Back to Journals » Drug Design, Development and Therapy » Volume 10
Plasmodium-infected erythrocytes (pRBC) induce endothelial cell apoptosis via a heme-mediated signaling pathway
Authors Liu M, Dickinson-Copeland C, Hassana S, Stiles J
Received 22 September 2015
Accepted for publication 27 October 2015
Published 3 March 2016 Volume 2016:10 Pages 1009—1018
DOI https://doi.org/10.2147/DDDT.S96863
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Mingli Liu, Carmen Dickinson-Copeland, Salifu Hassana, Jonathan K Stiles
Department of Microbiology, Biochemistry and Immunology, Morehouse School of Medicine, Atlanta, GA, USA
Abstract: Heme is cytotoxic to the plasmodium parasite, which converts it to an insoluble crystalline form called hemozoin (malaria pigment) in erythrocytes during replication. The increased serum levels of free heme cause tissue damage, activation of microvascular endothelial and glial cells, focal inflammation, activation of apoptotic pathways, and neuronal tissue damage. Several hypotheses have been proposed to explain how these causative factors exacerbate fatal malaria. However, none of them fully explain the detailed mechanisms leading to the high morbidity and mortality associated with malaria. We have previously reported that heme-induced brain microvascular endothelial cell (HBVEC) apoptosis is a major contributor to severe malaria pathogenesis. Here, we hypothesized that heme (at clinically relevant levels) induces inflammation and apoptosis in HBVEC, a process that is mediated by independent proinflammatory and proapoptotic signaling pathways. In this study, we determined the key signaling molecules associated with heme-mediated apoptosis in HBVEC in vitro using RT2 profiler polymerase chain reaction array technology and confirmed results using immunostaining techniques. While several expressed genes in HBVEC were altered upon heme stimulation, we determined that the apoptotic effects of heme were mediated through p73 (tumor protein p73). The results provide an opportunity to target heme-mediated apoptosis therapeutically in malaria-infected individuals.
Keywords: heme, endothelial cells, signaling pathways, cerebral malaria
Introduction
Malaria pathogenesis is characterized by the infection and destruction of erythrocytes by proliferating plasmodium parasites, resulting in the release of heme moieties in the bloodstream. Heme is cytotoxic to plasmodium, which converts it to an insoluble crystalline form called hemozoin (malaria pigment) in erythrocytes. In severe cases such as cerebral malaria (CM), parasitized red blood cells (pRBCs) in brain microcirculation are sequestered by adhering to other pRBCs and to microvascular endothelial cells (ECs) to evade splenic clearance of parasites. The pRBCs damaged by proliferation of erythrocytic stages of parasites release cytotoxic heme, which induces deleterious responses, including inflammation, activation, and apoptosis of microvascular ECs.1–12 In individuals with Plasmodium falciparum-induced CM, a combination of these factors disrupts the super structure of the blood–brain barrier, resulting in leakage into brain parenchyma. Subsequently, apoptosis occurs in ECs and in contiguous neuronal and glial cells. Recent studies in our laboratory and others demonstrate the increasing importance of elevated plasma concentrations of free heme as a key inducer of inflammation and damage to the host microvascular endothelium as well as the mortality associated with fatal CM.13,14 Heme induces human brain vascular endothelial cell (HBVEC) apoptosis that correlates with its concentration in plasma in a dose-dependent manner. Since brain microvascular ECs are key components of the blood–brain barrier and are severely disrupted during CM pathogenesis, we hypothesized that heme (at clinically relevant levels) induces inflammation and apoptosis in HBVEC and is mediated by independent proinflammatory and proapoptotic signaling pathways. Our goal was to utilize a surrogate in vitro system to investigate the mechanism underlying the apoptotic and inflammatory effects of heme on brain microvascular ECs.
Materials and methods
Antibodies and reagents
Antibody against FasL was purchased from Cell Signaling; antibodies against tumor protein p73 (TP73) and BCL-2 were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Antibody to β-actin used for Western blot was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). All secondary antibodies used for Western blot were purchased from EMD Millipore (Billerica, MA, USA). Hemin was purchased from Frontier Scientific, Inc. (Logan, UT, USA).
Cell culture and siRNA transfection
HBVECs (BioWhittaker, Walkersville, MD, USA) were cultured at 37°C with 5% CO2 in endothelial basal medium-2 (Lonza) supplemented with 5% fetal bovine serum (American Type Culture Collection [ATCC], Manassas, VA, USA), growth factors, and other supplements including human recombinant epidermal growth factor, hydrocortisone, GA-100 (Gentamicin, Amphotericin-B), human recombinant vascular endothelial growth factor (VEGF), recombinant human fibroblast growth factor-B, recombinant long R insulin-like growth factor, ascorbic acid, heparin, 100 U/mL of streptomycin, and 100 U/mL of penicillin (Lonza). The cells were harvested and passaged at ~70%–90% confluence as described earlier.15 HBVECs (2×105 cells/mL) were seeded in 35 mm tissue culture dish and incubated at 37°C in 5% CO2 for 24–48 hours for future use.
Small interfering RNA (siRNA) duplexes of p73 were designed and purchased from Santa Cruz Biotechnology Inc. A scrambled siRNA with no homology to any known sequences was used as control. Serum-starved HBVEC cells were transfected with 100 nM specific siRNA or control using Lipofectamine 3000 reagent (Thermo Fisher Scientific, Waltham, MA, USA) in a serum-free Opti-MEM1 medium (Thermo Fisher Scientific) according to the manufacturer’s instruction. After 48 hours of transfection, HBVEC cells were treated with heme for another 24 hours.
Western blotting
Cells were lysed with lysis buffer (50 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 150 mm NaCl, 1.5 mm MgCl2, 1 mm ethylene glycol tetraacetic acid, 10% glycerol, 1% Nonidet P-40, 100 mm NaF, 10 mm sodium pyrophosphate, 0.2 mm sodium orthovanadate, 1 mm phenylmethylsulfonyl fluoride, 10 μg/mL aproptinin, and 10 μg/mL leupeptin). Samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and separated proteins were transferred to nitrocellulose membranes and identified by immunoblotting. Primary antibodies were obtained from commercial sources (these antibodies were diluted at the ratio of 1:1,000 according to the manufacturer’s instruction), while secondary antibodies included horseradish peroxidase-conjugated antirabbit and antimouse antibodies obtained from Calbiochem. Blots were developed with SuperSignal Pico or Femto substrate (Pierce; Waltham, MA, USA). A densitometric analysis of the bands was performed with the ImageQuant program (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Ethics approval was not sought as the Morehouse School of Medicine Institutional Review Board does not require this for commercially obtained human cell lines.
Real-time reverse transcription-polymerase chain reaction analysis
Cell pellets were stored in Trizol reagent and homogenized in fresh Trizol. Total RNA was isolated from cells using an RNeasy Mini Kit (Qiagen NV, Venlo, the Netherlands). Total RNA was quantified using the Nanodrop N-1000 by Agilent Technologies (Santa Clara, CA, USA). cDNA was synthesized from the isolated RNA using iScript cDNA synthesis kit (Bio-Rad Laboratories Inc.). Reverse transcription was performed by using random hexamers at 25°C for 5 minutes, 42°C for 30 minutes, and 85°C for 5 minutes. Quantitative polymerase chain reaction (qPCR) was performed using iQ SYBR Green Supermix (Bio-Rad Laboratories Inc.) in a CFX96 Real-Time PCR System machine (Bio-Rad Laboratories Inc.). The data were analyzed using CFX96 Real-Time PCR System. Primer sequences for the genes are described in Table 1.
 | Table 1 Primers for real-time polymerase chain reaction |
Immunofluorescence staining
HBVECs (2×105 cells/mL) were seeded in 35 mm tissue culture dish and incubated at 37°C in 5% CO2 for 24 hours. Cells grown in monolayer cultures were fixed with 4% paraformaldehyde in phosphate-buffered saline, permeabilized with 0.2% Triton X-100, and blocked with 10% goat serum prior to antibody staining. For terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, the in situ cell death detection kit (TMR red; Boehringer Mannheim, Mannheim, Germany) was used. The cells were incubated with the TUNEL reaction solution for 60 minutes at 37°C in the dark. Cover slips were mounted onto slides with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole dihydrochloride (H-1200; Vector Laboratories, Inc.; Burlingame, CA, USA). Fluorescent images were collected by using a Zeiss LSM510 confocal microscope, and images were captured with LSM software, Version 2.3 (Carl Zeiss Meditec AG, Jena, Germany).
RT2 profiler PCR array
HBVECs were serum starved for 24 hours followed by treatment with 30 μM heme or with vehicle (as control). Total RNA was extracted and subjected to cDNA synthesis and then underwent RT2 profile PCR array assay. Briefly, total RNA extraction was performed using the RNeasy Mini Kit as described earlier. The first-strand cDNA synthesis was performed using an RT2 First-Strand cDNA Synthesis kit (Qiagen NV) and 2,000 ng of total RNA. cDNA was processed according to the manufacturer’s protocol. Briefly, the cDNA template was combined with RT2 Real-Time SYBR Green Master Mix and RNase-free water. A final reaction volume of 25 μL was added to each well of the human apoptosis signaling pathway PCR array (PAHS-012Z; SABiosciences; Valencia, CA, USA). Finally, pathway focused on mRNA was amplified following the manufacturer’s protocol. Housekeeping genes and reverse transcription and positive controls were included in this format. Reverse transcription-PCR data analysis was performed as follows: alterations in mRNA transcript levels at 24 hours with heme-treated and control (without heme treated) groups were initially analyzed using SABiosciences webportal software (http://www.sabiosciences.com/pcrarraydataanalysis.php). Fold changes were calculated, and P-values were calculated using Student’s t-test. A P-value <0.05 and a fold change >2.0 were considered to be a significant dysregulation.
Results
Heme induces apoptosis in HBVECs
To determine whether heme causes apoptosis of HBVECs, we performed TUNEL assay on HBVECs when the cells were treated with 30 μM of heme for 24 hours as described previously.13,14 To avoid the effects of heme and other factors in commercially acquired serum, we starved the cells with serum-free medium before treatment of heme to maximize the effects of heme. We then randomly chose ten fields to count the TUNEL-positive cells in slide using a 20× microscope objective. Apoptotic indices (% of TUNEL-positive cells/total cell nuclei ×100) were calculated after counting cells under a fluorescence microscope.16,17 The apoptotic cells were found to be increased by heme treatment (Figure 1) using TUNEL assay. This indicated that heme induces apoptosis in HBVEC. We next determined which apoptotic signaling pathways are involved during this process.
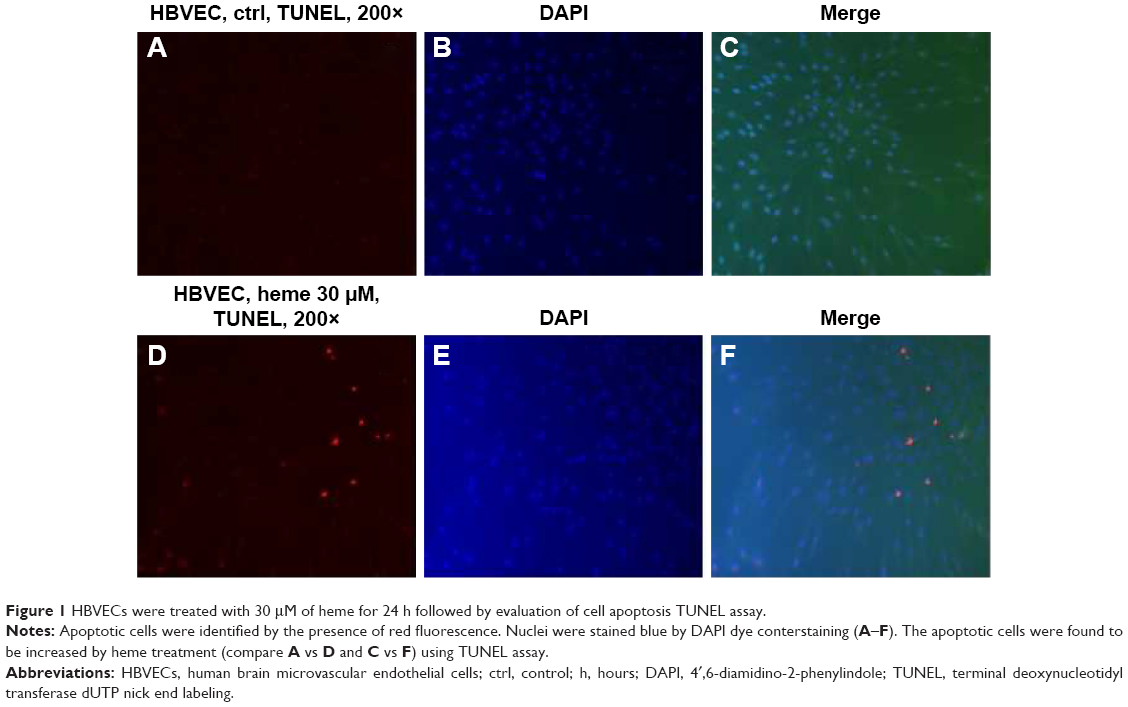 | Figure 1 HBVECs were treated with 30 μM of heme for 24 h followed by evaluation of cell apoptosis TUNEL assay. |
Analysis of heme-induced apoptotic genes (mRNA) involved in HBVEC programmed cell death using real-time RT2 profile PCR arrays
HBVECs were serum starved for 24 hours followed by treatment with 30 μM of heme or with vehicle (as control). Total RNA was extracted and subjected to cDNA synthesis and then underwent RT2 profile PCR array assay. To avoid the effects of heme and other factors in serum, we starved the cells with serum-free medium before treatment of heme to maximize the effects of heme. Apoptotic gene expression profiles induced upon heme treatment on HBVECs were assessed using real-time RT2 profile PCR arrays (PAHS-012Z). The results of real-time RT2 profile PCR array analysis are expressed as the fold changes in expression obtained by comparing HBVECs treated with heme or with vehicle as control. Figure 2A shows a list of upregulated and downregulated genes with a fold change >2 (a P<0.05 was considered to be significantly different). The upregulated genes include C-abl oncogene 1; nonreceptor tyrosine kinase (ABL1); BCL-2-like 10, apoptosis facilitator (BCL2L10); BCL-2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3); caspase-3; apoptosis-related cysteine peptidase (CASP3); caspase-4, apoptosis-related cysteine peptidase (CASP4); caspase-5, apoptosis-related cysteine peptidase (CASP5); CD40 ligand (CD40LG); CD70 molecule (CD70); cell death-inducing DFFA-like effector a (CIDEA); cytochrome, somatic (CYCS); Fas ligand (tumor necrosis factor [TNF] superfamily, member 6; FASLG); myeloid cell leukemia sequence 1; BCL-2-related (MCL1); tumor necrosis factor receptor superfamily, member 9 (TNFRSF9); TNF (ligand) superfamily, member 10 (TNFSF10); TP73; and TNF receptor-associated factor 3 (TRAF3). The downregulated genes consist of BCL-2-associate athanogene (BAG1), BCL-2-associated athanogene 3 (BAG3), B-cell CLL/lymphoma 2 (BCL-2), BCL-2-related protein A1 (BCL2A1), Baculoviral IAP repeat containing 3 (BIRC3), and BCL-2/adenovirus E1B 19 kDa interacting protein 3-like (BNIP3L). The scatterplot and heat map are shown in Figure 2B and C.
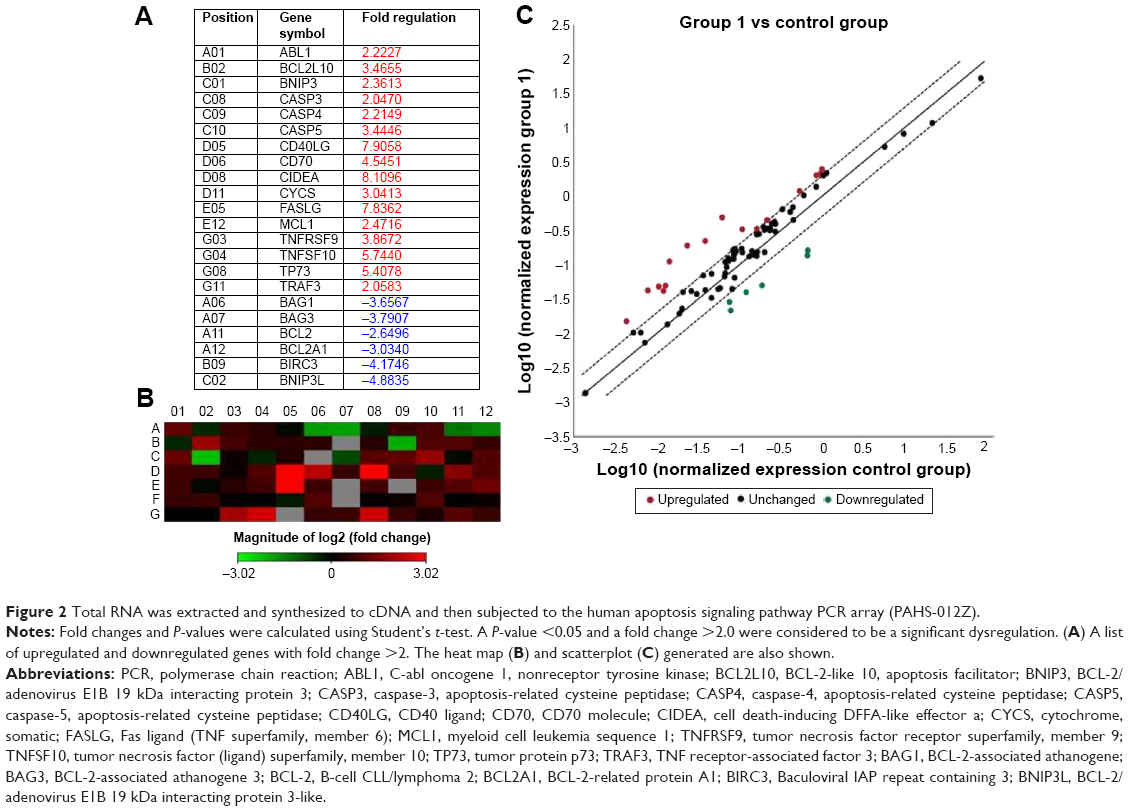 | Figure 2 Total RNA was extracted and synthesized to cDNA and then subjected to the human apoptosis signaling pathway PCR array (PAHS-012Z). |
Evaluation of mRNA expression of apoptotic-related genes by qPCR
Several genes with prominent proapoptotic function were upregulated in HBVEC cells after treatment with heme and confirmed by validation of qPCR (Figure 3), including BCL2L10, CD40LG, CD70, CIDEA, FASLG, TNFRSF9, TNFSF10, and TP73. Some genes for antiapoptosis were downregulated, such as BIRC3, BAG3, and BCL2A1.
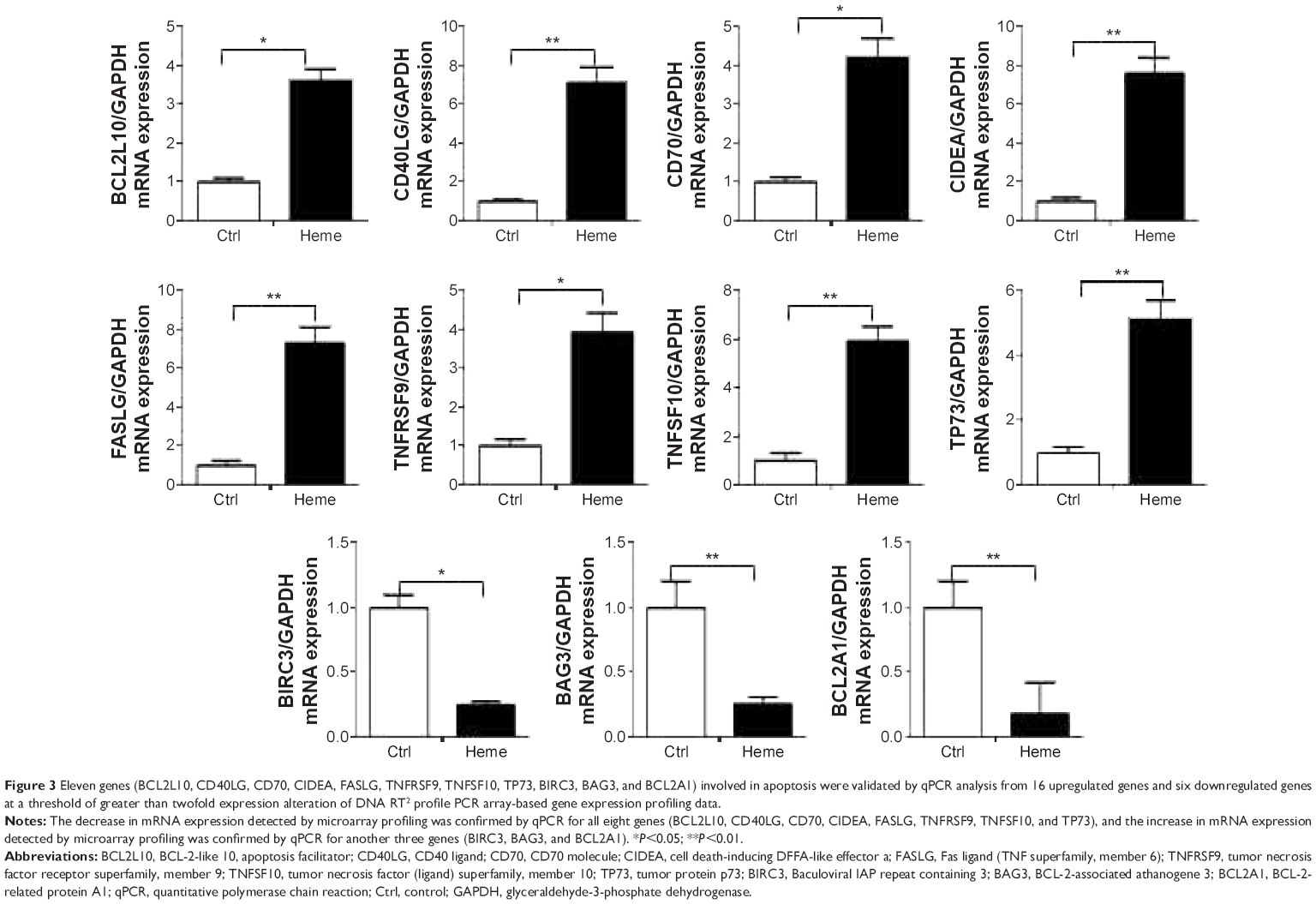 | Figure 3 Eleven genes (BCL2L10, CD40LG, CD70, CIDEA, FASLG, TNFRSF9, TNFSF10, TP73, BIRC3, BAG3, and BCL2A1) involved in apoptosis were validated by qPCR analysis from 16 upregulated genes and six downregulated genes at a threshold of greater than twofold expression alteration of DNA RT2 profile PCR array-based gene expression profiling data. |
Evaluation of apoptosis-related gene products (protein) by Western blot
Several gene products with prominent proapoptotic function were upregulated in HBVEC cells after treatment with heme and confirmed by Western blot (Figure 4). They include FasL and TP73. Some genes for antiapoptosis, such as BCL-2, were downregulated.
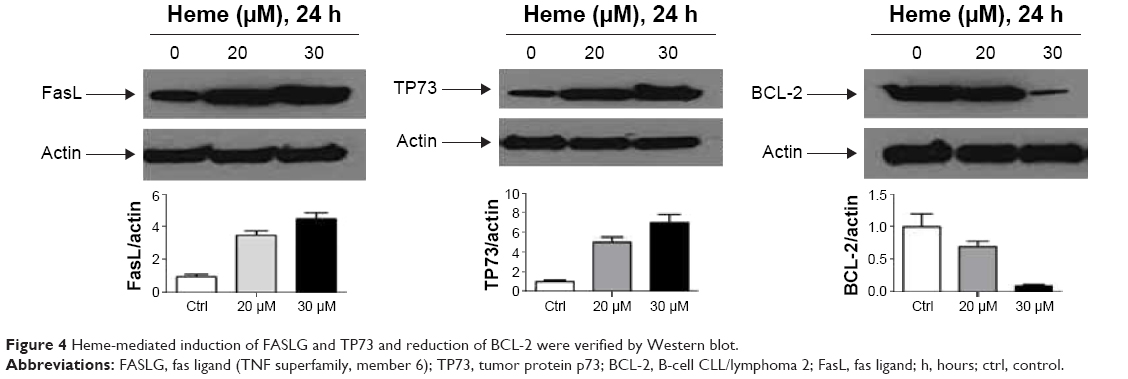 | Figure 4 Heme-mediated induction of FASLG and TP73 and reduction of BCL-2 were verified by Western blot. |
Apoptotic effects of heme on HBVEC are mediated through TP73
Treating HBVEC with heme led to the induction of TP73 (Figures 2–4). Early studies on human aortic endothelial cells (HAECs) have suggested that TP73 levels facilitate apoptosis.18 To test the role of this protein in heme-induced apoptosis of HBVECs, cells were transfected with siRNA to p73 or a nontargeting control siRNA. The transfected cells were stimulated with heme (30 μM) for 24 hours, and apoptosis was examined by TUNEL staining. Compared to control (Figure 5A–C), it was found that approximately fivefold increase of untransfected cells underwent apoptosis after heme treatment (Figure 5D–F and M). Silencing of p73 decreased apoptosis to 3.5-fold compared with control siRNA-transfected cells upon heme treatment. Silencing of TP73f protects HBVEC from heme-induced apoptosis, suggesting that p73 is an effector in mediating apoptosis induced by heme (Figure 5).
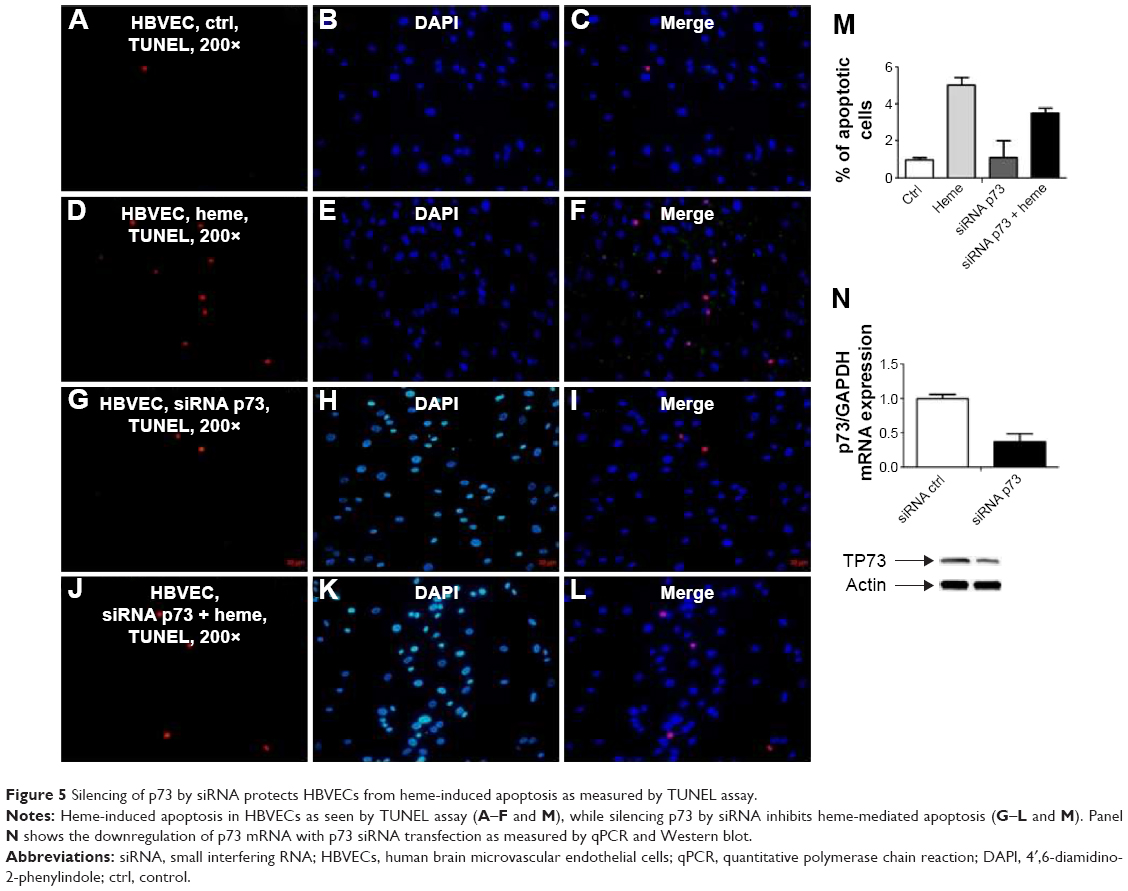 | Figure 5 Silencing of p73 by siRNA protects HBVECs from heme-induced apoptosis as measured by TUNEL assay. |
Discussion
We previously reported that heme induced 20%–50% of HBVEC cell death when treated with 10–40 μM of heme (consistent with serum heme levels in patients with severe malaria) for 24 hours with 20–30 μM causing maximum effects.14 Reduced cell viability due to heme was further determined to be caused by cell apoptosis14 and confirmed in the current study. In the present study, we used real-time RT2 profile PCR array for apoptosis to analyze apoptotic gene expression profiles of HBVEC cells after heme treatment for 24 hours. Our results showed that heme causes cell death in HBVEC via the classical apoptotic pathway. DNA RT2 profile PCR array-based gene expression profiling detected 16 upregulated genes and six downregulated genes at a threshold of greater than twofold expression alteration (P<0.05) in the heme-treated HBVEC cells. Heme-induced upregulation of apoptotic genes include ABL1, BCL2L10, BNIP3, CASP3, CASP4, CASP5, CD40LG, CD70, CIDEA, CYCS, FASLG, MCL1, TNFRSF9, TNFSF10, TP73, and TRAF3, while downregulated genes are BAG1, BAG3, BCL-2, BCL2A1, BIRC3, and BNIP3L. Eleven genes (BCL2L10, CD40LG, CD70, CIDEA, FASLG, TNFRSF9, TNFSF10, TP73, BIRC3, BAG3, and BCL2A1) involved in cellular apoptosis were chosen for validation by qPCR analysis. The decrease in mRNA expression detected by microarray profiling was confirmed by qPCR for all eight genes (BCL2L10, CD40LG, CD70, CIDEA, FASLG, TNFRSF9, TNFSF10, and TP73), and the increase in mRNA expression detected by microarray profiling was confirmed by qPCR for three other genes (BIRC3, BAG3, and BCL2A1). At the level of protein, heme-mediated induction of FASLG and TP73 and reduction of BCL-2 were verified by Western blot. We conclude that heme is potently apoptotic to human brain microvascular ECs through multiple signaling pathways. This indicates that reducing or inhibiting the production of excess free heme may potentially reduce the adverse outcomes associated with microvascular EC programmed cell death associated with severe forms of malaria such as CM, severe malaria anemia, and other hemolytic diseases.
The role of Fas/FasL in regulating ECs apoptosis and angiogenesis is well understood.19 The quiescent ECs express low levels of Fas; the expression of Fas and FasL is upregulated in activated ECs and when ECs are under hypoxic conditions or when stimulated by VEGF or TNF-α.20–23 Fas/FasL has no effect on basal apoptosis. Membrane-associated FasL is usually cleaved by upstream molecules and then releases proapoptotic sFasL. For instance, in primary human brain microvascular ECs, plasminogen is activated by converting into plasmin, which can cleave membrane-associated FasL to sFasL.19 sFasL binds to Fas receptor and FADD-activating caspase-8, the extrinsic apoptotic pathway, subsequently activating caspase-3 as well as poly (ADP-ribose) polymerase (PARP) and then causes cell apoptosis in vitro.19 The presence of plasminogen activator inhibitor-1 (PAI-1) in the pericellular space inhibits the activation of plasminogen into plasmin by cell-associated urokinase-type plasminogen activator at the surface of ECs, preventing ECs from FasL-mediated apoptosis.19 PAI-1 is the potential therapeutic target for both tumor angiogenesis and vascular diseases. In primary mouse choroidal ECs, α1(IV)NCl is a potent inhibitor of EC growth, proliferation, migration, and tumor formation. α1(IV)NCl facilitates both intrinsic and extrinsic apoptosis pathways in the ECs, the former is via activation of caspase-3 and PARP cleavage by suppressing KAK/p38-MAPK/Bcl-2 and Bcl-xL signaling pathway24 and the latter is through N- and C-terminal domains of α1(IV)NCl interacting with FasL and activating FasL as well as its downstream apoptotic molecules such as caspase-8, caspase-3, and PARP.25
BCL-2 is a growing family of apoptosis regulatory gene products, which includes either death blockers (Bcl-2, Bcl-XL, Bcl-w, Bfl-1, Brag-1, Mcl-1, and A1) or death promoters (Bax, Bak, Bcl-Xs, Bad, Bid, Bik, and Hrk).26 BCL-2 is localized at mitochondria, endoplasmic reticulum, and the nuclear envelope27,28 and has been shown to antagonize apoptosis without inducing cellular proliferation.29,30 Several studies have revealed that changes in BCL-2 levels in ECs are involved in physiological and pathological angiogenesis. VEGF can protect serum starvation-induced apoptosis of primary human umbilical vein endothelial cells through induction of BCL-2.31 When a cytomegalovirus-driven BCL-2 expression vector was transduced into HBVECs, the overexpressed BCL-2 was sufficient to inhibit EC apoptosis induced by serum starvation; thus, BCL-2 is a novel VEGF target gene.31
CD40LG (CD40L or CD154) is a member of the TNF family32 expressed on the surface of vascular ECs, vascular smooth muscle cells, immune cells such as activated CD4+ T cells and platelets. Vascular dysfunction associated with vascular inflammation, oxidative stress, and a prothrombotic state is regulated by CD40L and its receptor CD40 interaction.33–35 Both the membrane and a soluble CD40L (sCD40L) have similar proinflammatory effects on vascular cells. Nuclear factor-kappa B (NF-κB) is a crucial inter player in the CD40L-induced inflammatory network. CD40L activates the ECs, subsequently recruiting immune cells through NF-κB, while protein kinase C beta inhibition attenuated CD40L-induced NF-κB activation.35 NF-κB is implicated in the TNF-αCD40 expression in ECs, while sirtuin 1 decreases the expression and activation of acetylated NF-κB p65.
TP73 is well documented for its role in the induction of apoptosis and cell cycle arrest.36 In the present study, we found that heme had an effect on p73-mediated regulation of apoptosis in HBVECs, TP73 levels were found to be upregulated, and depletion of TP73 inhibited heme-induced apoptosis. TP73 maps to chromosome 1p36 and encodes a protein that is structurally homologous to the p53 protein.37,38 Several mechanisms by which TP73 induces endothelial apoptosis have been revealed. TNF-α is an inflammatory cytokine that shows dual roles in cardiac function and pathology.18 Rastogi et al18 found that upon TNF-α stimulation, apoptosis signal-regulating kinase 1 (ASK1) mediated Rb inactivation, promoting upregulation of the proapoptotic protein TP73α through E2F1 regulation. TNF-α can induce both cell survival and cell apoptosis pathways simultaneously; for instance, it activates cell program death in HAECs, whereas it enhances the proliferation of vascular smooth muscle cells using Rb-E2F signaling pathway.39,40 When HAECs were exposed to TNFα, ASK1 and TP73 levels were upregulated. Depletion of TP73α reduced TNF-α-induced apoptosis, implicating TP73α to be one of the primary contributors to EC apoptosis. Some studies documented that the apoptotic activity of TP73 needs the activated kinase-competent c-Abl kinase, which is located upstream for TP73 to phosphorylate TP73 due to DNA damaging factors.41,42 Salimath et al demonstrated that expression of VEGF gene is inhibited by TP73.37 As is well known, tumor cells and stromal cells express VEGF mRNA, while ECs express VEGF receptors VEGF-R1 and VEGF-R2. This expression pattern indicates that VEGF functions predominantly as a paracrine modulator of angiogenesis.37 By studying the effect of ectopically expressed TP73 on the regulation of the VEGF gene using a cell line (Saos-2, osteosarcoma cell line), which has a homozygous deletion at the p53 gene locus43 and has no detectable levels of endogenous TP73 protein,44 they concluded that TP73 regulates EC survival functions by inhibiting VEGF expression in tumor cells.37
Deciphering the mechanism(s) mediating fatal malaria pathogenesis continues to be a challenge to malaria researchers.45 Recent studies indicate that increased levels of circulating free heme in plasma during malaria infection induces system inflammation that results in the damage of host brain microvascular endothelium leading to fatal CM pathogenesis.12,46–48 Heme oxygenase (HO) is the rate-limiting step enzyme in the degradation of heme groups to biliverdin, carbon monoxide, and iron. HO-1 protects against cellular stress, including oxidative stress, heavy metal toxicity, UV radiation, and inflammation, and prevents deleterious effects of heme as well as mediates anti-inflammatory and antiapoptotic functions,49–52 which are necessary to facilitate the repair of injured tissues.53 Thus, we propose that agents that effectively elevate HO-1 and reduce free plasma heme in the infected host coupled with appropriate antimalaria treatments could significantly reduce or eliminate malaria-associated mortalities.13,14,54
Acknowledgment
This work was supported by the National Institutes of Health, grant numbers NIH/NINDS 1 R56 NS091616-01 and NIH/RCMI RR033062 (G12).
Disclosure
The authors report no conflicts of interest in this work.
References
Baptista FG, Pamplona A, Pena AC, Mota MM, Pied S, Vigário AM. Accumulation of Plasmodium berghei-infected red blood cells in the brain is crucial for the development of cerebral malaria in mice. Infect Immun. 2010;78(9):4033–4039. | ||
Clark IA, Rockett KA, Cowden WB. Proposed link between cytokines, nitric oxide and human cerebral malaria. Parasitol Today. 1991;7(8):205–207. | ||
Medana IM, Turner GD. Human cerebral malaria and the blood-brain barrier. Int J Parasitol. 2006;36(5):555–568. | ||
Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes decrease the integrity of human blood-brain barrier endothelial cell monolayers. J Infect Dis. 2007;195(7):942–950. | ||
Wassmer SC, Taylor T, Maclennan CA, et al. Platelet-induced clumping of Plasmodium falciparum-infected erythrocytes from Malawian patients with cerebral malaria-possible modulation in vivo by thrombocytopenia. J Infect Dis. 2008;197(1):72–78. | ||
White NJ, Turner GD, Medana IM, Dondorp AM, Day NP. The murine cerebral malaria phenomenon. Trends Parasitol. 2010;26(1): 11–15. | ||
Belnoue E, Potter SM, Rosa DS, et al. Control of pathogenic CD8+ T cell migration to the brain by IFN-gamma during experimental cerebral malaria. Parasite Immunol. 2008;30(10):544–553. | ||
Chen L, Sendo F. Cytokine and chemokine mRNA expression in neutrophils from CBA/NSlc mice infected with Plasmodium berghei ANKA that induces experimental cerebral malaria. Parasitol Int. 2001;50(2):139–143. | ||
Claser C, Malleret B, Gun SY, et al. CD8+ T cells and IFN-gamma mediate the time-dependent accumulation of infected red blood cells in deep organs during experimental cerebral malaria. PLoS One. 2011;6(4):e18720. | ||
Core A, Hempel C, Kurtzhals JA, Penkowa M. Plasmodium berghei ANKA: erythropoietin activates neural stem cells in an experimental cerebral malaria model. Exp Parasitol. 2011;127(2):500–505. | ||
Franklin BS, Ishizaka ST, Lamphier M, et al. Therapeutical targeting of nucleic acid-sensing Toll-like receptors prevents experimental cerebral malaria. Proc Natl Acad Sci U S A. 2011;108(9):3689–3694. | ||
Pamplona A, Ferreira A, Balla J, et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med. 2007;13(6):703–710. | ||
Liu M, Amodu AS, Pitts S, et al. Heme mediated STAT3 activation in severe malaria. PLoS One. 2012;7(3):e34280. | ||
Liu M, Wilson NO, Hibbert JM, Stiles JK. STAT3 regulates MMP3 in heme-induced endothelial cell apoptosis. PLoS One. 2013;8(8):e71366. | ||
Wilson NO, Huang MB, Anderson W, et al. Soluble factors from Plasmodium falciparum-infected erythrocytes induce apoptosis in human brain vascular endothelial and neuroglia cells. Mol Biochem Parasitol. 2008;162(2):172–176. | ||
Combes V, El-Assaad F, Faille D, Jambou R, Hunt NH, Grau GE. Microvesiculation and cell interactions at the brain-endothelial interface in cerebral malaria pathogenesis. Prog Neurobiol. 2010;91(2):140–151. | ||
Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect Immun. 2006;74(6):3262–3270. | ||
Rastogi S, Rizwani W, Joshi B, Kunigal S, Chellappan SP. TNF-alpha response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ. 2012;19(2):274–283. | ||
Bajou K, Peng H, Laug WE, et al. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell. 2008;14(4):324–334. | ||
Bouchet D, Tesson L, Ménoret S, et al. Differential sensitivity of endothelial cells of various species to apoptosis induced by gene transfer of Fas ligand: role of FLIP levels. Mol Med. 2002;8(10):612–623. | ||
Cardier JE, Schulte T, Kammer H, Kwak J, Cardier M. Fas (CD95, APO-1) antigen expression and function in murine liver endothelial cells: implications for the regulation of apoptosis in liver endothelial cells. FASEB J. 1999;13(14):1950–1960. | ||
Chavakis E, Dimmeler S. Regulation of endothelial cell survival and apoptosis during angiogenesis. Arterioscler Thromb Vasc Biol. 2002;22(6):887–893. | ||
Dimova EY, Kietzmann T. Cell type-dependent regulation of the hypoxia-responsive plasminogen activator inhibitor-1 gene by upstream stimulatory factor-2. J Biol Chem. 2006;281(5):2999–3005. | ||
Boosani CS, Nalabothula N, Munugalavadla V, et al. FAK and p38-MAP kinase-dependent activation of apoptosis and caspase-3 in retinal endothelial cells by alpha1(IV)NC1. Invest Ophthalmol Vis Sci. 2009;50(10):4567–4575. | ||
Verma RK, Gunda V, Pawar SC, Sudhakar YA. Extra cellular matrix derived metabolite regulates angiogenesis by FasL mediated apoptosis. PLoS One. 2013;8(12):e80555. | ||
Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3(6):614–620. | ||
Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53(19): 4701–4714. | ||
Monaghan P, Robertson D, Amos TA, Dyer MJ, Mason DY, Greaves MF. Ultrastructural localization of bcl-2 protein. J Histochem Cytochem. 1992;40(12):1819–1825. | ||
Hockenbery D, Nuñez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348(6299):334–336. | ||
Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335(6189):440–442. | ||
Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998;273(21):13313–13316. | ||
André P, Prasad KS, Denis CV, et al. CD40L stabilizes arterial thrombi by a beta3 integrin – dependent mechanism. Nat Med. 2002;8(3):247–252. | ||
Hausding M, Jurk K, Daub S, et al. CD40L contributes to angiotensin II-induced pro-thrombotic state, vascular inflammation, oxidative stress and endothelial dysfunction. Basic Res Cardiol. 2013; 108(6):386. | ||
Wang W, Bai L, Qiao H, et al. The protective effect of fenofibrate against TNF-alpha-induced CD40 expression through SIRT1-mediated deacetylation of NF-kappaB in endothelial cells. Inflammation. 2014;37(1):177–185. | ||
Wu Z, Zhao G, Peng L, et al. Protein kinase C beta mediates CD40 ligand-induced adhesion of monocytes to endothelial cells. PLoS One. 2013;8(9):e72593. | ||
Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 2006;13(6):962–972. | ||
Salimath B, Marme D, Finkenzeller G. Expression of the vascular endothelial growth factor gene is inhibited by p73. Oncogene. 2000;19(31):3470–3476. | ||
Jost CA, Marin MC, Kaelin WG Jr. p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature. 1997;389(6647):191–194. | ||
Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274(5288):782–784. | ||
Wang X, Yue TL, Barone FC, White RF, Gagnon RC, Feuerstein GZ. Concomitant cortical expression of TNF-alpha and IL-1 beta mRNAs follows early response gene expression in transient focal ischemia. Mol Chem Neuropathol. 1994;23(2–3):103–114. | ||
Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature. 1999;399(6738):809–813. | ||
Yuan ZM, Shioya H, Ishiko T, et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999;399(6738):814–817. | ||
Diller L, Kassel J, Nelson CE, et al. p53 functions as a cell cycle control protein in osteosarcomas. Mol Cell Biol. 1990;10(11):5772–5781. | ||
Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90(4):809–819. | ||
Zhu J, Wu X, Goel S, et al. MAPK-activated protein kinase 2 differentially regulates plasmodium falciparum glycosylphosphatidylinositol-induced production of tumor necrosis factor-{alpha} and interleukin-12 in macrophages. J Biol Chem. 2009;284(23):15750–15761. | ||
Hunt NH, Stocker R. Heme moves to center stage in cerebral malaria. Nat Med. 2007;13(6):667–669. | ||
Pamplona A, Hanscheid T, Epiphanio S, Mota MM, Vigário AM. Cerebral malaria and the hemolysis/methemoglobin/heme hypothesis: shedding new light on an old disease. Int J Biochem Cell Biol. 2009;41(4):711–716. | ||
Epiphanio S, Campos MG, Pamplona A, et al. VEGF promotes malaria-associated acute lung injury in mice. PLoS Pathog. 2010;6(5):e1000916. | ||
Datta D, Banerjee P, Gasser M, Waaga-Gasser AM, Pal S. CXCR3-B can mediate growth-inhibitory signals in human renal cancer cells by downregulating the expression of heme oxygenase-1. J Biol Chem. 2010;285(47):36842–36848. | ||
Geuken E, Buis CI, Visser DS, et al. Expression of heme oxygenase-1 in human livers before transplantation correlates with graft injury and function after transplantation. Am J Transplant. 2005;5(8):1875–1885. | ||
Bussolati B, Mason JC. Dual role of VEGF-induced heme-oxygenase-1 in angiogenesis. Antioxid Redox Signal. 2006;8(7–8):1153–1163. | ||
Pae HO, Oh GS, Choi BM, Kim YM, Chung HT. A molecular cascade showing nitric oxide-heme oxygenase-1-vascular endothelial growth factor-interleukin-8 sequence in human endothelial cells. Endocrinology. 2005;146(5):2229–2238. | ||
Datta D, Dormond O, Basu A, Briscoe DM, Pal S. Heme oxygenase-1 modulates the expression of the anti-angiogenic chemokine CXCL-10 in renal tubular epithelial cells. Am J Physiol Renal Physiol. 2007;293(4):F1222–F1230. | ||
Wilson NO, Solomon W, Anderson L, et al. Pharmacologic inhibition of CXCL10 in combination with anti-malarial therapy eliminates mortality associated with murine model of cerebral malaria. PLoS One. 2013;8(4):e60898. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.