.16 Polymer-drug conjugates, macromolecular prodrugs, which were first developed in the mid-1970s, have laid the foundation for new types of anticancer entities.17 Among them, poly(lactide-co-glycolide) (PLGA, 50:50 approved by the US Food and Drug Administration) has been used in the preparation of a wide variety of drug delivery systems, because it is biodegradable, biocompatible, capable of controlling the release of the incorporated entity at specific target sites and shows low toxicity.18,19 Moreover, these colloidal systems are able to extravasate solid tumors, in which the capillary endothelium is defective.20,21
We have devised an approach for loading bioactive catechins from black tea (theaflavin, TF) and green tea (epigallocatechin-3-gallate, EGCG) in a PLGA carrier so that their uptake, retention, and cytotoxicity in cancer cells can be enhanced in vitro and in vivo. We followed a PLGA drug development strategy to encapsulate TF and EGCG, anticipating that NP-mediated delivery of anticancer phytochemicals could be useful for enhancing the anticancer potency of chemotherapeutic drugs like CDDP and concomitantly limiting drug toxicity.
Materials and methods
Chemicals
PLGA 50:50 (molecular weight 40–75 kDa), polyvinyl alcohol (molecular weight 30 kDa), 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT), TF, EGCG, CDDP, propidium iodide, and β-actin (clone AC-74) were purchased from Sigma-Aldrich (St Louis, MO, USA). Caspase-3, caspase-9, cytochrome C, p-NF-κB, p-IκBα, p53, and Bcl-2 antibodies were sourced from Cell Signaling Technology (Beverly, MA, USA) while Bax antibody was obtained from Santa Cruz Biotechnology Inc (Santa Cruz, CA, USA). The rabbit anti-mouse and goat anti-rabbit horseradish peroxidase-conjugated secondary antibodies were obtained from Bangalore Genei (Bangalore, India). The polyvinylidene fluoride membrane was obtained from Millipore (Bedford, MA, USA). Caspase-3 inhibitor z-DEVD-fmk was purchased from Calbiochem (Boston, MA, USA). Fetal bovine serum, Dulbecco’s Modified Eagle’s Medium, and Roswell Park Memorial Institute medium were supplied by Invitrogen (Invitrogen, Carlsbad, CA, USA) and the antibiotics by Gibco (Lifetech, Karlsruche, Germany). 2′,7′-dichlorofluorescein diacetate (DCF-DA) and rhodamine 123 from BD Pharmingen (San Diego, CA, USA) were used. Other chemicals used were of analytical grade and sourced locally.
Preparation of TF/EGCG-loaded PLGA-NPs
PLGA-NPs loaded with TF and EGCG were prepared using the standard solvent evaporation method.22 Briefly, for the preparation of EGCG-encapsulated PLGA-NPs, the drug to polymer ratio was kept at 1:5. EGCG (20 mg) was dissolved in deionized water (0.5 mL), and emulsified with a solution of PLGA (100 mg) dissolved in dichloromethane (2 mL) in an ice bath on a magnetic stirrer operating at 1,600 rpm. The resulting w/o primary emulsion was stirred for 2 hours at room temperature and then added dropwise to a rapidly stirred aqueous solution of polyvinyl alcohol (1%, 4 mL, w/v) to obtain a w/o/w secondary emulsion. Stirring was continued until the organic solvent evaporated off. The solution was then subjected to centrifugation at 13,000 rpm for complete removal of the organic solvent and hardening of the NPs. The NPs obtained were suspended in water and again centrifuged. The process was repeated three times, and finally, the NPs were freeze-dried to obtain a solid dry powder. Likewise, TF-encapsulated PLGA-NPs were prepared and stored at 4°C under anhydrous conditions until used for further studies.
Characterization of NPs
Particle size and zeta potential measurements
The mean particle size and distribution as well as the zeta potential of the TF/EGCG-loaded NPs (1 mg/mL in H2O) were determined by a dynamic light scattering technique using a Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK) employing a 5 mW He-Ne laser operating at a 633 nm wavelength. Three measurements per sample were carried out, with an average of 14 runs per measurement; refractive index and viscosity of water were kept at 1.33 and 0.89 cP, respectively. The particle size reported is the average of three samples. Zeta potential measurements were carried out on the same instrument in triplicate using automatic mode, and the values are presented as the average value of 30 runs. The Smoluchowski approximation was used to calculate the zeta potential from the electrophoretic mobility.23
Determination of percentage yield
The dried solid NPs were weighed and the percent yield was calculated using the following equation:
|
|

Drug loading and encapsulation efficiency
Drug loading and encapsulation efficiency was determined by spectrophotometric estimation using an ultraviolet-visible spectrophotometer (Lambda Bio 20, Perkin Elmer, Boston, MA, USA). Briefly, an accurately weighed sample of drug-loaded NPs (~10–15 mg) were suspended in acetonitrile (1 mL), and after 10 minutes the suspension was centrifuged at 13,000 rpm for 30 minutes. The supernatant was removed, the pellet was resuspended in acetonitrile (1 mL), and then the process was repeated. The pellet was dried and dissolved in water (1 mL), and the absorbance of the solution was measured at 273 nm. The amount of drug (mg) was calculated from the standard curve drawn for a varying amount of drug (mg) and absorbance (optical density). Drug loading and encapsulation efficiency were determined from the following equation. All the measurements were conducted in triplicate.
|
|

In vitro release of TF/EGCG from PLGA-NPs
TF/EGCG-loaded NPs were evaluated for their in vitro release kinetics using the dialysis method, as reported previously by Asadishad et al.24 A weighed amount of TF/ECGC-encapsulated PLGA-NPs (~10 mg) was dispersed in 100 mM phosphate-buffered saline (PBS; pH 7.4, 1 mL), after which the solution was transferred to a dialysis bag (molecular weight cut-off 12 kDa) and allowed to dialyze against 15 mL of the same buffer at 37°C±0.5°C with stirring at 50 rpm in a shaking incubator (Unimax 1010, Heidolph, Schwabach, Germany). At predetermined time intervals, a 1 mL aliquot of the sample was withdrawn, its absorbance was measured at 273 nm, and the same amount of fresh medium was added to the dialysis container. The amount of drug released was then calculated using a previously prepared standard curve for the pure drug.
Colloidal stability of TF/EGCG-loaded NPs
The stability of TF/EGCG-loaded NPs was examined by dynamic light scattering in 100 mM phosphate buffer (pH 7.4) at 4°C by monitoring changes in the hydrodynamic diameter of the NPs during storage. At various time points, aliquots of the sample solution were withdrawn and the distribution of particle size was determined. This experiment was carried out three times.
In vitro studies
Cell lines
The cell lines, ie, A549 (human lung adenocarcinoma epithelial cells), HeLa (human epithelial cervical cancer cells), and THP-1 (human acute monocytic leukemia cells) were obtained from the National Centre for Cell Science (Pune, India). The HeLa and A549 cells were maintained in Dulbecco’s Modified Eagle’s Medium and the THP-1 cells were maintained in Roswell Park Memorial Institute medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Storage and culture of cell lines for this research did not require a license or ethical review, as cell lines are not “relevant material” according to the Human Tissue Act. Therefore, ethical approval was not needed.
Cell proliferation assay
The cytotoxic effects of different doses were determined by the MTT uptake method as described by us elsewhere.6 The IC50 value was determined from a plot of percentage cell survival versus drug concentration, where cells with no treatment were considered 100% viable.
Apoptosis assay
The effect of the treatments on the cell cycle distribution was determined by analysis of DNA content of cells following staining with propidium iodide. After treatment, the cells were washed with PBS and fixed in 70% ethanol overnight at 4°C. The cells were then treated with DNase-free RNaseA and propidium iodide (50 μg/mL) for 30 minutes in the dark at 4°C, and analyzed by flow cytometry (BD-LSR).6 A total of 10,000 events were analyzed.
Measurement of intracellular ROS production
For microscopic and flow cytometric detection of formation of reactive oxygen species (ROS), cells were grown on 6-well plates. At the end of treatment for 6, 12, 18, and 24 hours, the cells were incubated with DCF-DA (10 μM) for 30 minutes in the dark (37°C). After incubation, the cells were kept on ice and immediately fluorescence was measured with a fluorescence microscope (IX51, Olympus, Tokyo, Japan) and by flow cytometry.
Measurement of mitochondrial membrane potential
The untreated and treated cells (24 hours) were incubated with rhodamine 123 (5 μg/mL) for 30 minutes in the dark at 37°C, harvested, and resuspended in PBS.6 The mitochondrial membrane potential (MMP, ΔΨm) was measured using flow cytometry by the fluorescence intensity (FL-1, 530 nm) of 10,000 cells and the results are shown as mean fluorescence intensity. This analysis was performed using Cell Quest software.
Western blot analysis
A549 cells that received no treatment and cells that were treated with bulk TF/EGCG or TF/EGCG-loaded NPs were washed with PBS, resuspended in ice-cold lysis buffer (50 mM Tris-HCl, 150 nM NaCl, 1 mM EGTA, 1 mM EDTA, 20 mM NaF, 100 mM Na3VO4, 0.5% NP-40, 1% Triton X-100, 1 mM PMSF, 10 μg/mL aprotinin, 10 μg/mL leupeptin, pH 7.4) at 4°C for 30 minutes and then centrifuged at 12,000 g for 10 minutes at 4°C to obtain total cell lysates which were stored at −80°C until further use.25 Total nuclear/cytosolic extracts were prepared and their protein concentration was estimated. The cytosolic fraction (for caspase-3, caspase-9, cytochrome C, p-IκBα, p53, Bcl-2, Bax, and β-actin) and the nuclear fraction (for NF-κB) were used. For Western blotting, proteins (50 μg) were resolved on 10% sodium dodecyl sulfate–polyacrylamide gels followed by electrotransfer onto an immobile polyvinylidene difluoride membrane. The blots were blocked overnight with 5% non-fat dry milk, probed with the respective primary antibodies, then detected by horseradish peroxidase-conjugated anti-rabbit or anti-mouse immunoglobulin G using a chemiluminescence kit from Millipore, and visualized using a Versa Doc MP imaging system (Model 4000, BioRad, Hercules, CA, USA). The intensity was given in terms of relative pixel density for each band normalized to band of β-actin. The intensity of the bands was measured using UNSCAN-IT automated digital system version 5.1 software (Orem, UT, USA).
Reverse transcription polymerase chain reaction analysis
Untreated and treated A549 cells were lysed in TRIzol reagent after 24 hours in culture, and total RNA was prepared. First, 1 μg of total RNA was reverse-transcribed to cDNA according to the manufacturer’s instructions using the Fermentas kit, and 20 μL of polymerase chain reaction (PCR) mixture (containing 4 μL of 5× reaction buffer, 2 μL of dNTP mixture [10 mM], 20 units of RNase inhibitor, 200 units of avian-myeloblastosis virus reverse transcriptase enzyme, and 0.5 μg of oligo [dT] primer) was amplified. After amplification, the PCR products were resolved on 1.5% agarose gel. The PCR products were visualized as bands using an IS1000 image analysis system (Alpha Innotech, San Leandro, CA, USA). Negative controls without cDNA were also run. The ratio of the target gene to GAPDH gene band density was used for quantitative evaluation. Oligonucleotide sequences and PCR conditions are described in Table S1.
In vivo studies in mice bearing Ehrlich’s ascites carcinoma tumors
Swiss albino mice (20–25 g body weight) were obtained from the animal breeding colony at our institution and kept under standard experimental conditions (temperature 23°C±2°C, relative humidity 55%±5%). Prior ethical approval for the experiment was obtained from the institutional ethics committee. Ehrlich’s ascites carcinoma (EAC) cells were maintained as an ascitic tumor model in Swiss albino mice. In brief, tumors were induced by injecting EAC cells (1×106) into the intraperitoneal cavity of each mouse; this was taken as day 0 and treatments were started 24 hours after transplantation. The animals were randomly divided into 14 groups consisting of five animals each. Normal saline (0.9% w/v, 100 μL/mouse/day) was administered to a normal group (group 1) and an EAC-bearing positive control group (group 2). TF/EGCG (100 μg/mouse intraperitoneally twice in one week), CDDP, and different doses of TF/EGCG-loaded PLGA-NPs (10 and 20 μg/mouse) with or without CDDP were administered to mice in groups 3–13 for 15 days. CDDP alone (100 μg/mouse) was given intraperitoneally on days 1, 7, and 14 (total three doses)26 and in the combination treatment groups a 1/10th dose (10 μg/mouse) was given. Animals in all groups were examined daily for formation of ascites by measuring changes in body weight. The day of death and change in body weight were recorded. Mean survival time and percentage increase in the life span (% ILS) were also recorded for all untreated and treated EAC tumor-bearing mice.
|
|

where A is the animal weight on any particular day, and B is the animal weight on day 0. Mean survival time was calculated as follows:
Mean survival time:
|
|
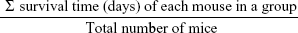
|
|

where T is the number of days the treated animals survived and C is the number of days the control animals survived.
Isolation of EAC from the peritoneal cavity
Two weeks after implantation of the EAC cells, EAC tumors were isolated from the peritoneal cavity of each mouse. In brief, 2–3 mL of sterile normal saline was injected into the peritoneal cavity and peritoneal fluid containing the tumor cells was withdrawn, collected in sterile Petri dishes, and incubated at 37°C for 2–3 hours. Cells of macrophage lineage adhered to the bottom of the Petri dishes. The nonadherent population was gently aspirated out, washed repeatedly with phosphate-buffered saline, and further processed. For the cell viability test, EAC cells (1×106) were harvested from all groups, washed repeatedly in PBS, and mixed with 5 μL of 0.5% trypan blue solution. Percent viability was determined using a cell counter chamber (Invitrogen).
Statistical analysis
The data are presented as the mean ± standard deviation of three independent experiments. For the in vitro studies, statistically significant differences were determined using the Student’s t-test. For the animal studies, the data analysis was done by one-way analysis of variance, followed by post hoc Dunnett test. A value of P<0.05 was considered to be statistically significant.
Results
Characteristics of TF/EGCG-loaded PLGA-NPs
TF/EGCG was encapsulated into PLGA-NPs using the standard double emulsion solvent evaporation method.27 The resulting NPs were washed, lyophilized, and evaluated for their percentage yield, size, shape, morphology, and surface charge. The NPs loaded with TF/EGCG were found to be 215 nm and 239 nm in size, respectively, with an excellent particle size distribution. The percentage yield of the TF-PLGA NPs and EGCG-PLGA NPs was found to be 79.2%±5.45% and 96.9%±4.16%, respectively. As expected, the zeta potential of these particles was found to be negative due to the negatively charged PLGA (Table 1).
 | Table 1 Characteristics of TF/EGCG-loaded PLGA nanoparticles |
In vitro release of TF/EGCG-loaded NPs
The PLGA-NP drug release kinetics were carried out in PBS at 37°C (Figure 1). The release profiles revealed that ~32% of the total ECGC load was released in the first 6 hours, whereas 23% of the total TF load was released in the first 3 hours, indicating an initial burst release. This was followed by a lag phase until 27 hours for EGCG-loaded NPs and 48 hours for TF-loaded NPs, until sufficient polymer erosion had taken place,28 and then by a secondary burst release, with about 90% of EGCG and 70% of TF released from the loaded NPs in 10 and 9 days, respectively.
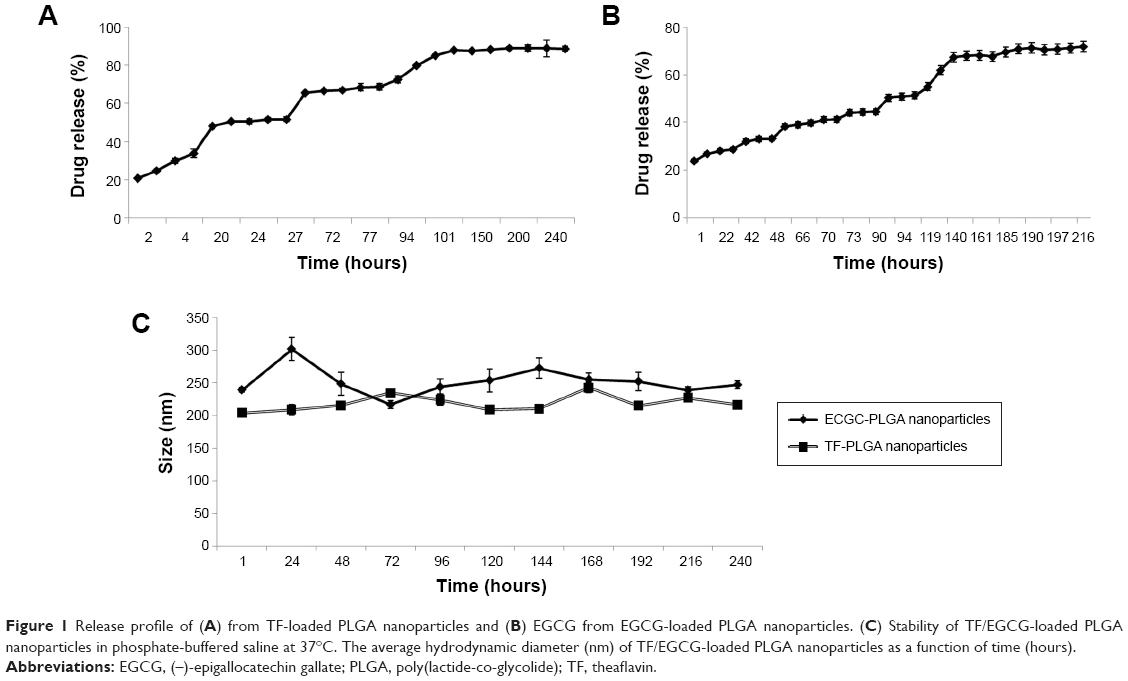 | Figure 1 Release profile of (A) from TF-loaded PLGA nanoparticles and (B) EGCG from EGCG-loaded PLGA nanoparticles. (C) Stability of TF/EGCG-loaded PLGA nanoparticles in phosphate-buffered saline at 37°C. The average hydrodynamic diameter (nm) of TF/EGCG-loaded PLGA nanoparticles as a function of time (hours). |
Kinetic stability of TF/EGCG-loaded NPs
Increasing the stability of drug-loaded NPs has been shown to improve their blood circulation time.29 The kinetic stabilty in a biological medium is an essential property for a drug nanocarrier. Changes in the hydrodynamic diameter of the TF/EGCG-loaded PLGA-NPs in PBS (pH 7.4) at 4°C were monitored by dynamic light scattering, and it was found that the NP size remained fairly stable after 10 days, indicating satisfactory stability (Figure 1). Therefore, TF/EGCG-loaded PLGA-NPs appear to be promising vehicles for drug delivery.
Antiproliferative activity of TF/EGCG-loaded NPs
The ability of both bulk TF/EGCG and TF/EGCG-loaded PLGA-NPs to inhibit proliferation of HeLa, A549, and THP-1 cells was investigated at 24 hours. TF-loaded PLGA-NPs significantly inhibited the proliferation of the three types of cells in a dose-dependent manner with an IC50 value of 6 μM (bulk IC50 45 μM) in A549 cells, 7 μM (bulk IC50 30 μM) in HeLa cells, and 15 μM (bulk IC50 65 μM) in THP-1 cells, suggesting that TF-NPs are more potent than bulk TF (Figures S1 and S2). EGCG-loaded PLGA-NPs inhibited cell proliferation, with an IC50 dose of 9 μM in A549 cells, 12 μM in HeLa cells, and 27 μM in THP-1 cells, while bulk IC50 doses of EGCG were 60 μM, 55 μM, and 93 μM, respectively (Figure S2). Thus, it appears that TF/EGCG-loaded NPs could allow a ~3–7-fold reduction in IC50 doses when compared with bulk doses of polyphenols (P<0.05). Treatment of the cells with PLGA-NPs alone had no significant effect, thereby confirming their lack of toxicity (P>0.05).
Antiproliferative activity of CDDP enhanced by TF/EGCG-loaded NPs
The cytotoxicity of CDDP doses was also evaluated, and the IC50 concentration of CDDP was 15 μM in A549 cells, 9 μM in HeLa cells, and 20 μM in THP-1 cells at 24 hours (Figure 2A). However, doses of TF/EGCG-loaded PLGA-NPs that were 1/5, 1/10, and 1/20th the dose of bulk TF/EGCG IC50 doses were found to significantly enhance the cytotoxicity of low doses of CDDP (IC20) by up to ~60% (Figure 2B–D, P<0.05). In further mechanistic studies, doses of bulk TF/EGCG and TF/EGCG-loaded PLGA-NPs (1/5, 1/10 and 1/20th of the TF/EGCG IC50 dose) were combined with a respective IC20 dose of CDDP (ie, 5 μM in A549 cells, 4 μM in HeLa cells, and 7 μM in THP-1 cells) for each cell type.
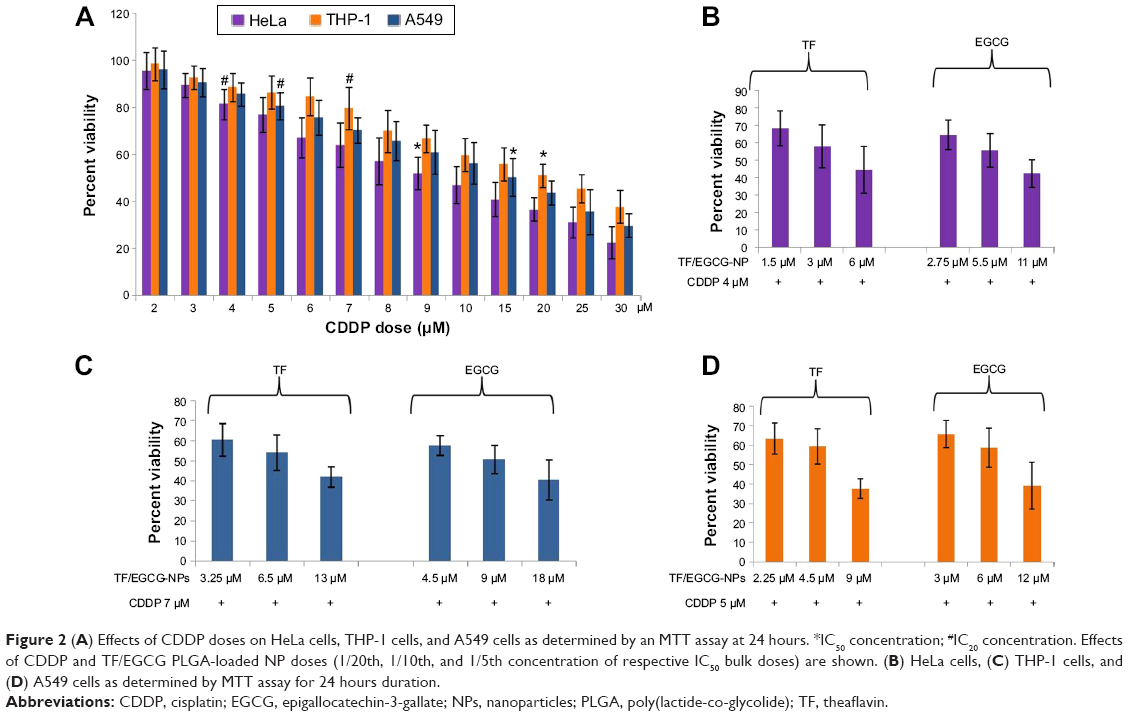 | Figure 2 (A) Effects of CDDP doses on HeLa cells, THP-1 cells, and A549 cells as determined by an MTT assay at 24 hours. *IC50 concentration; #IC20 concentration. Effects of CDDP and TF/EGCG PLGA-loaded NP doses (1/20th, 1/10th, and 1/5th concentration of respective IC50 bulk doses) are shown. (B) HeLa cells, (C) THP-1 cells, and (D) A549 cells as determined by MTT assay for 24 hours duration. |
Apoptosis-inducing potential of CDDP enhanced by TF/EGCG-loaded PLGA-NPs
The apoptosis-inducing potential of TF/EGCG and TF/EGCG-loaded PLGA-NPs alone and in combination with CDDP was determined by quantifying the percentage of cells with sub-G0/G1 DNA content. Exposure of HeLa, A549, and THP-1 cells to a combination of doses of tea-loaded PLGA-NPs and CDDP for 24 hours resulted in a statistically significant increase in the number of cells with subdiploid DNA content as compared with cells treated with TF/EGCG, TF/EGCG-loaded PLGA-NPs, or CDDP alone (Figure 3; P<0.05). A significant increase in sub-G1 phase was noted in cells treated with a combination of TF/EGCG-loaded PLGA-NPs and CDDP, with a 20-fold dose advantage over the IC50 dose of bulk TF/EGCG (P<0.05). A concentration-dependent increase in the sub-G1 population was also noted in cells treated with combinations of TF/EGCG-loaded PLGA-NPs and CDDP (Figure 3; P<0.05).
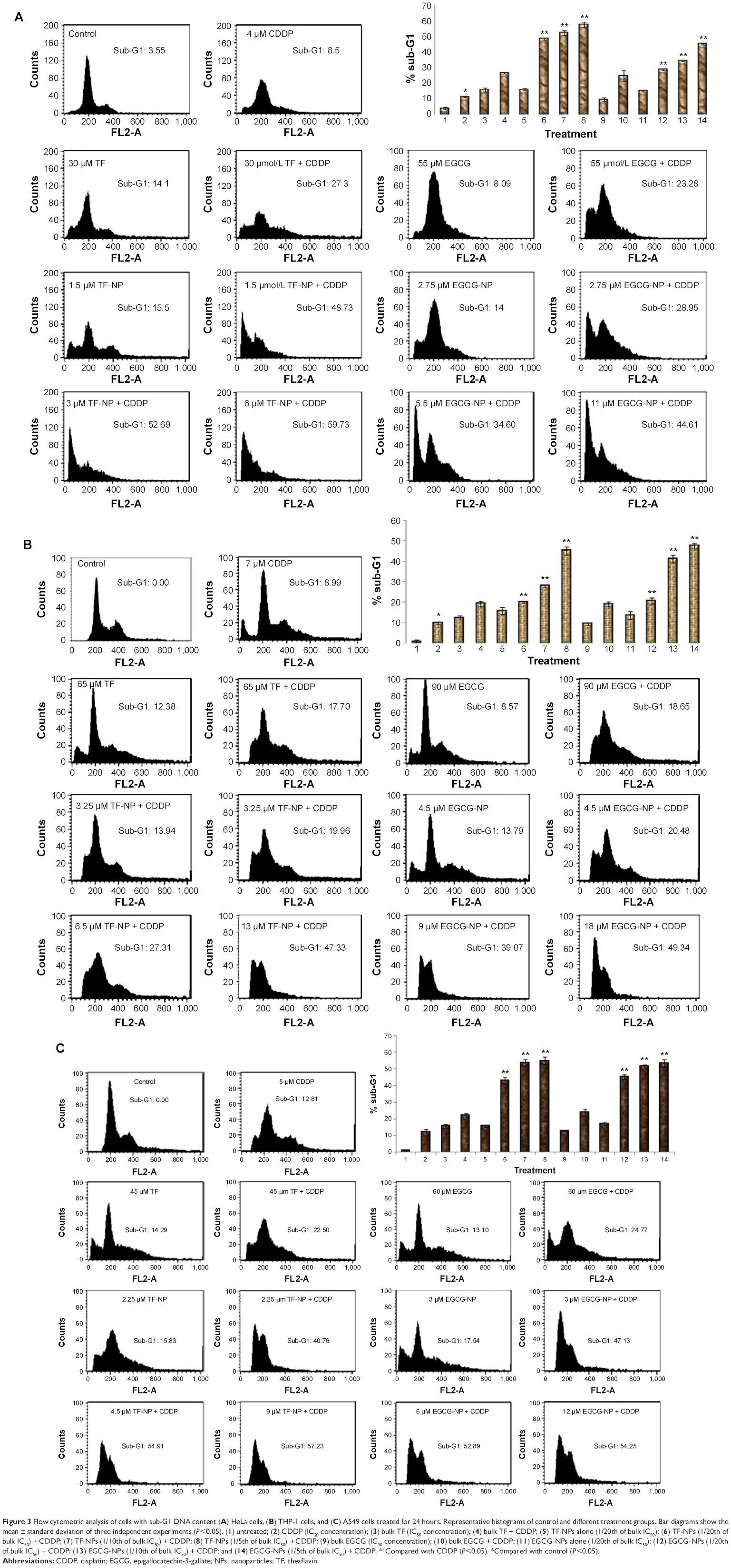 | Figure 3 Flow cytometric analysis of cells with sub-G1 DNA content (A) HeLa cells, (B) THP-1 cells, and (C) A549 cells treated for 24 hours. Representative histograms of control and different treatment groups. Bar diagrams show the mean ± standard deviation of three independent experiments (P<0.05). (1) untreated; (2) CDDP (IC20 concentration); (3) bulk TF (IC50 concentration); (4) bulk TF + CDDP; (5) TF-NPs alone (1/20th of bulk IC50); (6) TF-NPs (1/20th of bulk IC50) + CDDP; (7) TF-NPs (1/10th of bulk IC50) + CDDP; (8) TF-NPs (1/5th of bulk IC50) + CDDP; (9) bulk EGCG (IC50 concentration); (10) bulk EGCG + CDDP; (11) EGCG-NPs alone (1/20th of bulk IC50); (12) EGCG-NPs (1/20th of bulk IC50) + CDDP; (13) EGCG-NPs (1/10th of bulk IC50) + CDDP; and (14) EGCG-NPs (1/5th of bulk IC50) + CDDP. **Compared with CDDP (P<0.05). *Compared with control (P<0.05). |
ROS generation potential of CDDP enhanced by TF/EGCG-loaded PLGA-NPs in A549 cells
In the presence of intracellular hydrogen peroxide and related peroxides, non-fluorescent DCF-DA is hydrolyzed to DCFH to give highly fluorescent DCF-DA.30 This process is used to measure the rate of intracellular ROS generation as a part of the mechanism by which tea polyphenol-loaded PLGA-NPs induce apoptosis in cancer cells.4 Increased DCF fluorescence intensity was noted in cells treated with combinations of CDDP and TF/EGCG-loaded PLGA-NPs as compared with untreated cells and CDDP, TF/EGCG alone treated cells (Figure 4; P<0.05). Further, it was noted that tea polyphenol-loaded PLGA-NPs were much more efficient in generating ROS than their respective bulk forms (Figure 4C; P<0.05).
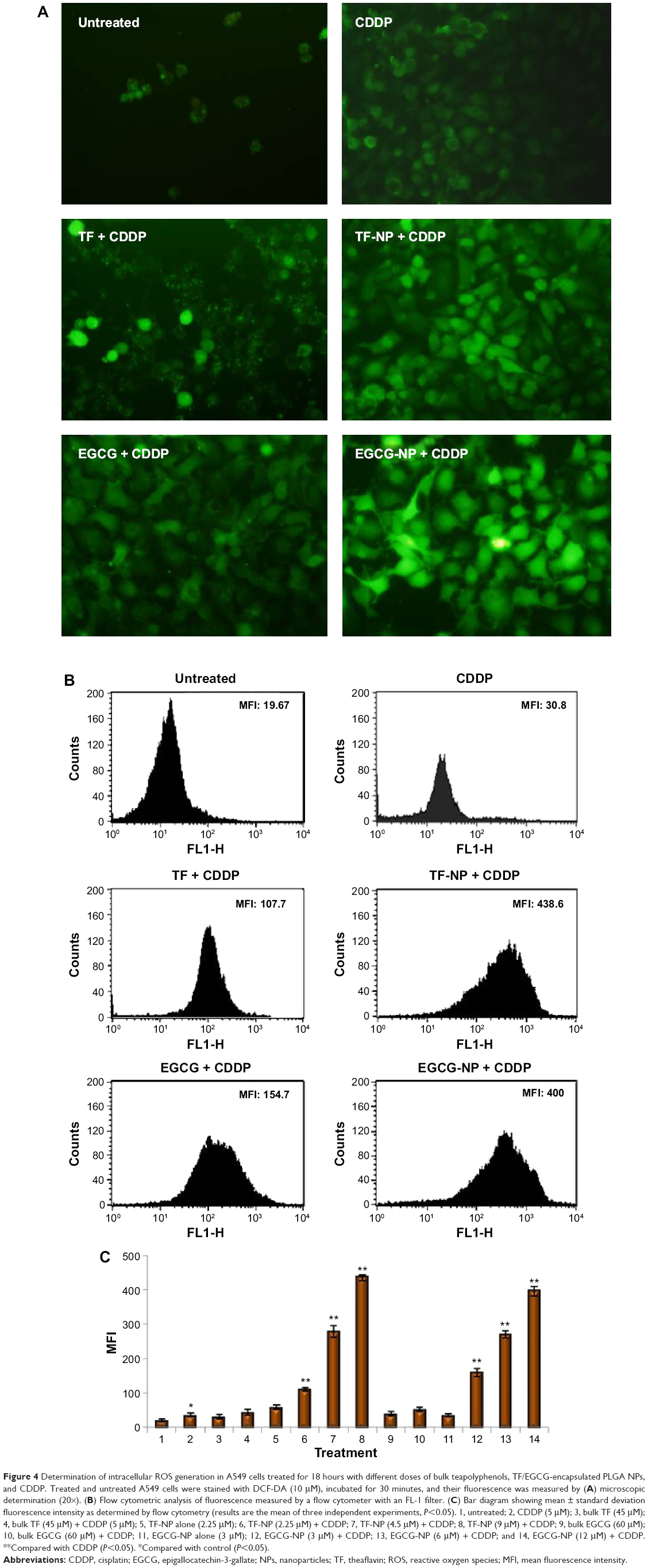 | Figure 4 Determination of intracellular ROS generation in A549 cells treated for 18 hours with different doses of bulk teapolyphenols, TF/EGCG-encapsulated PLGA NPs, and CDDP. Treated and untreated A549 cells were stained with DCF-DA (10 μM), incubated for 30 minutes, and their fluorescence was measured by (A) microscopic determination (20×). (B) Flow cytometric analysis of fluorescence measured by a flow cytometer with an FL-1 filter. (C) Bar diagram showing mean ± standard deviation fluorescence intensity as determined by flow cytometry (results are the mean of three independent experiments, P<0.05). 1, untreated; 2, CDDP (5 μM); 3, bulk TF (45 μM); 4, bulk TF (45 μM) + CDDP (5 μM); 5, TF-NP alone (2.25 μM); 6, TF-NP (2.25 μM) + CDDP; 7, TF-NP (4.5 μM) + CDDP; 8, TF-NP (9 μM) + CDDP; 9, bulk EGCG (60 μM); 10, bulk EGCG (60 μM) + CDDP; 11, EGCG-NP alone (3 μM); 12, EGCG-NP (3 μM) + CDDP; 13, EGCG-NP (6 μM) + CDDP; and 14, EGCG-NP (12 μM) + CDDP. **Compared with CDDP (P<0.05). *Compared with control (P<0.05). |
Enhancement of CDDP-induced mitochondrial damage by TF/EGCG-loaded PLGA-NPs in A549 cells
Functional changes can occur in the mitochondria due to changes in the MMP (ΔΨm). Thus, to clarify the possible role of the MMP in TF/EGCG-loaded PLGA-NP-induced apoptosis, ΔΨm was evaluated using rhodamine 123. A significant decrease in MMP was observed after treatment with TF/EGCG-loaded PLGA-NPs alone as well as in combination with CDDP in comparison with untreated cells and cells treated with CDDP alone (Figure 5; P<0.05). Additionally, PLGA-NPs treated with tea polyphenols were found to be more able to enhance the CDDP-induced decrease in ΔΨm than bulk and CDDP combination (Figure 5B).
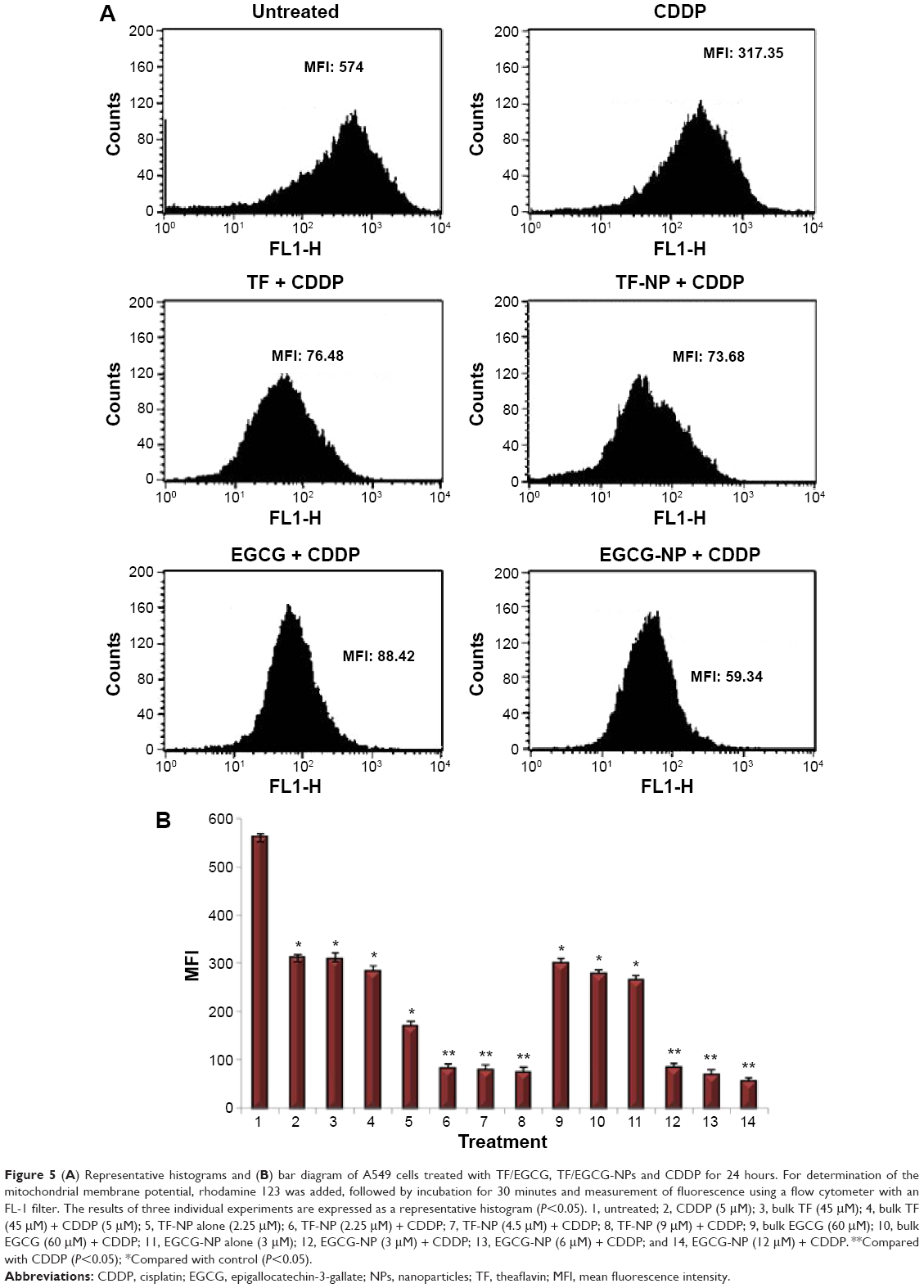 | Figure 5 (A) Representative histograms and (B) bar diagram of A549 cells treated with TF/EGCG, TF/EGCG-NPs and CDDP for 24 hours. For determination of the mitochondrial membrane potential, rhodamine 123 was added, followed by incubation for 30 minutes and measurement of fluorescence using a flow cytometer with an FL-1 filter. The results of three individual experiments are expressed as a representative histogram (P<0.05). 1, untreated; 2, CDDP (5 μM); 3, bulk TF (45 μM); 4, bulk TF (45 μM) + CDDP (5 μM); 5, TF-NP alone (2.25 μM); 6, TF-NP (2.25 μM) + CDDP; 7, TF-NP (4.5 μM) + CDDP; 8, TF-NP (9 μM) + CDDP; 9, bulk EGCG (60 μM); 10, bulk EGCG (60 μM) + CDDP; 11, EGCG-NP alone (3 μM); 12, EGCG-NP (3 μM) + CDDP; 13, EGCG-NP (6 μM) + CDDP; and 14, EGCG-NP (12 μM) + CDDP. **Compared with CDDP (P<0.05); *Compared with control (P<0.05). |
Enhancement of CDDP-induced apoptosis by TF/EGCG-loaded PLGA-NPs via mitochondrial pathway in A549 cells
The Bax and bcl-2 genes play an important role in regulation of apoptosis.30 Treatment of A549 cells for 24 hours with doses of TF, EGCG, and CDDP each administered alone independently achieved an increase in Bax protein levels and a decrease in Bcl-2 protein levels, and these effects were further amplified by combined treatment with TF/EGCG-loaded PLGA-NPs and CDDP (Figure S3; P<0.05). Tea polyphenol-loaded PLGA-NPs were found to be more efficient in enhancing the CDDP-induced changes than the bulk forms. Further, the ratio of Bax to Bcl-2 was also increased after treatment in a dose-dependent manner, indicating that apoptosis was occurring (Figure 3D).
Release of cytochrome c from the mitochondria into the cytosol is a key event in mitochondria-dependent apoptosis, and Bax has been reported to induce cytochrome c release and subsequent activation of caspase-9.31–33 The results of our study show that there was a marked increase in release of cytochrome c in the A549 cells upon exposure to TF, EGCG and CDDP alone (Figure 3). Additionally, increases in caspase-9 (37 kDa) and caspase-3 (17 kDa), corresponding to the active forms, were also detected in A549 cells following the alone doses of each compound, but not in untreated cells (Figure S3). CDDP-induced expression of cytochrome c, caspase-9 and caspase-3 was further enhanced by doses of tea-loaded PLGA-NPs as compared to bulk doses of tea polyphenols (Figure 3; P<0.05).
In compliance with these annotations, the apoptosis-inducing effects of TF/EGCG-NPs in A549 cells were also significantly attenuated upon treatment with z-DEVD-fmk, confirming the involvement of caspase-3 in cell death mediated by TF/EGCG-loaded PLGA-NPs (Figure 3B).
TF/EGCG-loaded PLGA-NPs enhance CDDP-induced apoptosis by suppression of NF-kB
Activation of NF-κB is linked to inflammation and proliferation of tumor cells and has been shown to suppress apoptosis induced by chemotherapeutic agents like CDDP.34 CDDP is reported to enhance the activity of NF-κB,35 and tea polyphenols are reported to suppress the action of NF-κB.7 The test concentration of CDDP was found to enhance activation of NF-κB in a time-dependent manner (data not shown). To determine whether being loaded into PLGA-NPs enhances the ability of TF/EGCG to suppress CDDP-induced NF-κB, the cells were incubated with different concentrations of bulk TF/EGCG or TF/EGCG-loaded NPs for 24 hours alone or in combination with a selected dose of CDDP. Neither TF/EGCG nor TF/EGCG-NPs alone activated NF-κB (data not shown), but both significantly abolished CDDP-induced NF-κB activation in a concentration-dependent manner (Figure 6A).
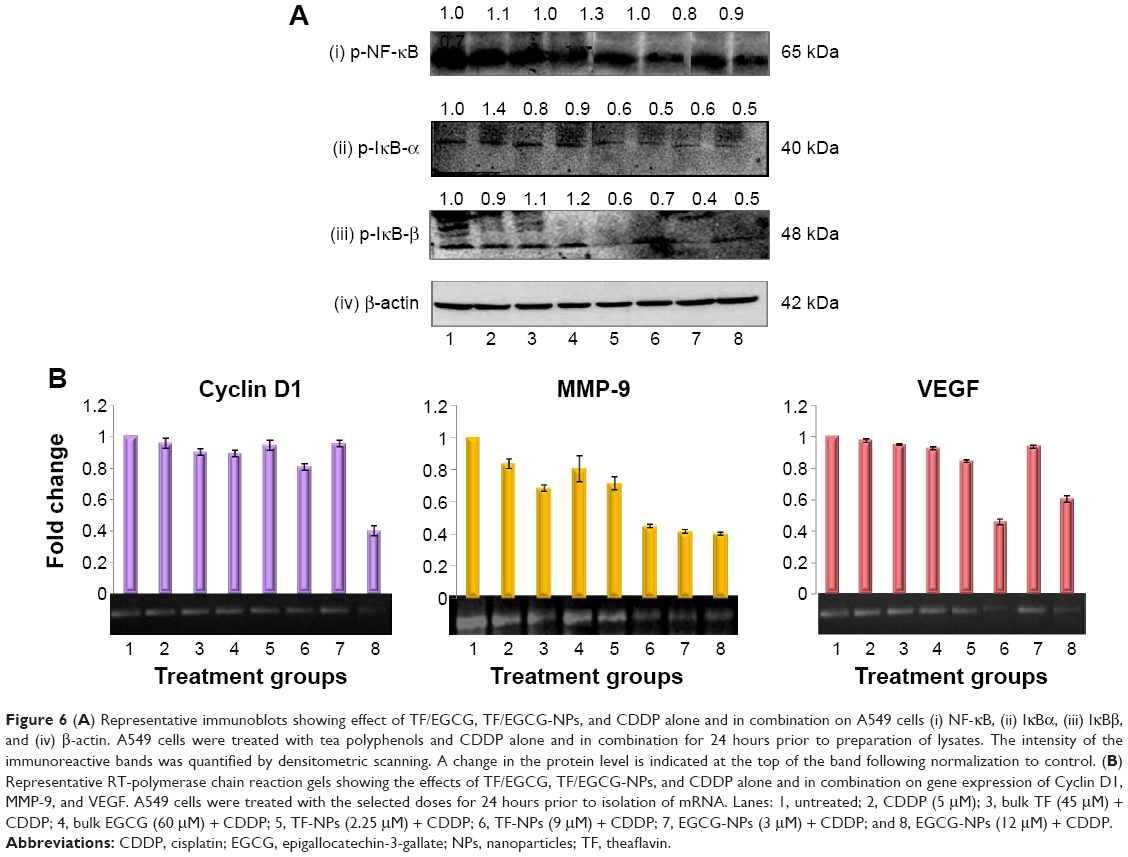 | Figure 6 (A) Representative immunoblots showing effect of TF/EGCG, TF/EGCG-NPs, and CDDP alone and in combination on A549 cells (i) NF-κB, (ii) IκBα, (iii) IκBβ, and (iv) β-actin. A549 cells were treated with tea polyphenols and CDDP alone and in combination for 24 hours prior to preparation of lysates. The intensity of the immunoreactive bands was quantified by densitometric scanning. A change in the protein level is indicated at the top of the band following normalization to control. (B) Representative RT-polymerase chain reaction gels showing the effects of TF/EGCG, TF/EGCG-NPs, and CDDP alone and in combination on gene expression of Cyclin D1, MMP-9, and VEGF. A549 cells were treated with the selected doses for 24 hours prior to isolation of mRNA. Lanes: 1, untreated; 2, CDDP (5 μM); 3, bulk TF (45 μM) + CDDP; 4, bulk EGCG (60 μM) + CDDP; 5, TF-NPs (2.25 μM) + CDDP; 6, TF-NPs (9 μM) + CDDP; 7, EGCG-NPs (3 μM) + CDDP; and 8, EGCG-NPs (12 μM) + CDDP. |
TF/EGCG-loaded PLGA-NPs enhanced suppression of NF-kB-dependent expression of genes involved in cell proliferation, invasion, and angiogenesis
Several gene products that mediate cellular proliferation, invasion, and angiogenesis, including cyclin D1, MMP-9, and VEGF, have NF-κB-binding sites in their promoters.36 The ability of TF/EGCG-loaded PLGA-NPs and bulk TF/EGCG alone and in combination with CDDP to modulate expression of these genes was assessed (Figure 6B). While expression of the cyclin D1, MMP-9, and VEGF genes was partially inhibited by TF/EGCG or CDDP, dose-dependent complete inhibition was observed with the combination of TF/EGCG-NP and CDDP, and was consistent with a decrease in NF-κB activity.
TF/EGCG-loaded PLGA-NPs enhanced antitumor potential in vivo
Our in vitro studies showed that TF/EGCG-loaded PLGA-NPs have significantly better anticancer properties and cancer chemosensitization potential than the bulk forms. To confirm our in vitro findings, we treated EAC-bearing mice with TF/EGCG-loaded PLGA-NPs alone and in combination with CDDP, and examined whether the NP formulation have better antitumor potential than the bulk forms. The effects of the combination of TF/EGCG-loaded PLGA-NPs and CDDP on body weight, viable and non-viable EAC cell count (Table 2), mean survival time, and % ILS were studied and compared with the other treatment groups. The results of our in vivo study indicated that the combination of TF/EGCG-loaded PLGA-NPs (10 and 20 μg/animal) and CDDP (10 μg/animal) had significant antitumor activity in EAC-bearing mice (Figure 7, P<0.05) when compared with bulk TF/EGCG (100 μg/animal) and CDDP (10 μg/animal). Moreover, the combination of NPs and CDDP prolonged the % ILS in treated EAC tumor-bearing mice, when compared with the untreated controls and bulk-treated mice (Figure 7C, P<0.05). A significant increase in mean survival time in mice treated with a combination of TF/EGCG-loaded PLGA-NPs and CDDP was also noted as compared with other groups of bulk treatment (Figure 7D, P<0.05).
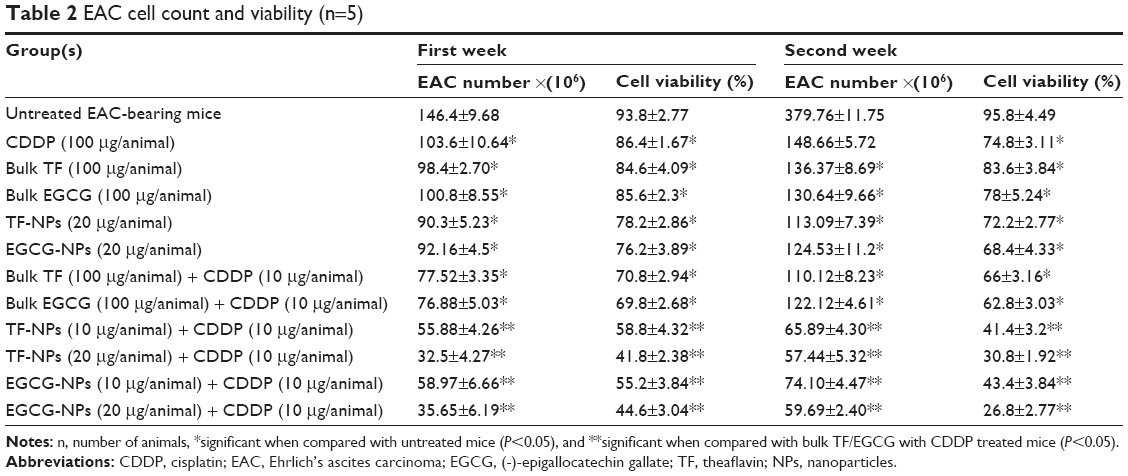 | Table 2 EAC cell count and viability (n=5) |
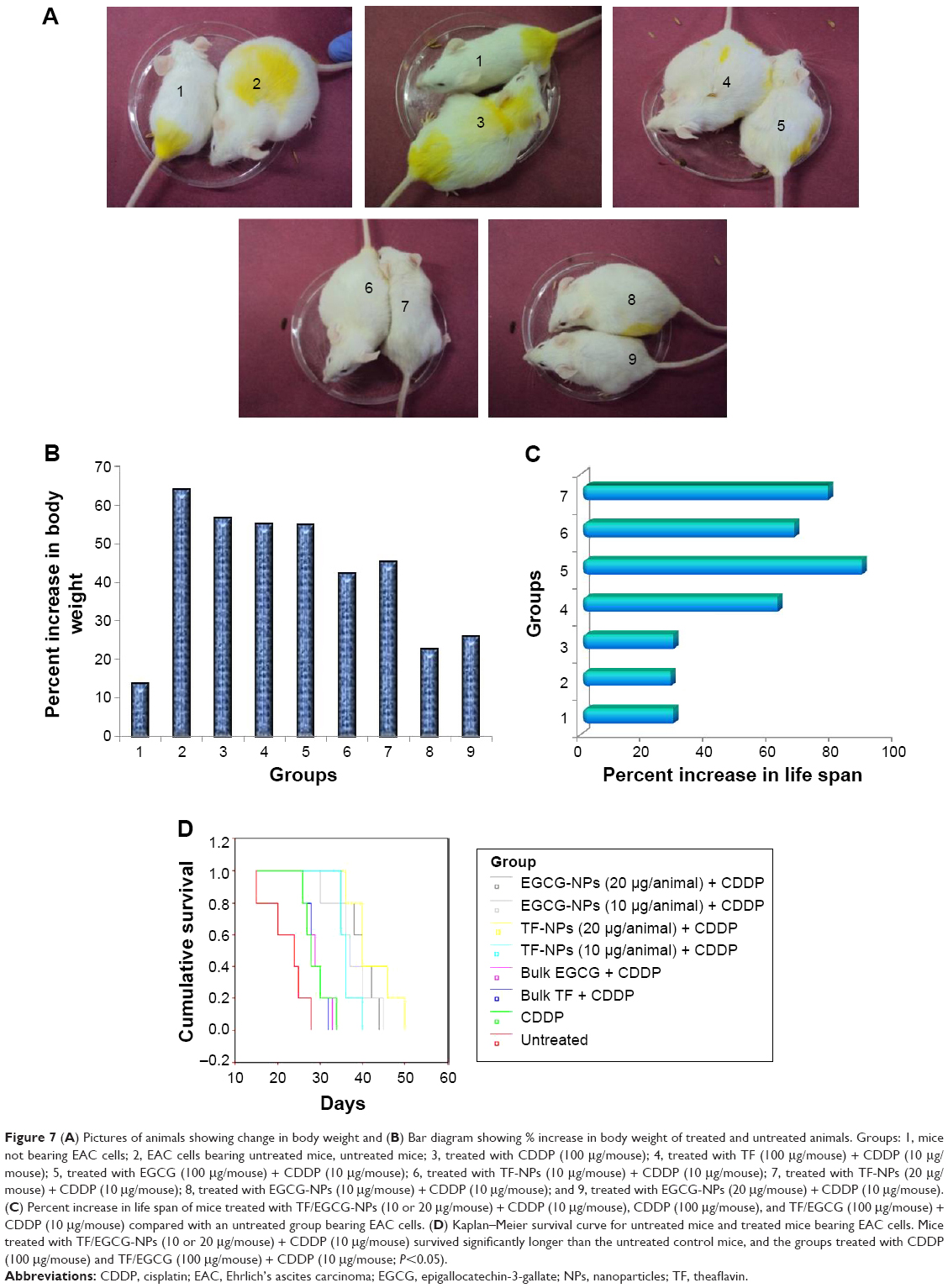 | Figure 7 (A) Pictures of animals showing change in body weight and (B) Bar diagram showing % increase in body weight of treated and untreated animals. Groups: 1, mice not bearing EAC cells; 2, EAC cells bearing untreated mice, untreated mice; 3, treated with CDDP (100 μg/mouse); 4, treated with TF (100 μg/mouse) + CDDP (10 μg/mouse); 5, treated with EGCG (100 μg/mouse) + CDDP (10 μg/mouse); 6, treated with TF-NPs (10 μg/mouse) + CDDP (10 μg/mouse); 7, treated with TF-NPs (20 μg/mouse) + CDDP (10 μg/mouse); 8, treated with EGCG-NPs (10 μg/mouse) + CDDP (10 μg/mouse); and 9, treated with EGCG-NPs (20 μg/mouse) + CDDP (10 μg/mouse). (C) Percent increase in life span of mice treated with TF/EGCG-NPs (10 or 20 μg/mouse) + CDDP (10 μg/mouse), CDDP (100 μg/mouse), and TF/EGCG (100 μg/mouse) + CDDP (10 μg/mouse) compared with an untreated group bearing EAC cells. (D) Kaplan–Meier survival curve for untreated mice and treated mice bearing EAC cells. Mice treated with TF/EGCG-NPs (10 or 20 μg/mouse) + CDDP (10 μg/mouse) survived significantly longer than the untreated control mice, and the groups treated with CDDP (100 μg/mouse) and TF/EGCG (100 μg/mouse) + CDDP (10 μg/mouse; P<0.05). |
Discussion
Drug bioavailability whether in vitro or in vivo, is critical to achieving optimal efficacy at the cell level. Nanotechnology has recently emerged as a way of improving drug bioavailability and the ability of drugs to target tumor cells.37,38 In the present study, we prepared TF/EGCG-loaded PLGA-NPs and investigated their ability to sensitize cancer cells to a low dose (IC20) of CDDP for inhibition of cancer cell proliferation, induction of apoptosis, and suppression of NF-κB activity. The NPs had a sustained-release capacity and an up to 7-fold greater ability to suppress proliferation of A549 cancer cells as compared with bulk doses, perhaps due to enhanced drug uptake. Siddiqui et al38 noted an over 10-fold dose advantage in the IC50 value of EGCG-NPs (at 24 hours) as compared with non-capsulated EGCG in prostate cancer cells. Studies from a number of laboratories have suggested that enhanced uptake of phytochemicals when formulated with NPs correlates with their enhanced antitumor activity.39,40 These effects are made possible by the efficient uptake of tea-NPs by cancer cells, suggesting that nanoencapsulation removes the barriers to penetration at the cell surface, because the NPs adsorb serum proteins nonspecifically onto their surface and enter the cell via receptors on the cell membrane.38 Further, in our study, we observed that these effects were not specific to lung cancer cells and were also observed with leukemia and cervical cancer cells, indicating that such effects are not limited to specific types of cancer cells; however, the efficacy of targeting is different.
Apoptosis is a major mechanisms by which various anticancer agents destroy tumor cells. Monitoring the sub-G1 phase of the cell cycle showed that TF/EGCG-loaded NPs were again more potent than the bulk forms in selected cell types. It was noted that even the smallest (1/20th of bulk IC50 dose) amount of tea-NPs was equally potent at inducing a sub-G1 peak than non-nanosized tea polyphenols. Further, the chemosensitization ability of CDDP was significantly enhanced by TF/EGCG-encapsulated PLGA NPs, as evidenced by both cytotoxic effects and induction of sub-G1 phase as compared to bulk tea polyphenols. These results provide further evidence that TF/EGCG loaded NPs have greater anticancer activity than bulk TF/EGCG. This improved anticancer efficacy in selected cancer cell lines may be due to sustained and long-term delivery of tea-loaded PLGA-NPs.38
In order to determine if tea-NPs retain their chemosensitization ability at the molecular level, we selected several molecular entities that have already been shown to be modulated by tea polyphenols.4 Like bulk TF/EGCG, treatment of A549 cells with a combination of TF/EGCG-loaded NPs and CDDP resulted in a significant increase in proapoptotic Bax, with a concomitant decrease in antiapoptotic Bcl-2, thereby increasing the Bax/Bcl-2 ratio and apoptosis.41 Further, an increase in cleavage of PARP, a protein involved in a number of cellular processes, mainly DNA repair and programmed cell death,42 was also found and was much more pronounced than with bulk doses. The combination of nanoformulated tea polyphenols and CDDP resulted in enhanced loss of ΔΨm and generation of ROS; this combination of events leads to release of cytochrome c from the intermembrane space into the cytosol, culminating in activation of the caspase cascade and thus the apoptotic cell death pathway.43 Excessive generation of ROS and a decline in ΔΨm has been documented in cancer cells treated with tea polyphenols.4,6,7 Notably, in our study, these results were also observed at a low concentration of TF/EGCG-NPs (1/20th, 1/10th and 1/5th of the respective bulk IC50 dose), again confirming the marked dose advantage achieved when TF/EGCG is delivered using a NP formulation.
Activation of NF-κB appears to be a major pathway involved in proliferation of tumor cells, chemoresistance, and inflammation.44 Inhibition of NF-κB is reported to increase the efficacy of a number of chemotherapeutic agents, including CDDP.35 We have already documented the potential of tea polyphenols in inhibition of NF-κB activity.6,7 In our present study, we noted the significant effect of a low concentration of TF/EGCG-NPs on chemosensitivity to CDDP (IC20 dose) via inhibition of NF-κB and its corresponding gene. It was found that low doses of TF/EGCG-loaded NPs (1/20th, 1/10th, and 1/5th of the respective bulk IC50 dose) suppressed NF-κB activation more efficiently in combination with CDDP than did the bulk TF/EGCG (IC50 dose). When examined for its ability to suppress NF-κB-regulated genes (cyclin D1, MMP-9, and VEGF), once again the TF/EGCG-NP and CDDP combination was found to be more effective than combination of their respective bulk forms. It is very likely that the enhanced antiproliferation activity afforded by the TF/EGCG-loaded NPs was due to enhanced suppression of expression of cyclin D1 protein, which is intimately related to the G1/S transition.7 It is well documented that NF-κB activates genes involved in cell proliferation, angiogenesis, and metastasis, thereby promoting tumor growth.45–47 Our study also showed that TF/EGCG-loaded NPs augmented antimetastatic and antiangiogenic activity, given that expression of both MMP-9 and VEGF was downregulated by selected low doses of these NPs in combination with CDDP.
To corroborate our in vitro findings, we used a transplantable murine EAC tumor model to investigate the antitumor activity, if any, of tea-NPs used alone and in combination with CDDP. Tea-NPs in combination with a low dose of CDDP significantly reduced tumor volume and viable EAC cell counts, simultaneous with an increased mean survival time and % ILS in EAC-bearing mice. These in vivo observations again show the efficacy of tea-NPs as an effective cancer chemosensitizing agent. Prolongation of survival in tumor-bearing animals is a well established method of evaluate the anticancer potential of a drug.48,49 Further, a reduction in the number of ascitic tumor cells is an indication of the effect of a drug on peritoneal macrophages and other components of the immune system.49 In this respect, it is very much convincing to consider that the increased killing of tumor cells by tea-NPs might be happening in the same manner in an EAC model as in in vitro system and therefore, slow tumor growth and enhanced survival of animals were observed in groups treated with nanoformulation as compared to bulk treated groups. Increased accumulation of tea-NPs in defective, leaky, and highly permeable vascular tumor tissue was possible due to the enhanced permeability and retention effect,50,51 allowing nanosized anticancer compounds to act more effectively and with reduced side effects from CDDP, thereby prolonging survival in EAC tumor-bearing mice.
Conclusion
Tea-NPs improve intracellular uptake and can be made available for a wide range of biological targets due to their small size and more rapid mobility and penetration as compared with bulk forms. The use of TF/EGCG-loaded PLGA-NPs as described here should have superior effects when compared with bulk TF/EGCG in the chemosensitization of anticancer drugs. Our data indicate that NP-mediated delivery of anticancer phytochemicals could serve as a basic strategy for both enhancing the bioavailability and limiting the unwanted toxicity of chemotherapeutic agents. More animal studies are presently needed to clarify the potential of TF/EGCG-loaded NPs, hopefully proceeding to clinical trials in the future.
Acknowledgment
The authors are grateful to the Council of Scientific and Industrial Research (India) for its financial support.
Disclosure
The authors report no conflicts of interest in this study.
References
Momekov G, Bakalova A, Karaivanova M. Novel approaches towards development of non-classical platinum-based antineoplastic agents: design of platinum complexes characterized by an alternative DNA-binding pattern and/or tumor-targeted cytotoxicity. Curr Med Chem. 2005;12:2177–2219. | ||
Kartalou M, Essiqmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478:23–43. | ||
Stewart DJ. Mechanism of resistance to cisplatin and carboplatin. Crit Rev Oncol Hematol. 2007;63:12–31. | ||
Singh M, Bhui K, Singh R, Shukla Y. Tea polyphenols enhance cisplatin chemosensitivity in cervical cancer cells via induction of apoptosis. Life Sci. 2013;93:7–16. | ||
Chanvorachote P, Pongrakhananon V, Wannachaiyasit S, Luanpitpong S, Rojanasakul Y, Nimmannit U. Curcumin sensitizes lung cancer cells to cisplatin-induced apoptosis through superoxide anion-mediated Bcl-2 degradation. Cancer Invest. 2009;27:624–635. | ||
Singh M, Tyagi S, Bhui K, Prasad S, Shukla Y. Regulation of cell growth through cell cycle arrest and apoptosis in HPV 16 positive human cervical cancer cells by tea polyphenols. Invest New Drugs. 2010;28:216–224. | ||
Singh M, Singh R, Bhui K, Tyagi S, Mahmood Z, Shukla Y. Tea polyphenols induce apoptosis through mitochondrial pathway and by inhibiting nuclear factor kappa B and Akt activation in human cervical cancer cells. Oncol Res. 2011;19:245–257. | ||
Roy P, Nigam N, Singh M, et al. Tea polyphenols inhibit cyclooxygenase-2 expression and block activation of nuclear factor-kappa B and Akt in diethylnitrosoamine induced lung tumors in Swiss mice. Invest New Drugs. 2010;28:466–471. | ||
Yang WH, Fong YC, Lee CY, et al. Epigallocatechin-3-gallate induces cell apoptosis of human chondrosarcoma cells through apoptosis signal-regulating kinase 1 pathway. J Cell Biochem. 2011;112:1601–1611. | ||
Chen X, Li Y, Lin Q, et al. Tea polyphenols induced apoptosis of breast cancer cells by suppressing the expression of survivin. Sci Rep. 2014;4:4416. | ||
Singh M, Bhatnagar P, Srivastava AK, Kumar P, Shukla Y, Gupta KC. Enhancement of cancer chemosensitization potential of cisplatin by tea polyphenols poly(lactide-co-glycolide) nanoparticles. J Biomed Nanotechnol. 2011;7:202. | ||
Periasamy VS, Alshatwi AA. Tea polyphenols modulate antioxidant redox system on cisplatin-induced reactive oxygen species generation in a human breast cancer cell. Basic Clin Pharmacol Toxicol. 2013;112:374–384. | ||
Kidd PM. Bioavailability and activity of phytosome complexes from botanical polyphenols: the silymarin, curcumin, green tea, and grape seed extracts. Altern Med Rev. 2009:14:226–246. | ||
Nishiyama N. Nanomedicine: nanocarriers shape up for long life. Nat Nanotechnol. 2007;2:203–204. | ||
Freitas RA Jr. What is nanomedicine? Nanomedicine. 2005;1:2–9. | ||
Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long circulating polymeric nanospheres. Science. 1994;263:1600–1603. | ||
Ringsdrorf H. Structure and properties of pharmacologically active polymers. J Polym Sci Polymer Symp. 1975;51:135–153. | ||
Hu S, Zhang Y. Endostar-loaded PEG-PLGA nanoparticles: in vitro and in vivo evaluation. Int J Nanomedicine. 2010;5:1039–1048. | ||
Yallapu MM, Gupta BK, Jaggi M, Chauhan SC. Fabrication of curcumin encapsulated PLGA nanoparticles for improved therapeutic effects in metastatic cancer cells. J Colloid Interface Sci. 2010;351:19–29. | ||
Barratt GM. Therapeutic applications of colloidal drug carriers. Pharm Sci Technolo Today. 2000;3:163–171. | ||
Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54:631–651. | ||
Esmaeili F, Hosseini-Nasr M, Malekshahi R, Samadi N, Atyabi F, Dinarvand R. Preparation and antibacterial activity evaluation of rifampicin-loaded poly lactide-co-glycolide nanoparticles. Nanomedicine. 2007;3:161–167. | ||
Swami A, Aggarwal A, Pathak A, et al. Imidazolyl-PEI modified nanoparticles for enhanced gene delivery. Int J Pharm. 2007;335:180–192. | ||
Asadishad B, Vossoughi M, Alamzadeh I. In vitro drug release behaviour and cytotoxicity of doxorubicin-loaded gold nanoparticles in cancerous cells. Biotechnol Lett. 2010;32:649–654. | ||
Siddiqui IA, Adhami VM, Afaq F, Ahmad N, Mukhtar H. Modulation of phosphatidylinositol-3-kinase/protein kinase B- and mitogen-activated protein kinase-pathways by tea polyphenols in human prostate cancer cells. J Cell Biochem. 2004;91:232–242. | ||
Park HR, Ju EJ, Jo SK, Jung U, Kim SH, Yee ST. Enhanced antitumor efficacy of cisplatin in combination with HemoHIM in tumor-bearing mice. BMC Cancer. 2009;9:85. | ||
Katare YK, Muthukumaran T, Panda AK. Influence of particle size, antigen load, dose and additional adjuvant on the immune response from antigen loaded PLA microparticles. Int J Pharm. 2005;301:150–160. | ||
Vert M, Li S, Garreau H. More about degradation of LA/GA derived matrices in aqueous media. J Control Release. 1991;16:15–26. | ||
Cabral H, Nishiyama N, Okazaki S, Koyama H, Kataoka K. Preparation and biological properties of dichloro(1,2-diaminocyclohexane)platinum(II) (DACHPt)-loaded polymeric micelles. J Control Release. 2005;101:223–232. | ||
Chen CY, Liu TZ, Liu YW, et al. 6-Shogaol (alkanone from ginger) induces apoptotic cell death of human hepatoma p53 mutant Mahlavu subline via an oxidative stress-mediated caspase-dependent mechanism. J Agric Food Chem. 2007;55:948–954. | ||
Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human Bax gene. Cell. 1995;80:293–299. | ||
Fleury C, Mignotte B, Vayssière JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84:131–141. | ||
Kroemer G. Mitochondrial implication in apoptosis. Towards an endosymbiont hypothesis of apoptosis evolution. Cell Death Differ. 1997;4:443–456. | ||
Aggarwal BB, Takada Y, Oommen OV. From chemoprevention to chemotherapy: common targets and common goals. Expert Opin Investig Drugs. 2004;13:1327–1338. | ||
Kim SB, Kim JS, Lee JH, et al. NF-κB activation is required for cisplatin-induced apoptosis in head and neck squamous carcinoma cells. FEBS Lett. 2006;580:311–318. | ||
Sethi G, Ahn KS, Aggarwal BB. Targeting nuclear factor-kappa B activation pathway by thymoquinone: role in suppression of antiapoptotic gene products and enhancement of apoptosis. Mol Cancer Res. 2008;6:1059–1070. | ||
Von Maltzahn G, Ren Y, Park JH, Min DH, Kotamraju VR, Jayakumar J. In vivo tumor cell targeting with “click” nanoparticles. Bioconjug Chem. 2008;19:1570–1578. | ||
Siddiqui IA, Adhami VM, Bharali DJ, et al. Introducing nanochemoprevention as a novel approach for cancer control: proof of principle with green tea polyphenol epigallocatechin-3-gallate. Cancer Res. 2009;69:1712–1716. | ||
Anand P, Nair HB, Sung B, et al. Design of curcumin-loaded PLGA nanoparticles formulation with enhanced cellular uptake, and increased bioactivity in vitro and superior bioavailability in vivo. Biochem Pharmacol. 2010;79:330–338. | ||
Yadav VR, Prasad S, Kannappan R, et al. Cyclodextrin-complexed curcumin exhibits anti-inflammatory and antiproliferative activities superior to those of curcumin through higher cellular uptake. Biochem Pharmacol. 2010;80:1021–1032. | ||
Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. | ||
Bursztajn S, Feng JJ, Berman SA, Nanda AR. Poly (ADP-ribose) polymerase induction is an early signal of apoptosis in human neuroblastoma. Brain Res Mol Brain Res. 2000;76:363–376. | ||
Chung YM, Bae YS, Lee SY. Molecular ordering of ROS production, mitochondrial changes, and caspase activation during sodium salicylate-induced apoptosis. Free Radic Biol Med. 2003;34:434–442. | ||
Venkatraman M, Anto RJ, Nair A, Varghese M, Karunagaran D. Biological and chemical inhibitors of NF-κB sensitize SiHa cells to cisplatin-induced apoptosis. Mol Carcinog. 2005;44:51–59. | ||
Baldwin AS Jr. The NF-kB and IkB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. | ||
Gutteridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr. NF-kB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–5799. | ||
Perkins ND. The Rel/NF-kB family: friend and foe. Trends Biochem Sci. 2000;25:434–440. | ||
Panyama J, Labhasetwara V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55:329–347. | ||
Prasad SB, Giri A. Antitumor effect of cisplatin against murine ascites Dalton’s lymphoma. Indian J Exp Biol. 1994;32:155–162. | ||
Banerjee SS, Aher N, Patil R, Khandare J. Poly(ethylene glycol)-prodrug conjugates: concept, design, and applications. J Drug Deliv. 2012;2012:103973. | ||
Yoo JW, Doshi N, Mitragotri S. Adaptive micro and nanoparticles: temporal control over carrier properties to facilitate drug delivery. Adv Drug Deliv Rev. 2011;63:1247–1256. |
Supplementary materials
 | Table S1 Oligonucleotide sequences |
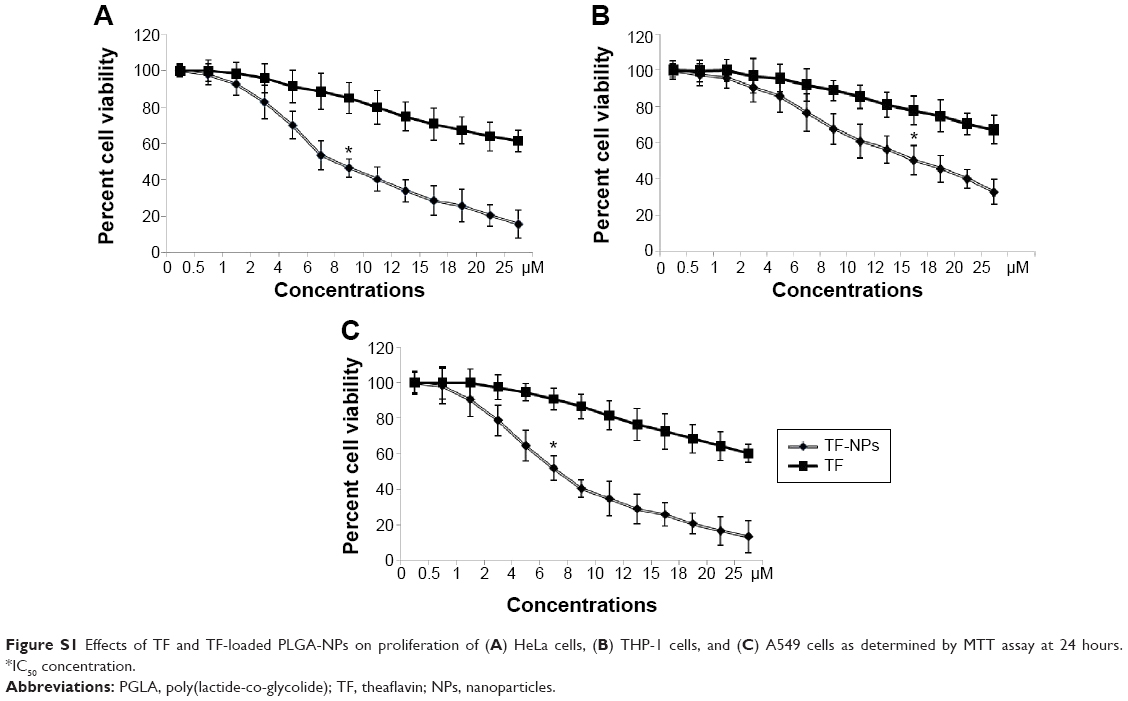 | Figure S1 Effects of TF and TF-loaded PLGA-NPs on proliferation of (A) HeLa cells, (B) THP-1 cells, and (C) A549 cells as determined by MTT assay at 24 hours. *IC50 concentration. |
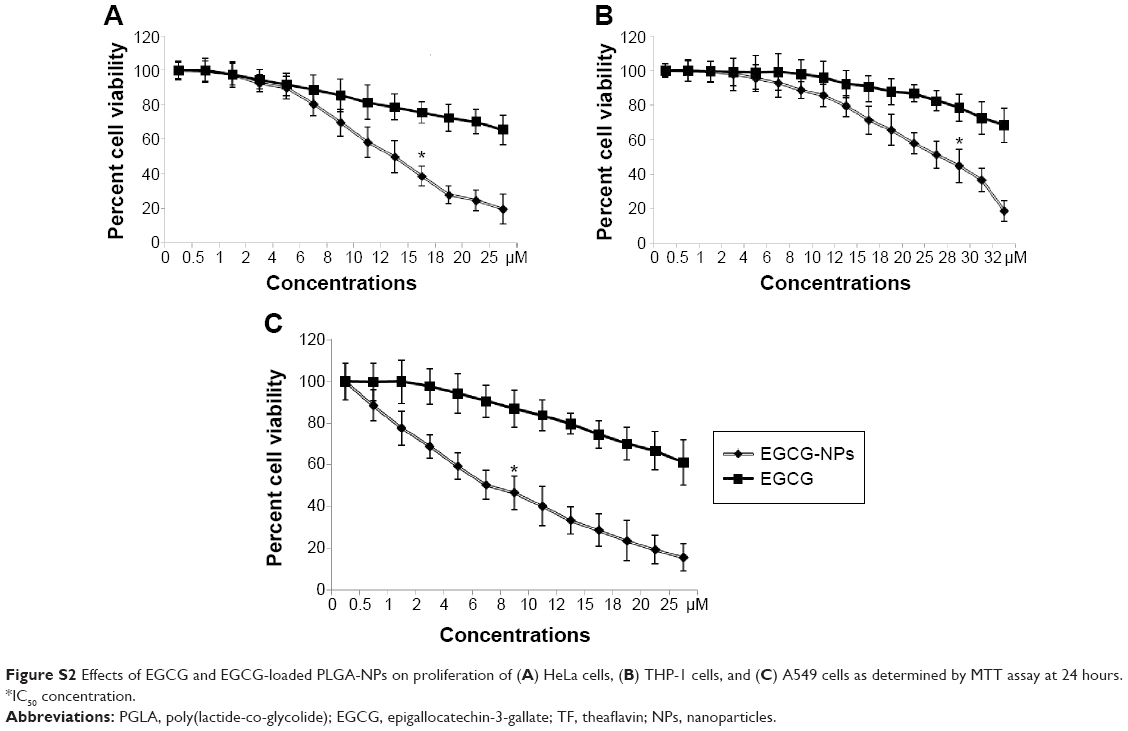 | Figure S2 Effects of EGCG and EGCG-loaded PLGA-NPs on proliferation of (A) HeLa cells, (B) THP-1 cells, and (C) A549 cells as determined by MTT assay at 24 hours. *IC50 concentration. |
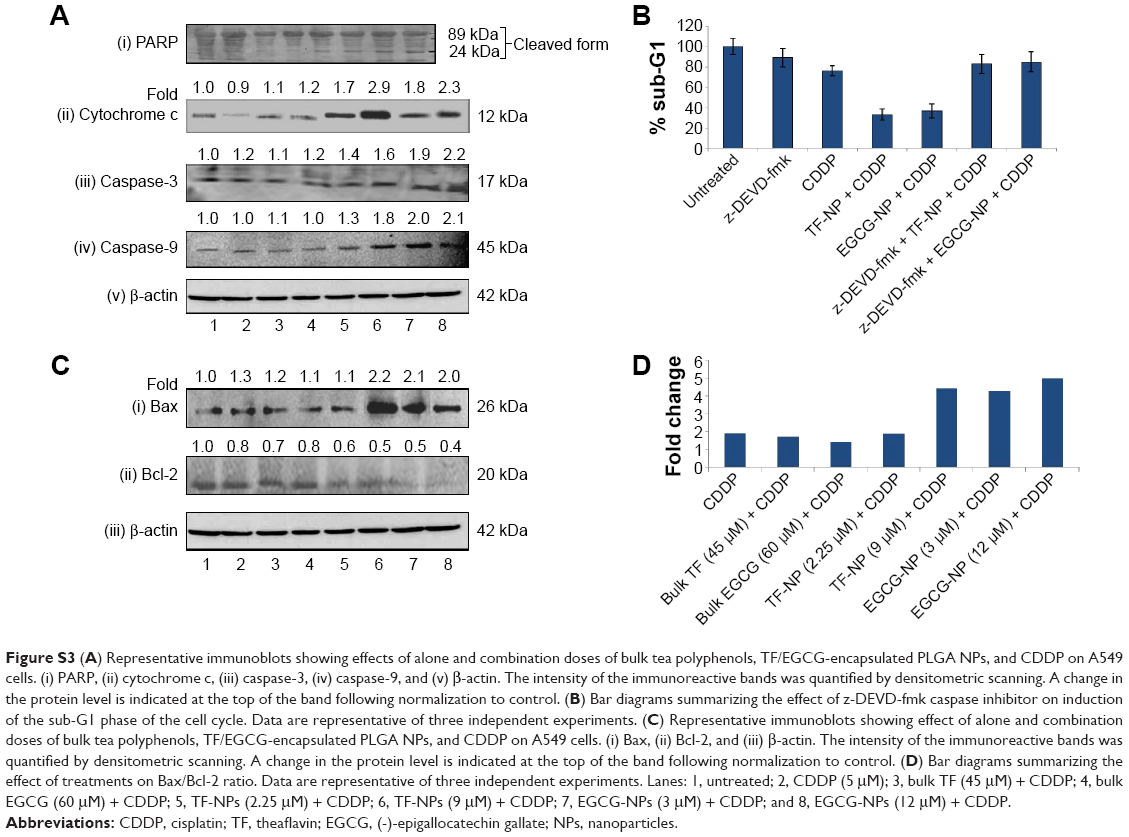 | Figure S3 (A) Representative immunoblots showing effects of alone and combination doses of bulk tea polyphenols, TF/EGCG-encapsulated PLGA NPs, and CDDP on A549 cells. (i) PARP, (ii) cytochrome c, (iii) caspase-3, (iv) caspase-9, and (v) β-actin. The intensity of the immunoreactive bands was quantified by densitometric scanning. A change in the protein level is indicated at the top of the band following normalization to control. (B) Bar diagrams summarizing the effect of z-DEVD-fmk caspase inhibitor on induction of the sub-G1 phase of the cell cycle. Data are representative of three independent experiments. (C) Representative immunoblots showing effect of alone and combination doses of bulk tea polyphenols, TF/EGCG-encapsulated PLGA NPs, and CDDP on A549 cells. (i) Bax, (ii) Bcl-2, and (iii) β-actin. The intensity of the immunoreactive bands was quantified by densitometric scanning. A change in the protein level is indicated at the top of the band following normalization to control. (D) Bar diagrams summarizing the effect of treatments on Bax/Bcl-2 ratio. Data are representative of three independent experiments. Lanes: 1, untreated; 2, CDDP (5 μM); 3, bulk TF (45 μM) + CDDP; 4, bulk EGCG (60 μM) + CDDP; 5, TF-NPs (2.25 μM) + CDDP; 6, TF-NPs (9 μM) + CDDP; 7, EGCG-NPs (3 μM) + CDDP; and 8, EGCG-NPs (12 μM) + CDDP. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.