Back to Journals » Drug Design, Development and Therapy » Volume 10
Plasmid pORF-hTRAIL targeting to glioma using transferrin-modified polyamidoamine dendrimer
Authors Gao S, Li J, Jiang C, Hong B, Hao B
Received 6 September 2015
Accepted for publication 19 November 2015
Published 17 December 2015 Volume 2016:10 Pages 1—11
DOI https://doi.org/10.2147/DDDT.S95843
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Song Gao,1,* Jianfeng Li,2 Chen Jiang,2 Bo Hong,3 Bing Hao4,*
1Department of Clinical Laboratory, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, 2Key Laboratory of Smart Drug Delivery, Ministry of Education, Department of Pharmaceutics, School of Pharmacy, Fudan University, Shanghai, 3Department of Pathology, The Second Affiliated Hospital, 4Key Laboratory of Combined Multi-Organ Transplantation, Ministry of Public Health, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: A gene drug delivery system for glioma therapy based on transferrin (Tf)-modified polyamidoamine dendrimer (PAMAM) was prepared. Gene drug, tumor necrosis factor-related apoptosis-inducing ligand (hTRAIL)-encoding plasmid open reading frame (pORF-hTRAIL, Trail), was condensed by Tf-modified PAMAM to form nanoparticles (NPs). PAMAM-PEG-Tf/DNA NPs showed higher cellular uptake, in vitro gene expression, and cytotoxicity than PAMAM-PEG/DNA NPs in C6 cells. The in vivo targeting efficacy of NPs was visualized by ex vivo fluorescence imaging. Tf-modified NPs showed obvious glioma-targeting trend. Plasmid encoding green fluorescence protein (GFP) was also condensed by modified or unmodified PAMAM to evaluate the in vivo gene expression level. The PAMAM-PEG-Tf/plasmid encoding enhanced green fluorescence protein (pEGFP) NPs exhibited higher GFP expression level than PAMAM-PEG/pEGFP NPs. TUNEL assay revealed that Tf-modified NPs could induce much more tumor apoptosis. The median survival time of PAMAM-PEG-Tf/Trail-treated rats (28.5 days) was longer than that of rats treated with PAMAM-PEG/Trail (25.5 days), temozolomide (24.5 days), PAMAM-PEG-Tf/pEGFP (19 days), or saline (17 days). The therapeutic effect was further confirmed by magnetic resonance imaging. This study demonstrated that targeting gene delivery system had potential application for the treatment of glioma.
Keywords: gene therapy, BBB, hTRAIL, PAMAM, C6 glioma
Introduction
Malignant gliomas, which have higher morbidity among brain tumors of adults, have a median survival time of approximately 12–15 months.1 The prognosis of this brain tumor remains poor due to the insufficient response to the available treatments, including surgical intervention, radiation, and chemotherapy.2 One of the obstacles for glioma therapies is the infiltrative nature of glioma cells, which could invade into normal brain parenchyma. Due to the importance of normal brain function, excessive surgical resection is restricted which could lead to residual of tumor cells.3,4 Another obstacle is the non-specificity of adjuvant treatment modalities including radiotherapy and chemotherapy, which cannot specifically reach the widely spread tumor cells. These modalities are also related to toxicities in terms of both systemic illness and cognitive impairment.5 Thus, new approaches are critical to be developed to treat this devastating disease with reduced side effects.
The tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) could induce apoptosis via the p53-independent pathway by binding to the death receptors 4 (TRAIL-R1) and 5 (TRAIL-R2/KILLER) at the cell surface.6 The specificity of cytotoxicity could be achieved by the differences of receptor expressions. On one hand, many glioma cells express the agonist TRAIL receptors with no or undetectable levels of the antagonist receptors.7 On the other hand, normal cells also express antagonist TRAIL receptors.8,9 Therefore, TRAIL protein may allow killing tumor cells alone without affecting the normal brain cells. TRAIL protein has been reported to induce tumor regression in a glioma xenograft model via intravenous administration.10,11 However, some disadvantages, including the short half-life, immunogenicity, and enzymolysis, limit the usage of protein drugs. Gene therapy with genes encoding therapeutic proteins like TRAIL may offer therapeutic promise including long-lasting expression of therapeutic proteins.
Due to the invasive nature, tumor cells could penetrate into normal brain tissue. The tumor vasculatures in the infiltrated region possess some features of normal brain including blood–brain barrier (BBB).12–14 Gene drugs cannot reach these tumor regions due to the impermeability of BBB to the large molecules.15–18 Therefore, there is an urgent need for developing smart carriers for gene delivery targeting glioma. Learning from the nature, many specific receptors including transferrin (Tf) receptor and insulin receptor are overexpressed on BBB that can potentially be utilized for facilitating brain targeting.19–21 Among these receptors, Tf receptor is a very promising one because of the restricted expression in brain capillaries than other tissues.22 Besides, the expression level of Tf receptor on tumor cells is much higher than that of normal cells.23,24 Thus, Tf is selected as the glioma-targeting ligand.
Non-viral vectors have been used to deliver therapeutic genes into the brain with higher safety. Among the vectors reported, polyamidoamine dendrimer (PAMAM) has proven to be a promising gene carrier.25 PAMAM-PEG-Tf has been successfully synthesized previously and showed great brain-targeting efficiency.26 Besides, it has been reported that Tf-conjugated polymersomes achieved preferential C6 glioma targeting in vivo and Tf-modified cisplatin liposome were identified to accumulate in malignant glioma cells in bEnd3/C6 co-culture BBB models.27,28
In this study, hTRAIL-encoding plasmid (pORF-hTRAIL, Trail) was selected as the therapeutic gene for glioma treatment. PAMAM-PEG-Tf was synthesized, and incorporated with pORF-hTRAIL to form nanoparticles (NPs). The antitumor effect of the NPs was evaluated in C6 glioma tumors-bearing Sprague Dawley (SD) rats.
Materials and methods
The plasmids encoding enhanced green fluorescence protein N2 (pEGFP-N2; Clontech, Palo Alto, CA, USA) and pORF-hTRAIL (Invitrogen, San Diego, CA, USA) were purified by QIAGEN Plasmid Mega Kit (Qiagen GmbH, Hilden, Germany). Human holo-Tf and methyl green were purchased from Sigma-Aldrich (St Louis, MO, USA). PAMAM G5 dendrimer was purchased from Dendritech, Inc (Midland, MI, USA). A-Maleimidyl-u-N-hydroxysuccinimidyl polyethyleneglycol (NHS-PEG-MAL, molecular weight 3,500) was obtained from Jenkem Technology (Beijing, People’s Republic of China). 4,6-diamidino-2-phenylindole (DAPI) and ethidium monoazide bromide (EMA) were purchased from Molecular Probes (Eugene, OR, USA).
Male SD rats (4–5 weeks) of 200–220 g body weight were purchased from Sino-British SIPPR/BK Lab Animal Ltd. (Shanghai, People’s Republic of China) and maintained under standard housing conditions. All animal experimental protocols were in accordance with guidelines evaluated and approved by the ethics committee of Zhejiang University.
The rat C6 glioma cell line was kindly provided by Prof Feng L (Institute of Neuroscience, Shanghai Institutes for Biological Sciences). In addition, this cell line was purchased from Shanghai Cell Bank, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China. C6 cells were expanded and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum, 100 U/mL penicillin, and 100 mg/mL streptomycin and cultured at 37°C under a humidified atmosphere containing 5% CO2.
Synthesis of PAMAM derivatives
Synthesis of PAMAM derivate was carried out as previously reported.26 In brief, PAMAM was reacted with NHS-PEG-MAL at a mole ratio of 1:2 in phosphate-buffered saline (PBS) pH 8.0 for 2 hours at room temperature (RT). The primary amino groups of PAMAM were specifically reacted with NHS groups of NHS-PEG-MAL. The resulting conjugate, PAMAM-PEG-MAL, was purified by ultrafiltration (cutoff: 5 kDa) and the solution was changed to PBS 7.0. Tf was thiolated by the Traut’s reagent, and the extent of thiolation was analyzed by the Ellmann’s reagent. Then PAMAM-PEG-MAL was reacted with Tf at a mole ratio of 1:1 in PBS 7.0 for 24 hours at RT. The MAL groups of PAMAM-PEG-MAL were specifically reacted with thiol groups of Tf. Theoretically, there are totally 128 free amine groups in G5 PAMAM and two of them were modified with polyethyleneglycol (PEG). One molecular Tf was attached to the end of one PEG.
Preparation and characterization of dendrimers/DNA NPs
Dendrimers (PAMAM-PEG-Tf) were diluted to desired concentrations in PBS 7.4. DNA solution was added to obtain appropriate weight ratios (0.1:1, 0.5:1, 1:1, 3:1, PAMAM to pORF-hTRAIL) and then vortexed for 30 seconds at RT. Agarose gel electrophoresis was performed to evaluate the retardation of DNA in the NPs. Freshly prepared NPs with weight ratio of 3:1 were used in the subsequent experiments. The morphology of NPs was examined under a transmission electron microscope (TEM) (JEM-2010, JEOL, Tokyo, Japan).
Cellular uptake of NPs in C6 cells
Plasmid DNA (pORF-hTRAIL) was covalently labeled with a fluorescent dye, EMA. Fresh plasmid DNA solution (0.1 mg/mL) and EMA (1 mg/mL) were incubated at equal volume for 0.5 hour in the dark. The complex was then exposed to ultraviolet (UV) light (365 nm) for 1 hour. The resulting solution was precipitated by adding ethanol to a final concentration of 30% (v/v). The precipitate was collected and dissolved in 50 mM sodium sulfate solution. This EMA-DNA solution was used to prepare the NPs as described earlier.
C6 cells were seeded at a density of 5×103/well in 24-well plates (Corning-Coaster, Tokyo, Japan), incubated for 48 hours, and checked under the microscope for morphology. When reaching 90% confluence, cells were incubated with naked DNA (EMA-labeled pORF-hTRAIL, Trail), PAMAM-PEG/Trail, and PAMAM-PEG-Tf/Trail at a dose of 5 μg/well DNA. After 2-hour incubation, the medium was removed, and the cells were washed twice with PBS. Then, the cells were visualized and imaged using a fluorescence microscope.
In vitro gene expression
C6 cells were seeded at a density of 5×103/well in 24-well plates, incubated for 24 hours, and checked under the microscope for morphology. When reaching 60% confluence, cells were incubated with PBS, naked DNA (pEGFP-N2), PAMAM-PEG/pEGFP, and PAMAM-PEG-Tf/pEGFP at a dose of 5 μg/well DNA. After 2-hour incubation, the medium was removed, and the cells were washed twice with PBS. After incubation for another 48 hours, cells were washed and imaged using a fluorescence microscope.
In vitro cell apoptosis
C6 cells were seeded at a density of 1×104/well in six-well plates, incubated for 24 hours, and checked under the microscope for morphology. When reaching 60% confluence, cells were incubated with PBS, naked DNA (Trail), PAMAM-PEG/Trail, and PAMAM-PEG-Tf/Trail at a dose of 10 μg/well DNA. After 2-hour incubation, cells were washed and further cultured in DMEM medium containing 10% fetal bovine serum for 48 hours. Then, the cells were incubated with Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit following the manufacturer’s protocol (KeyGEN, Nanjing, People’s Republic of China). Briefly, the collected cells were washed twice with PBS and incubated with Annexin V-FITC and propidium iodide for 15 minutes in the dark at RT. The cells were washed twice and then analyzed within 60 minutes by flow cytometry (BD FACSCalibur™; BD Biosciences, San Jose, CA, USA).
In vivo distribution of NPs in C6 glioma xenografted SD rat brain
C6 model: C6 cells (1×105 per well) were cultured in petri dish until 90% confluence was reached. Cells (2×104/5 μL in PBS) were implanted into the right striatum (3 mm lateral, 1 mm before the bregma, and 5 mm of depth) of SD rats by using a stereotactic fixation device with a rat adaptor. Eight days after implantation, rats received PAMAM-PEG/Trail and PAMAM-PEG-Tf/Trail NPs via the tail vein at a dose of 200 μg pDNA/rat, and 2 hours later were anesthetized with diethyl ether and killed by decapitation. Distribution of NPs in brain was investigated using Maestro™ (PerkinElmer Inc., Waltham, MA, USA) 2 in vivo imaging system 2 hours after injection.
Qualitative evaluation of in vivo gene expression
The PAMAM-PEG/pEGFP and PAMAM-PEG-Tf/pEGFP NPs (10:1, PAMAM to DNA, w/w) were injected into the tail vein of rats at a dose of 200 mg DNA/rat 10 days after C6 implantation. After 48 hours, animals were anesthetized with 10% chloral hydrate and sacrificed by the perfusion of saline and 4% paraformaldehyde. The brains were fixed in 4% paraformaldehyde and dehydrated in 15% and 30% sucrose solution. Frozen sections of rat gliomas with 20 μm thickness were prepared with a cryotome Cryostat (Leica, CM 1900, Leica Microsystems, Wetzlar, Germany) and stained with 300 nM DAPI for 15 minutes at RT. After washing twice with PBS, the sections were examined under the fluorescence microscope.
Immunohistochemistry of apoptosis markers
At day 8, 10, 12, 14, and 16 after implantation, rats received PAMAM-PEG/Trail, PAMAM-PEG-Tf/pEGFP, PAMAM-PEG-Tf/Trail NPs, or saline (control) via intravenous at a dose of 200 μg DNA/rat. Temozolomide was administrated via intragastric administration at day 8, 9, 10, 11, and 12 (one course of treatment). At day 18 after implantation, rats were sacrificed and used to prepare brain glioma frozen sections (20 μm) as described earlier. Terminal deoxynucleotidyl transferase-Biotin-dUTP nick end labeling (TUNEL) was used to detect apoptosis.
Therapeutic effect evaluation by survival monitoring and MRI imaging
After the same treatment course as described earlier, the survival of rats was monitored every day (n=10). Magnetic resonance imaging (MRI) was performed using a Bruker Biospec 4.7 T/30 cm scanner as previously suggested.29 Imaging was performed using a custom-built 50 mm diameter send-receive birdcage volume coil. Gradients were 12 cm diameter at 25 G/cm. Heartbeat rates were monitored so as to keep it at 60–120 times per minute by adjusting the ratio of isoflurane/oxygen. T2-weighted images were performed with the following parameters: slice thickness =1.00 mm, FOV (field of view) =2 cm ×2 cm, echo time =11 ms, repetition time =3,000 ms, number of averages =8, and matrix size =128×128. MRI was acquired before and after the same therapeutic regimen mentioned earlier.
Results
Preparation of dendrimers/DNA NPs
The ability of PAMAM-PEG-Tf to condense DNA was evaluated by electrophoretic analysis at different weight ratios. Complete retardation was achieved at the weight ratio of 3:1 (Figure 1A). This formulation was used in the following studies. The TEM results showed a narrow size distribution of NPs and the average diameter was 119±13 nm, data is presented as mean ± standard deviation (Figure 1B).
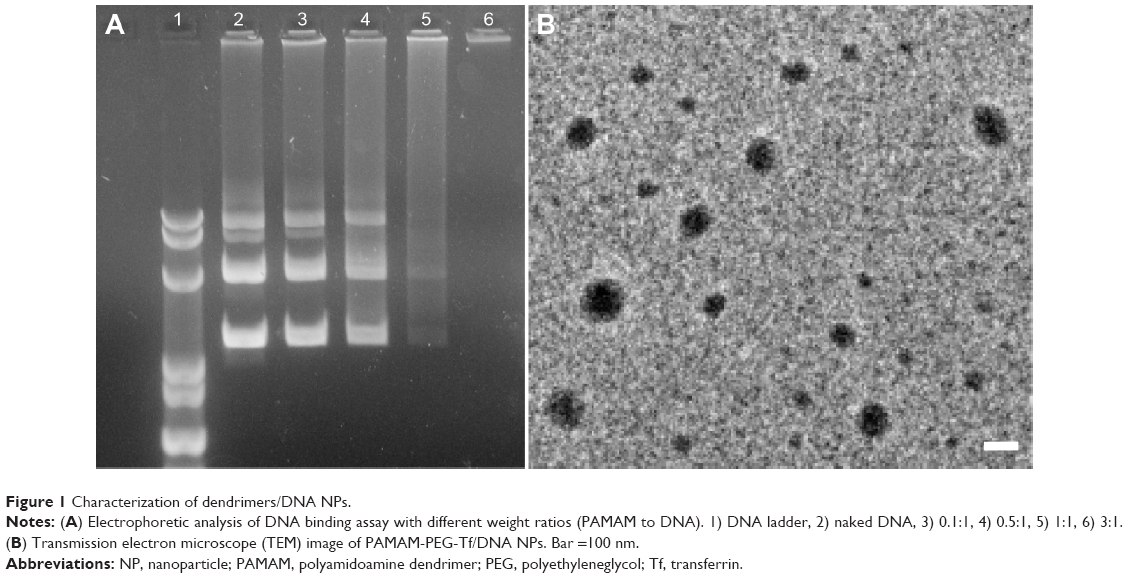 | Figure 1 Characterization of dendrimers/DNA NPs. |
Cellular uptakes of NPs
Trail was previously labeled with EMA for intracellular tracking. The cellular uptake of naked Trail, PAMAM-PEG/Trail, and PAMAM-PEG-Tf/Trail into C6 cells was compared by fluorescent imaging (Figure 2). Because of the negative charge, naked Trail showed little uptake into cells. When condensed into NPs, the cellular uptake was greatly increased. Besides, targeting NPs PAMAM-PEG-Tf/Trail exhibited significantly higher uptake than non-targeting NPs PAMAM-PEG/Trail. Tf conjugation facilitated the cellular uptake of NPs in a specific manner.
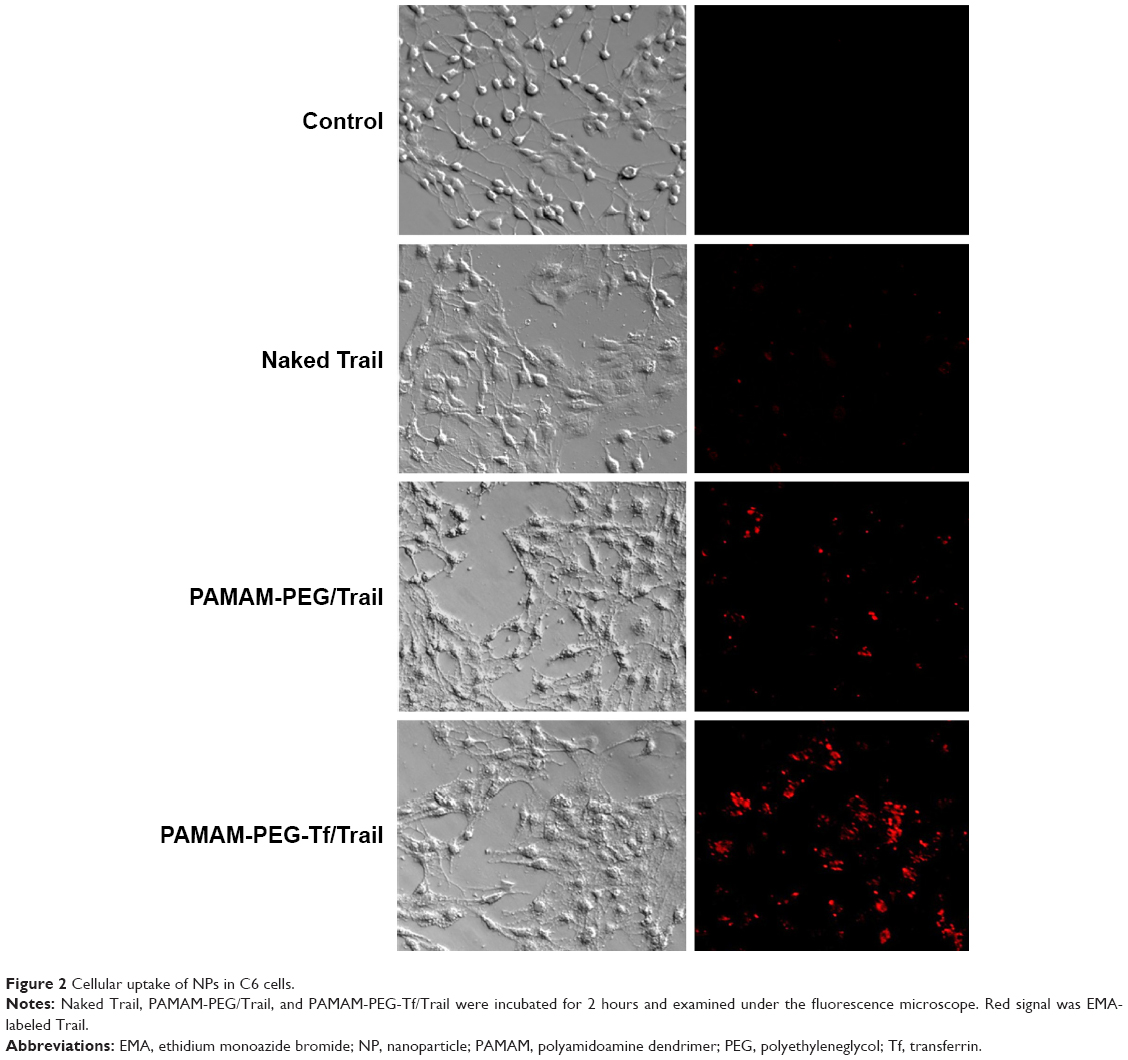 | Figure 2 Cellular uptake of NPs in C6 cells. |
In vitro gene expression
The expression of pEGFP was utilized to demonstrate the transfection efficiency of NPs. Due to the poor cellular uptake, naked pEGFP-treated cells showed little green fluorescence protein (GFP) expression as the blank control (Figure 3A and B). NP-treated cells had more GFP expression than naked pEGFP-treated ones, which contributed to the condensation of positively charged PAMAM (Figure 3C and D). Tf modification further increased GFP expression due to the overexpression of Tf receptors, which resulted in enhanced cellular uptake.
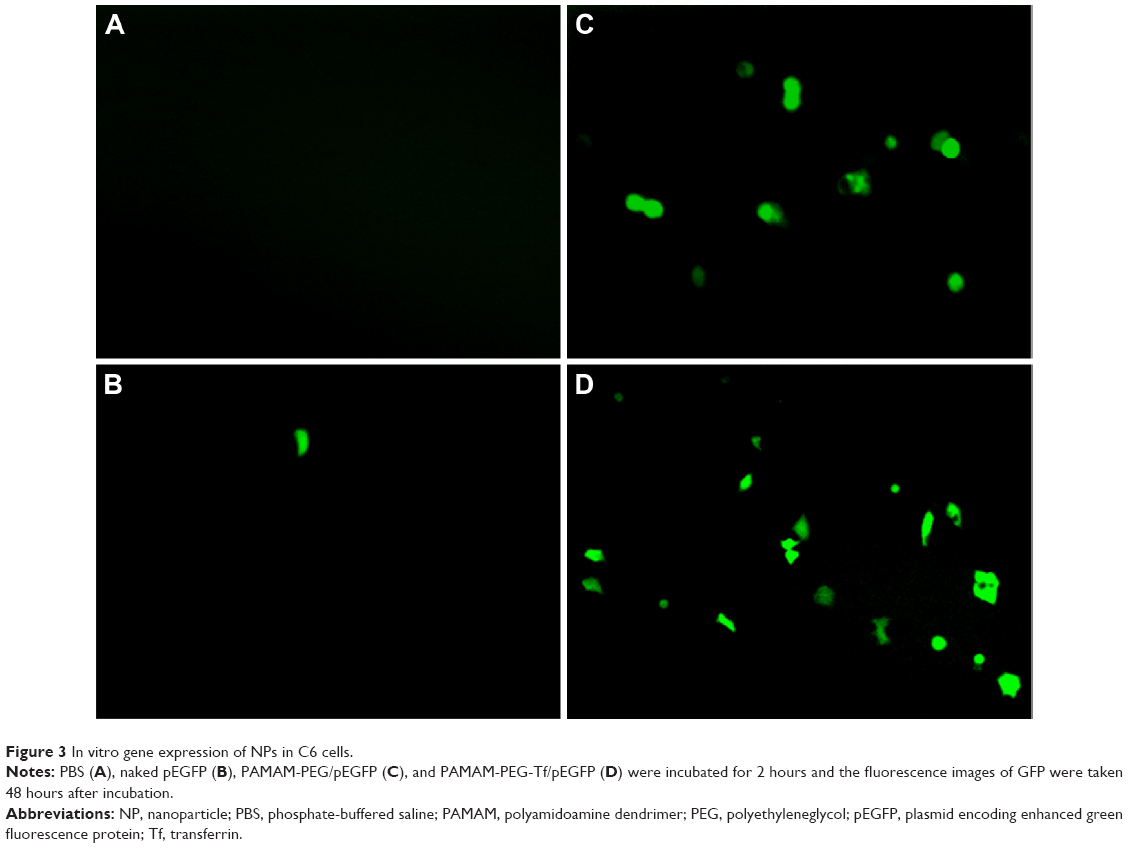 | Figure 3 In vitro gene expression of NPs in C6 cells. |
In vitro cell apoptosis
When the transfection efficiency was confirmed, the cell apoptosis of NPs containing therapeutic gene was evaluated by Annexin V-FITC staining. Annexin V-FITC could label the membrane phospholipid phosphatidylserine, which is translocated from the inner to the outer side of cell membranes in early cell apoptosis. Propidium iodide is a molecular probe that can penetrate through destructed membranes and label nucleus.30 UL stands for dead cells, UR for late apoptosis cells, LL for live cells, and LR for early apoptotic cells. Naked Trail induced total apoptosis rate (UR + LR) similar to that of PBS-treated group (Figure 4A and B). PAMAM-PEG/Trail-treated group had a total apoptosis rate of 19.22% (Figure 4C). In addition, PAMAM-PEG-Tf/Trail induced most significant cell apoptosis of 40.24% (Figure 4D), which demonstrated the superiority of dendrimer condensing and Tf modification.
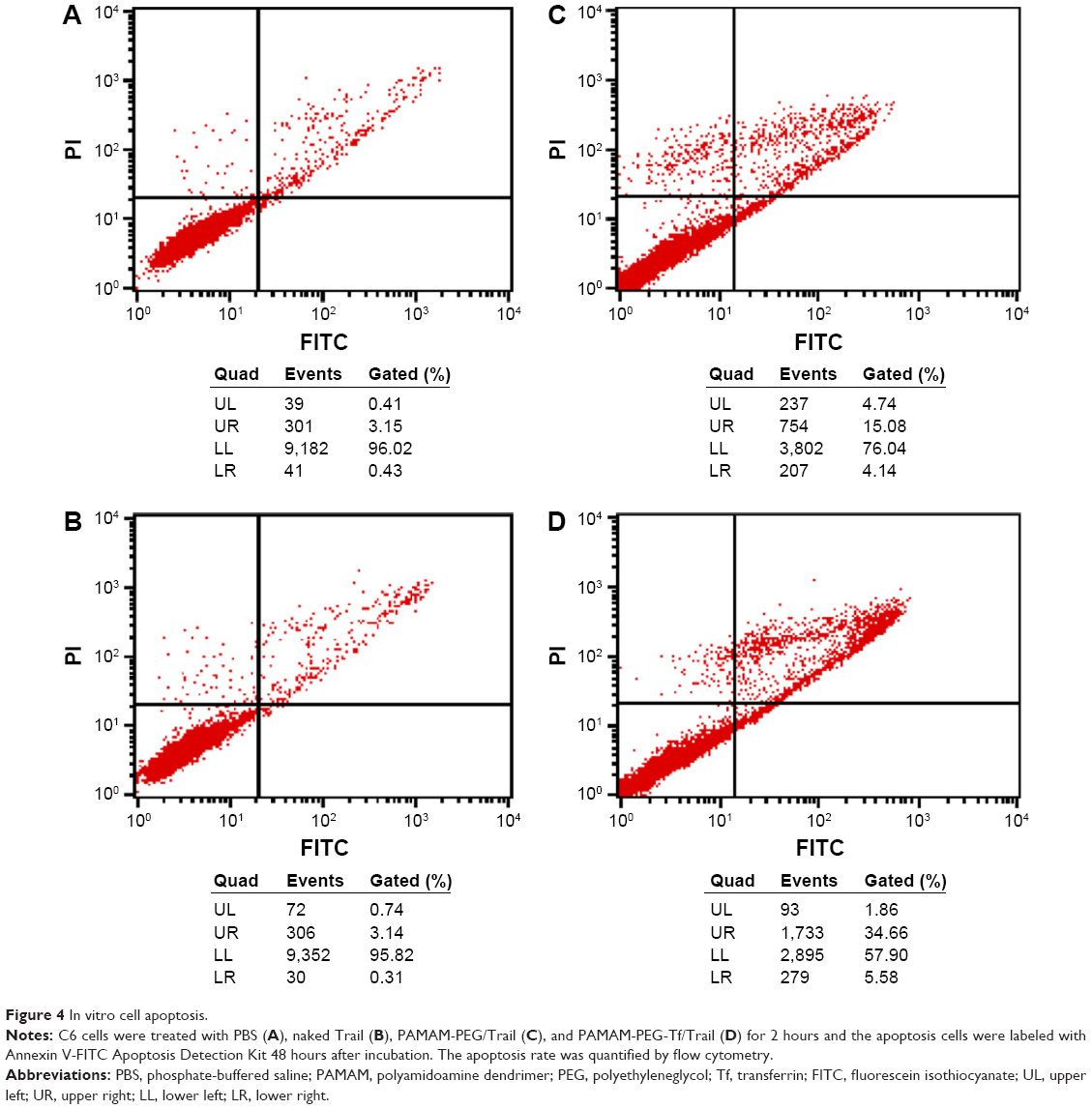 | Figure 4 In vitro cell apoptosis. |
In vivo distribution of NPs in C6 glioma-bearing rat brain
C6 glioma-bearing rats were injected with PAMAM-PEG-Tf/EMA-labeled DNA NPs and PAMAM-PEG/DNA NPs. Ex vivo fluorescent images of brain were taken 2 hours after injection. EMA-labelled DNA was obviously accumulated in the brain tumor, which treated with PAMAM-PEG-Tf/DNA NPs (Figure 5B and D), while that in the brain tumor treated with PAMAM/DNA NPs was less concentrated (Figure 5A and C).
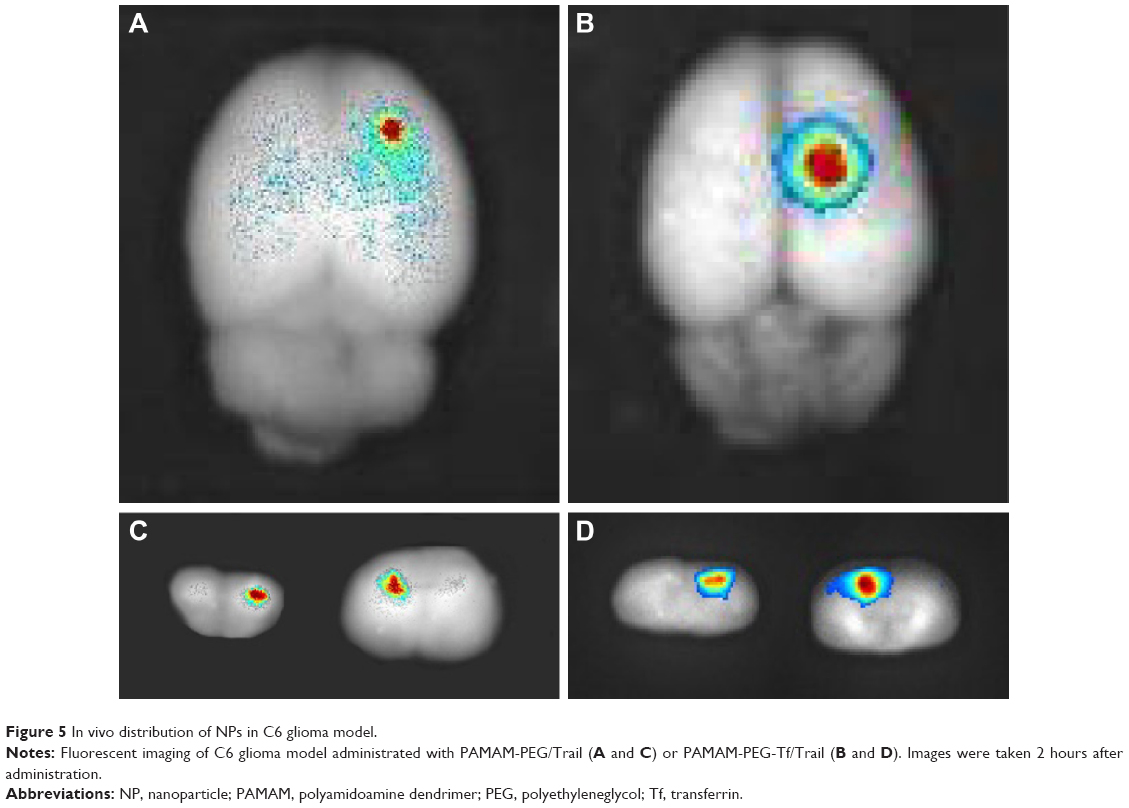 | Figure 5 In vivo distribution of NPs in C6 glioma model. |
Qualitative evaluation of in vivo gene expression
GFP expression in the brain tumor was examined 48 hours after administration (Figure 6). In PAMAM-PEG/pEGFP NP-treated group, GFP was expressed near the margin of the glioma region. In PAMAM-PEG-Tf/pEGFP NP-treated group, GFP was generally found both near the margin and inside of the tumor and was much more than that of PAMAM-PEG/pEGFP NP-treated group.
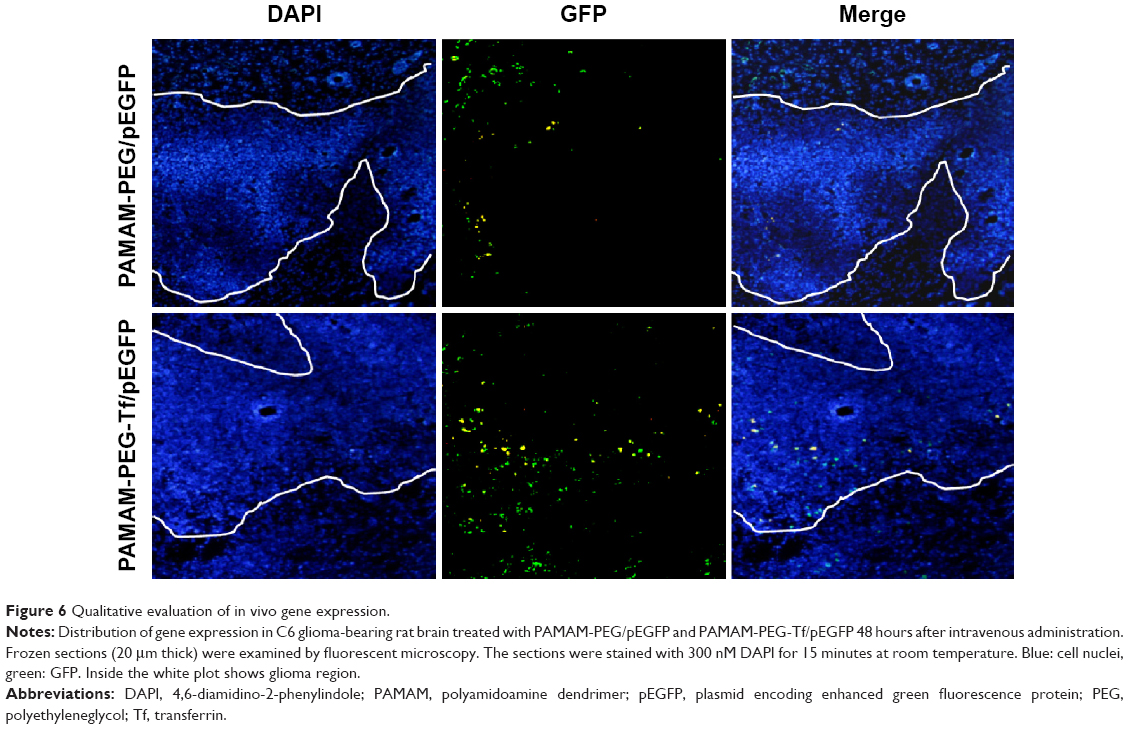 | Figure 6 Qualitative evaluation of in vivo gene expression. |
Immunohistochemistry of apoptosis markers
DNA fragmentation, a marker of late apoptosis, in nuclei of tumor cells was detected in margin region (Figure 7A) and central region (Figure 7B) using TUNEL assay. Virtually, little apoptotic cells were detected after PAMAM-PEG-Tf/pEGFP NPs and saline treatments in neither margin nor central region. PAMAM-PEG/Trail NPs and Temozolomide showed comparable apoptosis levels in margin region, but little tumor apoptosis was found in the central region. PAMAM-PEG-Tf/Trail NPs induced most tumor apoptosis in both two regions. This was consistent with the GFP expression trend.
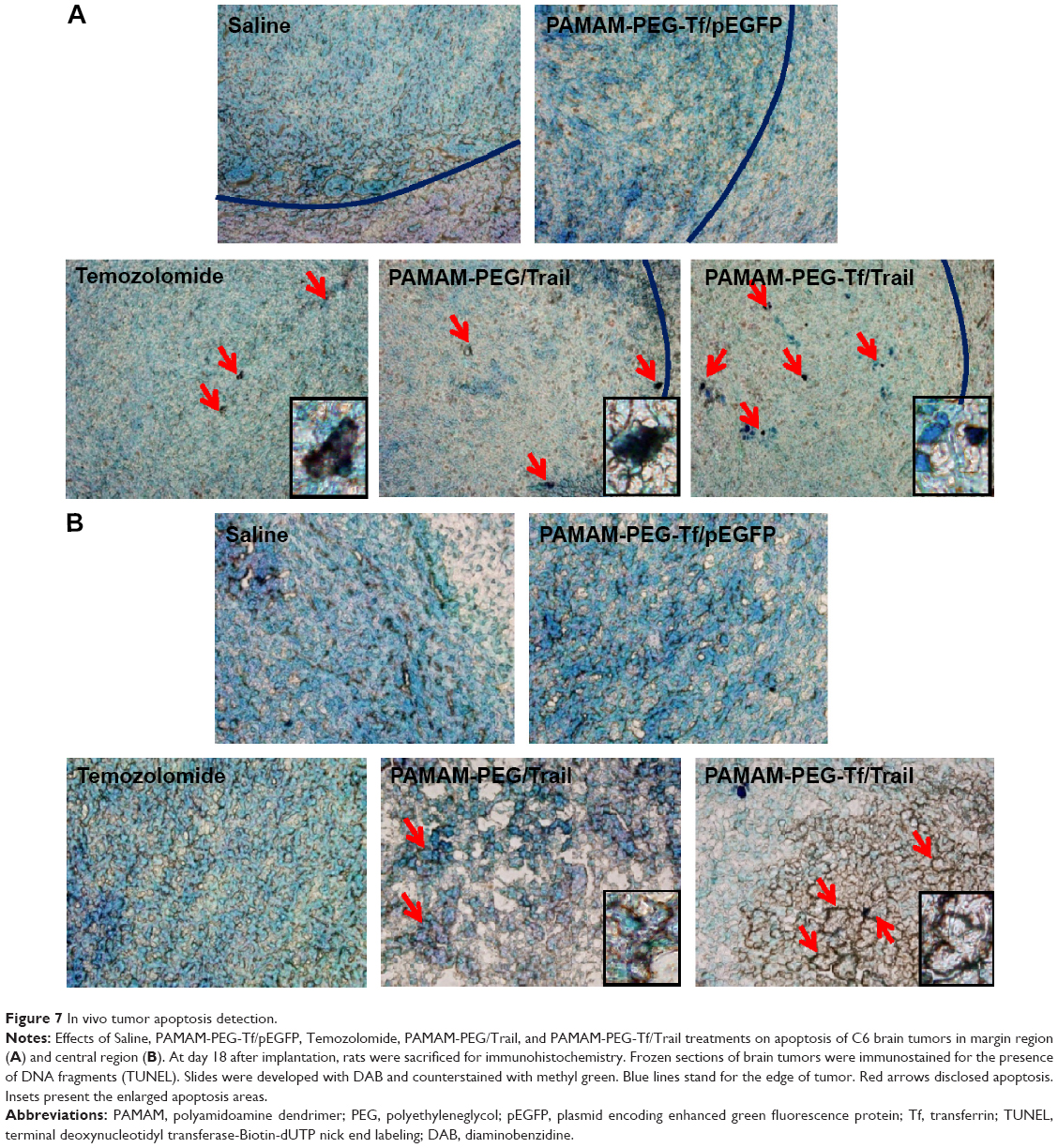 | Figure 7 In vivo tumor apoptosis detection. |
Therapeutic effect evaluation by survival monitoring and MRI imaging
The antitumor effect of PAMAM-PEG-Tf/Trail was also reflected in the median survival time of rats bearing glioma (Figure 8A). After one treatment course, the median survival time of PAMAM-PEG-Tf/Trail-treated rats (28.5 days) was longer than that of rats treated with PAMAM-PEG/hTRAIL (25.5 days), Temozolomide (24.5 days) PAMAM-PEG-Tf/pEGFP (19 days), or saline (17 days).
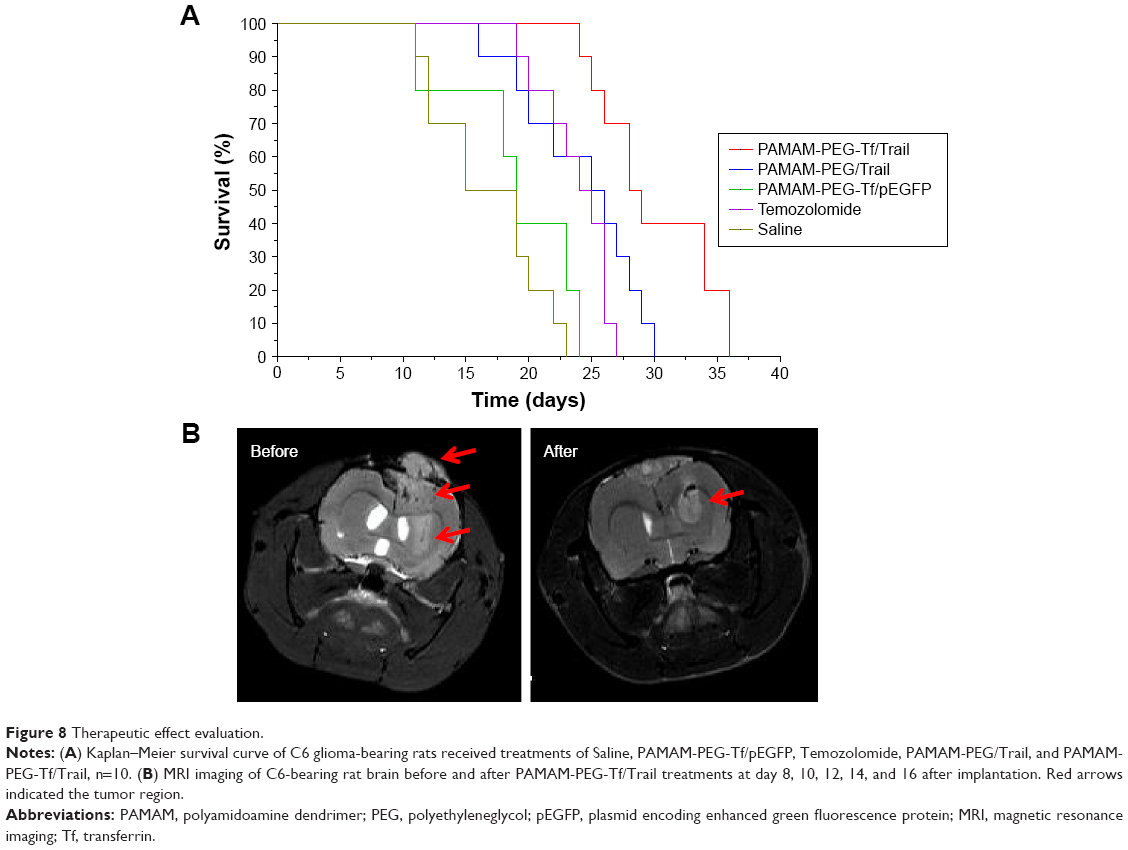 | Figure 8 Therapeutic effect evaluation. |
The anti-tumor effect of PAMAM-PEG-Tf/Trail was also confirmed by MRI imaging (Figure 8B). Tumor volume of PAMAM-PEG-Tf/hTRAIL-treated mice significantly decreased, which then prolonged the median survival time.
Discussion
Glioma is the most common malignant tumor of the central nervous system. Patients with malignant glioma have a poor prognosis due to the resistance to radiotherapy, chemotherapy, or immunotherapy.31 The feature of invasive growing of glioma made it impossible for surgical resection. Gene therapy of Trail was proven to be a novel one.32–34 We had successfully synthesized the targeting dendrimer PAMAM-PEG-Tf for efficient gene delivery into brain.26 It was demonstrated that Tf receptors expressed on the BBB and especially on tumor cells.35,36 Thus, this system was introduced to facilitate Trail crossing the BBB and targeting to the tumor.
A fundamental requirement for successful gene delivery was that dendrimer should be able to condense DNA efficiently to form NPs at a rational ratio. NPs should possess positive charge to enter the negatively charged cell membrane easily. At the weight ratio of PAMAM to DNA (3:1), PAMAM-PEG-Tf and DNA could form stable NPs with suitable size for enhanced permeability and retention (EPR) effect. The following cellular uptake and in vitro gene expression confirmed that PAMAM-PEG-Tf/DNA could enter the cells efficiently and yield high GFP expression. Theologically, the high PAMAM ratio could result in more stable NPs. However, cytotoxicity increased along with the PAMAM concentration due to the presence of surface amine groups.37–40 In the dendrimer design strategy, PEG could not only serve as a linker between PAMAM and Tf, but also reduce the toxicity by consuming some surface amines groups. Furthermore, PEG could provide a protective shell to avoid the reticulo-endothelial system, thus improving the half-life of NPs. Taken these together, 3:1 of PAMAM to DNA was optimized with tolerable toxicity and applied in the whole studies.
The glioma-targeting efficiency was also evaluated at the in vivo level. The fluorescent imaging showed that PAMAM-PEG-Tf/DNA NPs were more concentrated in glioma-bearing model than PAMAM-PEG/DNA NPs. PAMAM-PEG/DNA NPs could accumulate in tumor via EPR effects.41 Whereas, PAMAM-PEG-Tf/DNA NPs could also accumulate in tumor via the receptor-mediated mechanism. The result of the following in vivo gene expression study was consistent with the aforementioned results. Tf-modified NPs expressed more GFP in tumor. The defective vascular architecture near the tumor margin was created due to the rapid vascularization necessary to serve fast-growing and invasive glioma. Thus, GFP expression of unmodified NPs, which was affected only by EPR effect, was found near the tumor margin. The distribution of Tf-modified NPs was affected by both EPR effect and Tf receptor. Thus, GFP expression can be also found in the inner tumor region. This demonstrated the targeting efficacy and superiority of PAMAM-PEG-Tf. The following investigation provided more evidence of this hypothesis. TUNEL assay results had the same trend with GFP expression that Tf-modified NPs had more apoptosis cells in both margin and central regions. Temozolomide, a commercial superiority drug for glioma, was selected as a positive control. It inactivated O6-alkylguanine-DNA alkyltransferase activity to make the DNA mismatched and lead to cytotoxicity, which was effective especially for cell division.42 This was coincidental with the fact that Temozolomide treatment-induced apoptosis was only found in the margin region where the cells were proliferated rapidly. This difference of antitumor outcome between NP-based gene therapy and Temozolomide-based chemotherapy could provide us a new way to treat glioma more efficiently.
Conclusion
In conclusion, this study showed the feasibility of systemic administration of PAMAM-PEG-Tf/Trail for gene therapy of glioma. The current outcomes encourage further investigations into the application of gene therapy against tumor.
Acknowledgments
The authors would like to thank Mr XueFeng Yang for his technical support. This project has been funded in part with funds from the: Natural Science Foundation of Zhejiang Province (No. LY15H080002); Science and Technology Project of Zhejiang Province Department of Science and Technology (No. 2013C33230); Medical Science and Technology project of Zhejiang Province (No. 2012KYB088); Medical Science and Technology project of Zhejiang Province (No. 201337756); and Research project of Zhejiang education department (No. Y201328079).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegelin MD, Gaiser T, Habel A, et al. Daidzein overcomes TRAIL-resistance in malignant glioma cells by modulating the expression of the intrinsic apoptotic inhibitor, bcl-2. Neurosci Lett. 2009;454(3):223–228. | ||
Rich JN, Bigner DD. Development of novel targeted therapies in the treatment of malignant glioma. Nat Rev Drug Discov. 2004;3(5):430–446. | ||
Giese A. Glioma invasion--pattern of dissemination by mechanisms of invasion and surgical intervention, pattern of gene expression and its regulatory control by tumorsuppressor p53 and proto-oncogene ETS-1. Acta Neurochir Suppl. 2003;88:153–162. | ||
Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23(10):2411–2422. | ||
DeAngelis LM. Benefits of adjuvant chemotherapy in high-grade gliomas. Semin Oncol. 2003;30(6 Suppl 19):15–18. | ||
Oldenhuis CN, Stegehuis JH, Walenkamp AM, et al. Targeting TRAIL death receptors. Curr Opin Pharmacol. 2008;8(4):433–439. | ||
Rieger J, Naumann U, Glaser T, et al. APO2 ligand: a novel lethal weapon against malignant glioma? FEBS Lett. 1998;427(1):124–128. | ||
Keane MM, Ettenberg SA, Nau MM, et al. Chemotherapy augments TRAIL-induced apoptosis in breast cell lines. Cancer Res. 1999;59(3):734–741. | ||
Pan G, O’Rourke K, Chinnaiyan AM, et al. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276(5309):111–113. | ||
Hawkins CJ. TRAIL and malignant glioma. Vitam Horm. 2004;67:427–452. | ||
Roth W, Isenmann S, Naumann U, et al. Locoregional Apo2L/TRAIL eradicates intracranial human malignant glioma xenografts in athymic mice in the absence of neurotoxicity. Biochem Biophys Res Commun. 1999;265(2):479–483. | ||
Fukumura D, Xu L, Chen Y, et al. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001;61(16):6020–6024. | ||
Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95(8):4607–4612. | ||
Monsky WL, Fukumura D, Gohongi T, et al. Augmentation of transvascular transport of macromolecules and nanoparticles in tumors using vascular endothelial growth factor. Cancer Res. 1999;59(16):4129–4135. | ||
Pardridge W. Gene targeting technology and gene therapy of the brain. Drug Discov Today. 2001;6(3):125–126. | ||
Pardridge WM. Drug and gene delivery to the brain: the vascular route. Neuron. 2002;36(4):555–558. | ||
Newton HB. Advances in strategies to improve drug delivery to brain tumors. Expert Rev Neurother. 2006;6(10):1495–1509. | ||
Groothuis DR. The blood-brain and blood-tumor barriers: a review of strategies for increasing drug delivery. Neuro Oncol. 2000;2(1):45–59. | ||
Qian ZM, Li H, Sun H, et al. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev. 2002;54(4):561–587. | ||
Hatakeyama H, Akita H, Maruyama K, et al. Factors governing the in vivo tissue uptake of transferrin-coupled polyethylene glycol liposomes in vivo. Int J Pharm. 2004;281(1–2):25–33. | ||
Penichet ML, Kang YS, Pardridge WM, et al. An antibody-avidin fusion protein specific for the transferrin receptor serves as a delivery vehicle for effective brain targeting: initial applications in anti-HIV antisense drug delivery to the brain. J Immunol. 1999;163(8):4421–4426. | ||
Jefferies WA, Brandon MR, Hunt SV, et al. Transferrin receptor on endothelium of brain capillaries. Nature. 1984;312(5990):162–163. | ||
Tortorella S, Karagiannis TC. Transferrin receptor-mediated endocytosis: a useful target for cancer therapy. J Membr Biol. 2014;247(4):291–307. | ||
Dixit S, Novak T, Miller K, et al. Transferrin receptor-targeted theranostic gold nanoparticles for photosensitizer delivery in brain tumors. Nanoscale. 2015;7(5):1782–1790. | ||
Bielinska AU, Chen C, Johnson J, et al. DNA complexing with polyamidoamine dendrimers: implications for transfection. Bioconjug Chem. 1999;10(5):843–850. | ||
Huang RQ, Qu YH, Ke WL, et al. Efficient gene delivery targeted to the brain using a transferrin-conjugated polyethyleneglycol-modified polyamidoamine dendrimer. FASEB J. 2007;21(4):1117–1125. | ||
Pang Z, Gao H, Yu Y, et al. Enhanced intracellular delivery and chemotherapy for glioma rats by transferrin-conjugated biodegradable polymersomes loaded with doxorubicin. Bioconjug Chem. 2011;22(6):1171–1180. | ||
Lv Q, Li LM, Han M, et al. Characteristics of sequential targeting of brain glioma for transferrin-modified cisplatin liposome. Int J Pharm. 2013;444(1–2):1–9. | ||
Zhan C, Gu B, Xie C, et al. Cyclic RGD conjugated poly(ethylene glycol)-co-poly(lactic acid) micelle enhances paclitaxel anti-glioblastoma effect. J Control Release. 2010;143(1):136–142. | ||
Li J, Guo Y, Kuang Y, et al. Choline transporter-targeting and co-delivery system for glioma therapy. Biomaterials. 2013;34(36):9142–9148. | ||
Avgeropoulos NG, Batchelor TT. New treatment strategies for malignant gliomas. Oncologist. 1999;4(3):209–224. | ||
Lu W, Sun Q, Wan J, et al. Cationic albumin-conjugated pegylated nanoparticles allow gene delivery into brain tumors via intravenous administration. Cancer Res. 2006;66(24):11878–11887. | ||
Han L, Huang R, Li J, et al. Plasmid pORF-hTRAIL and doxorubicin co-delivery targeting to tumor using peptide-conjugated polyamidoamine dendrimer. Biomaterials. 2011;32(4):1242–1252. | ||
Huang S, Li J, Han L, et al. Dual targeting effect of Angiopep-2-modified, DNA-loaded nanoparticles for glioma. Biomaterials. 2011;32(28):6832–6838. | ||
Mohsenikia M, Alizadeh AM, Khodayari S, et al. The protective and therapeutic effects of alpha-solanine on mice breast cancer. Eur J Pharmacol. 2013;718(1–3):1–9. | ||
Tortorella S, Karagiannis TC. The significance of transferrin receptors in oncology: the development of functional nano-based drug delivery systems. Curr Drug Deliv. 2014;11(4):427–443. | ||
Naha PC, Davoren M, Lyng FM, et al. Reactive oxygen species (ROS) induced cytokine production and cytotoxicity of PAMAM dendrimers in J774A.1 cells. Toxicol Appl Pharmacol. 2010;246(1–2):91–99. | ||
Naha PC, Davoren M, Casey A, et al. An ecotoxicological study of poly(amidoamine) dendrimers-toward quantitative structure activity relationships. Environ Sci Technol. 2009;43(17):6864–6869. | ||
Naha PC, Byrne HJ. Generation of intracellular reactive oxygen species and genotoxicity effect to exposure of nanosized polyamidoamine (PAMAM) dendrimers in PLHC-1 cells in vitro. Aquat Toxicol. 2013;132–133:61–72. | ||
Maher MA, Naha PC, Mukherjee SP, et al. Numerical simulations of in vitro nanoparticle toxicity – the case of poly(amido amine) dendrimers. Toxicol In Vitro. 2014;28(8):1449–1460. | ||
Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2004;56(11):1649–1659. | ||
Tolcher AW, Gerson SL, Denis L, et al. Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules. Br J Cancer. 2003;88(7):1004–1011. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.