Back to Journals » Cancer Management and Research » Volume 13
Plakophilin-2 Promotes Lung Adenocarcinoma Development via Enhancing Focal Adhesion and Epithelial–Mesenchymal Transition
Authors Wu Y, Liu L ![]() , Shen X, Liu W, Ma R
, Shen X, Liu W, Ma R
Received 24 September 2020
Accepted for publication 5 January 2021
Published 22 January 2021 Volume 2021:13 Pages 559—570
DOI https://doi.org/10.2147/CMAR.S281663
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Seema Singh
Yang Wu,1 Lu Liu,2 Xiaoyu Shen,1 Wenjing Liu,1 Rui Ma1
1Medical Oncology Department of Thoracic Cancer (2), Cancer Hospital of China Medical University, Liaoning Cancer Hospital & Institute, Shenyang 110042, Liaoning, People’s Republic of China; 2Institute of Cancer Stem Cell, Dalian Medical University, Dalian 116044, Liaoning, People’s Republic of China
Correspondence: Rui Ma
Medical Oncology Department of Thoracic Cancer (2), Cancer Hospital of China Medical University, Liaoning Cancer Hospital & Institute, Shenyang 110042, Liaoning, People’s Republic of China
Email [email protected]
Background: Lung cancer is one of the most aggressive tumors with high incidence and mortality, which could be classified into lung squamous cell carcinoma (LUSC) and lung adenocarcinoma (LUAD). Overexpression of Plakophilin-2 (PKP2) has been reported in multiple malignancies. However, the expression and function mechanism of PKP2 in LUAD remain illusive.
Methods: Real-time PCR (RT-PCR) was conducted to assess the expression of PKP2 in LUAD cells and tissues. An integrated analysis of PKP2 expression in The Cancer Genome Atlas (TCGA) was further performed. The effect of PKP2 on cell proliferation and invasion potential were then evaluated with loss-of-function assays in vitro. Xenograft nude mouse models were used to determine the role of PKP2 in LUAD tumorigenicity in vivo. Bioinformatics prediction, immunohistochemistry and Western blot were performed to examine whether PKP2 promoted LUAD development via enhancing focal adhesion and epithelial–mesenchymal transition.
Results: PKP2 expression was highly expressed in LUAD tissues compared with that in normal tissues and predicated poor prognosis of LUAD patients. TCGA LUAD cohort analysis also showed that high expression of PKP2 indicated unfavorable outcomes in LUAD patients. PKP2 expression was also upregulated in lung cancer cells. Functionally, knockdown of PKP2 suppressed lung cancer cell proliferation and invasion in vitro, while inhibited xenograft lung tumor development in vivo. Mechanistically, we demonstrated that high expression of PKP2 in LUAD was correlated with enhanced EMT and focal adhesion. Knockdown of PKP2 inhibited the expression of EMT-related Vimentin and N-cadherin and focal adhesion-associated expression of BMP4, ICAM1, and VCAM1 in xenograft tumors and lung cancer cells.
Conclusion: In summary, our findings indicate that PKP2 functions as an oncogene in LUAD, which could be utilized as a novel diagnostic and therapeutic marker for LUAD treatment.
Keywords: PKP2, focal adhesion, EMT, lung adenocarcinoma
Introduction
Lung cancer is one of the most aggressive tumors with high incidence and mortality around the world, which could be classified into lung squamous cell carcinoma (LUSC) and lung adenocarcinoma based on genetic and molecular character (LUAD).1,2 The development of LUAD is a complicated, multiple-stage process, and various factors including genetic mutations, cigarette smoking, and environment toxins contribute to the tumorigenesis.3–5 The advances in diagnosis and therapy greatly improve the outcome of LUAD patients.6 However, the prognosis of LUAD patients remains poor due to the tumor metastasis and recurrence, while the 5-year survival rate of LUAD is only around 15%.7,8 Thus, it is of great importance to identify new diagnostic biomarkers and develop novel therapeutic strategies to treat LUAD patients.
Plakophilin-2 (PKP2) belongs to the family of plaque-bound plakophilins and is widely expressed in epithelial cells.9 PIKP2 has been demonstrated to a play a critical role in heart morphogenesis and cardiac junction formation.10,11 In bladder cancer, high expression of PKP2 is associated with enhanced tumor cell invasion and metastasis, but not cell proliferation.12 Arimoto et al reported that PKP2 could promote tumor progression via inducing phosphorylation of epidermal growth factor receptor (EGFR) and increasing its dimerization and activation.13,14 The oncogenic role of PKP2 has also been studied in glioma and knockdown of PKP2 inhibits the glioma cell proliferation and migration.15 Nevertheless, the expression, function and clinical implication of PKP2 in LUAD are still not fully explored.
In this study, we aimed to study the expression profile and function of PKP2 in LUAD. We found that PKP2 expression was highly expressed in LUAD, which predicted the poor outcome of LUAD patients. TCGA LUAD cohort analysis also showed that high expression of PKP2 indicated unfavorable outcomes in LUAD patients. Functionally, knockdown of PKP2 suppressed lung cancer cell proliferation and invasion in vitro, while inhibited xenograft lung tumor development in vivo. Mechanistically, we demonstrated that high expression of PKP2 in LUAD was correlated with enhanced EMT and focal adhesion. In summary, our findings indicate that PKP2 functions as an oncogene in LUAD, which could be utilized as a novel diagnostic and therapeutic marker for LUAD treatment.
Materials and Methods
LUAD Patient Tissue Samples
LUAD tissue samples and adjacent normal tissues were collected from LUAD patients who underwent surgery at Cancer Hospital of China Medical University (CHCMU) from the year 2004 to 2018. The patient specimens were snap-frozen and stored in liquid nitrogen until further examination. Written informed consent was obtained from each patient. All protocols were approved by the Institutional Ethical Review Board of Cancer Hospital of China Medical University. The collection of patient specimens is carried out in accordance with the Declaration of Helsinki. The details of patients are presented in Supplementary Table 1. The welfare of the laboratory animals was followed by guidelines for the ethical review of laboratory animal welfare People’s Republic of China National Standard (GB/T 35892‐2018) (DOI:10.1002/ame2.12111).
Cell Lines and Cell Culture
Lung cancer cell lines (H1299, H226, HCC827, H1975, PC-9 and A549) and human bronchial epithelial control cell line (16HBE) were obtained from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco’s Modified Eagle’s medium with 10% fetal bovine serum (Gibco, USA), 1% penicillin-streptomycin (Gibco, USA). Cells were cultured in a humidified cell incubator at 37°C with 5% CO2.
Transfection
The PKP2 shRNAs and negative control were obtained from Shanghai GenePharma (Shanghai, China). Transfection was performed using TransIT-2020 (Mirus, USA) following the manufacturer’s instruction.
Quantitative Real-Time PCR (qRT-PCR)
To analyze PKP2 mRNA expression, total RNA was extracted from tissue samples or cultured cells using Trizol (Invitrogen, USA) and reverse-transcribed into cDNA using a Prime Script RT master kit (Takara, Japan). Quantitative RT-PCR analysis was performed using the SYBR master mix (Takara, Japan) and the 2−ΔΔCT method was used to calculate relative mRNA expression. The primer sequences used for PKP2 and GAPDH were listed below: PKP2 (amplicon size: 253 bp, Tm: 54°C), 5ʹ-AGATTACCAGCCAGATGACA-3ʹ (forward) and 5ʹ-ATGCCACAGCCACTCCAC-3ʹ (reverse); GAPDH (amplicon size: 231 bp, Tm: 54°C), 5ʹ- GAGAAGGCTGGGGCTCATTT −3ʹ (forward) and 5ʹ- AGTGATGGCATGGACTGTGG −3ʹ (reverse);
Western Blot
To analyze PKP2 protein expression, total protein was extracted from tissue samples or cultured cells using RIPA buffer (Cell Signaling Tech, USA) and separated by SDS-PAGE. The electrophoretic protein was transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, USA) and blocked with 5% BSA/TBST for 1 hour. The membranes were incubated with primary antibodies overnight at 4°C, followed by incubation with secondary antibody at room temperature for 1 hour. The protein bands were detected using a chemiluminescence reaction kit (Pierce, USA) and densitometry were analyzed using Quantity One (Bio-Rad, USA). The primary antibodies used in the study were listed as follows: PKP2 (Proteintech, Ag24163), E-cadherin (Proteintech, 60335-1-Ig), vimentin (Proteintech, 10366-1-AP), N-cadherin (Proteintech, 22018-1-AP), BMP4 (Proteintech, 12492-1-AP), ICAM1 (Proteintech, 10831-1-AP), VCAM1 (Proteintech, 11444-1-AP) and GAPDH (Proteintech, 60004-1-Ig).
5-Ethynyl-20-Deoxyuridine (EdU) Incorporation Assay
Cells were seeded into 96-well plates (3000 cells/well) and 50 mM EdU solution (Ribobio, China) was added to the cell culture. After 48 hours, cells were fixed with 4% formaldehyde and permeabilized with Triton X-100. EdU staining was performed using an EdU staining Kit (Ribobio, China) and cells were counterstained with DAPI. The number of EdU incorporated cells was counted under a fluorescence microscopy.
Colony Formation Assay
Five hundred cells were seeded into 6-well plates and cultured for 14 days. Cell colonies were fixed with methanol, stained with 1% crystal violet, and counted.
Transwell Assay
Transwell assay was performed to assess the cell invasion using a transwell chamber with pre-coated Matrigel (8 μm pore size, Corning, USA). 5 × 104 cells were suspended in serum free medium and seeded into the upper chamber. 500 μL medium supplemented with 10% FBS was added to the lower chamber. After incubation for 48 hours, the invaded cells were fixed, stained with 1% crystal violet, and counted under a light microscope.
Tissue Microarray (TMA) Construction
The lung cancer tissue microarray (TMA) was constructed using the 90-paired lung cancer specimens and adjacent normal control tissues. Briefly, for each patient, a 1-mm diameter core of the tissue was punched from formalin-fixed paraffin-embedded tissues and arranged into the TMA blocks. The expression of PKP2 in patient specimens was scored based on the staining intensity (range 1+ to 4+, negative to intensively strong).
Immunohistochemical (IHC) Staining
Xenograft tumor tissue sections were deparaffinized, rehydrated and blocked with 1% BSA/PBS. Then, tissue sections were incubated with anti-Ki-67 antibody (Proteintech, 27309-1-AP), PKP2 antibody (Proteintech, Ag24163), overnight at 4°C, followed by incubation with secondary antibody. The sections were stained with 3ʹ30-diaminobenzidine (DBA) and hematoxylin.
Xenograft Tumor Model
Xenograft tumor model was established by inoculating A549 cells (5 × 106) stably transfected with sh-PKP2 or mock control into the right flank of male nude mice (6 weeks old). Tumor growth was measured every week. Mice were euthanized 5 weeks post inoculation. Xenograft tumors were dissected and weighted. The animal experiments were approved by the Institutional Animal Care and Use Committee of Cancer Hospital of China Medical University.
Statistical Analysis
All data were presented as mean ± standard deviation (SD) of at least three experiments. The statistical analysis was carried out using GraphPad Prism (V6, Prism, USA). Student’s t-test or one-way ANOVA followed by a Bonferroni post hoc test was used to compare the differences between two or more groups. A P value <0.05 was considered statistical significance.
Results
PKP2 Expression is Upregulated in LUAD and High PKP2 Expression Predicts Poor Outcome of LUAD Patients
Previous studies indicate that PKP2 functions as an oncogene in various tumors.13,16,17 To study the function of PKP2 in LUAD, we first examined the expression of PKP2 in lung LUAD specimen and adjacent normal specimen. We found that PKP2 expression was significantly higher in LUAD tissues than that in adjacent normal tissues (Figure 1A). Western blot also confirmed that PKP2 protein expression was enhanced in LUAD tissues (Figure 1B). In addition, we demonstrated that mRNA and protein expression of PKP2 were markedly higher in lung cancer cell lines (H1299, H226, HCC827, H1975, PC-9 and A549) than that in human bronchial epithelial control 16HBE cells (Figure 1C and D). We also performed IHC staining of PKP2 and scored LUAD tissue microarray (MTA) based on the PKP2 IHC staining intensity (Figure 2A). LUAD tissues had remarkably higher scores of PKP2 IHC staining in comparison with that of normal tissues (Figure 2B). Intriguingly, LUAD patients with high PKP2 levels had a worse prognosis, with lower overall survival (OS) and disease-free survival (DFS) than that in LUAD patients with low PKP2 levels (Figure 2C and D). Furthermore, PKP2 expression was found to be independent prognostic factors by univariate and multivariate analysis (Supplementary Table 2). These findings suggest that PKP2 is highly expressed in LUAD and predicts unfavorable outcome in LUAD patients.
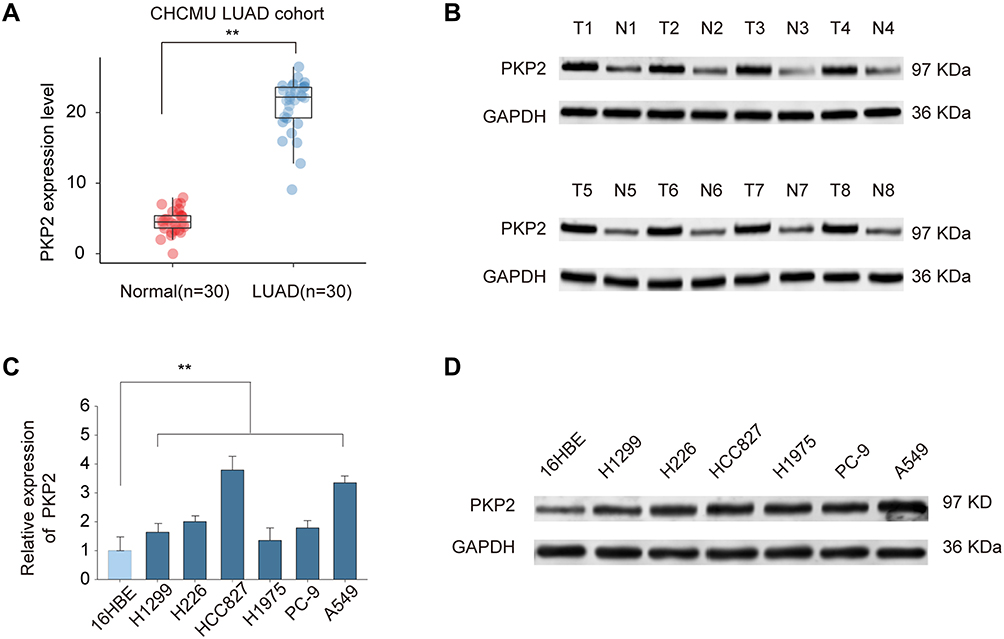 |
Figure 1 PKP2 expression is upregulated in LUAD. (A) The relative PKP2 mRNA expression levels of PKP2 in LUAD tissues and adjacent normal tissues were analyzed by qPCR. (B) The protein expression of PKP2 in 8-paired LUAD and normal control tissues were analyzed by Western blot. (C and D) The mRNA expression (C) and protein expression (D) of PKP2 in lung cancer cell lines (H1299, H226, HCC827, H1975, PC-9 and A549) and human bronchial epithelial control (16HBE) were analyzed by qPCR and Western blot. **P < 0.01. |
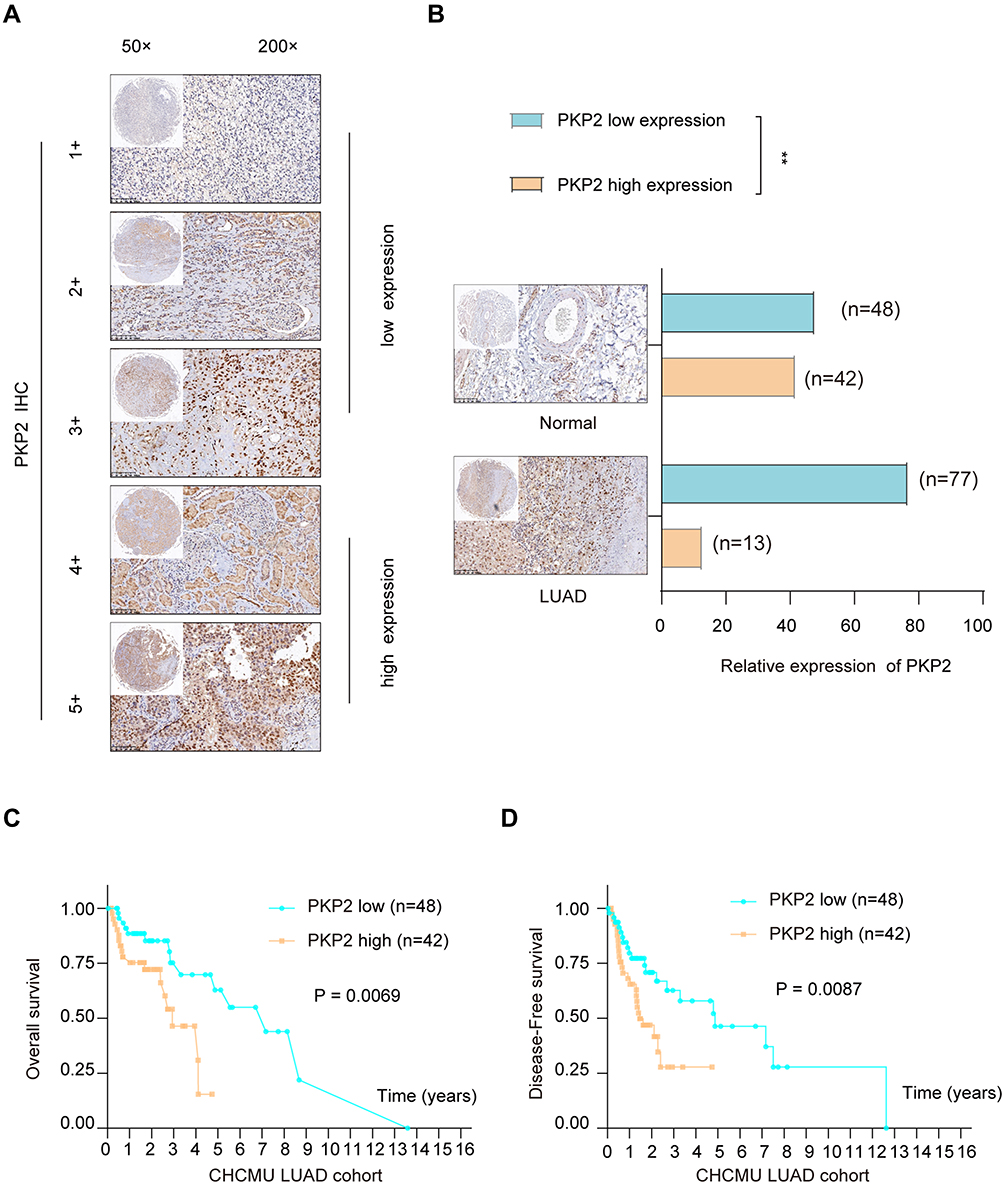 |
Figure 2 High expression of PKP2 predicts poor prognosis in LUAD patients. (A) LUAD tissue microarray (TMA) was prepared and IHC staining of PKP2 was performed. The representative staining of PKP2 was shown and the expression of PKP2 was scored as 1–5+ based on the IHC staining intensity. (B) The distribution of PKP2 IHC staining scores in LUAD tissues and adjacent normal tissues was analyzed. (C) Kaplan-Meier analysis was performed to analyze the overall survival (OS) in LUAD patients with low or high-expression of PKP2. (D) Kaplan–Meier analysis was performed to analyze the disease-free survival (DFS) in LUAD patients with low or high-expression of PKP2. **P < 0.01. |
High Expression of PKP2 Indicates Unfavorable Outcome of LUAD Patients in TCGA Cohort
To further characterize the expression of PKP2, we performed bioinformatics analysis to examine the expression of PKP2 in pan-cancers. As shown in Figure 3A, PKP2 was highly expressed in most malignancies including LUAD. In addition, we demonstrated that PKP2 expression was significantly higher in LUAD than that in adjacent normal tissues in LUAD TCGA cohort (Figure 3B). Further analysis revealed that the expression of PKP2 in advanced TNM stages (TNM-III and IV) was markedly higher than that in LUAD with TNM stage I and II (Figure 3C, P < 0.05). Consistently, in LUAD TCGA cohort, patients with high PKP2 expression had worse OS and DFS compared with patients with low PKP2 levels (Figure 3D and E).
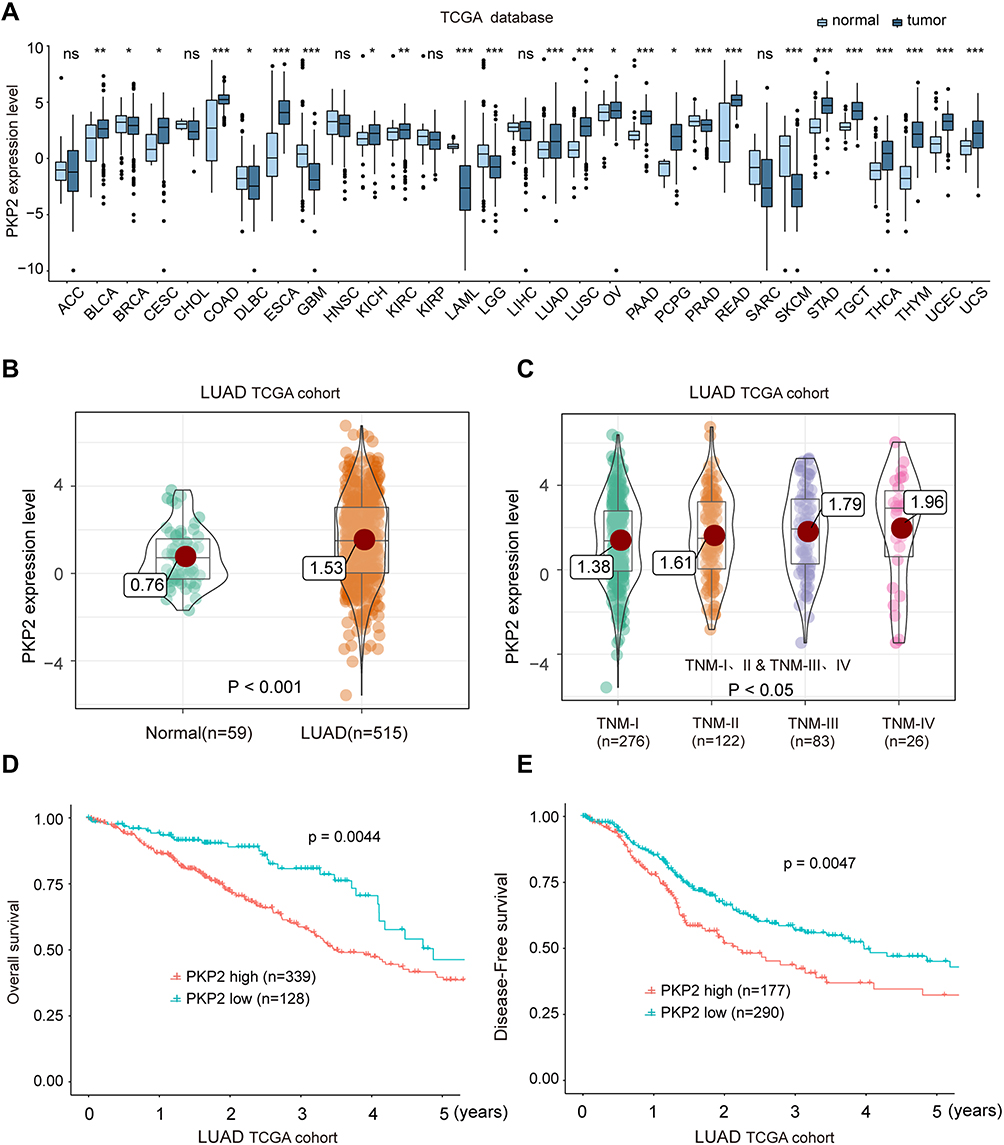 |
Figure 3 High expression of PKP2 indicates unfavorable outcome of LUAD patients in TCGA cohort. (A) The mRNA expression of PKP2 in a series of tumor tissues and the corresponding normal tissues was analyzed in TCGA database. (B) The mRNA expression of PKP2 in LUAD tissues and normal control tissues was analyzed in TCGA LUAD cohort. (C) The mRNA expression of PKP2 in LUAD patients at different TNM stages was analyzed in LUAD TCGA cohort. (D and E) Kaplan–Meier analysis was performed to analyze the OS and DFS in LUAD patients with low or high-expression of PKP2. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: ACC, adenoid cystic carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast cancer; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon cancer; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; ESCA, esophagus cancer; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG, brain lower grade glioma; LIHC, liver cancer; LUAD, lung cancer; LUSC, lung squamous cell carcinoma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectal cancer; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach cancer; TGCT, testicular germ cell tumors; THCA, thyroid cancer; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma. |
Knockdown of PKP2 Suppresses Cell Proliferation and Invasion of Lung Cancer Cells
To study the function of PKP2 in lung cancer, we performed RNA interference to knockdown the expression of PKP2 in HCC827 and A549 cells. Transfection of shRNA targeting PKP2 significantly inhibited the mRNA and protein expression of PKP2 in lung cancer cells (Figure 4A and B). The most efficient sh-PKP2-3 was used for the subsequent experiments. EdU incorporation and colony formation assays demonstrated that knockdown of PKP2 suppressed cell proliferation and colony formation (Figure 4C and D). In addition, inhibition of PKP2 dampened the invasive capability of lung cancer cells, as demonstrated by transwell assay (Figure 4E). On the contrary, Over-expression of PKP2 promoted lung cancer cell proliferation and metastasis in vitro (Supplementary Figure S1).
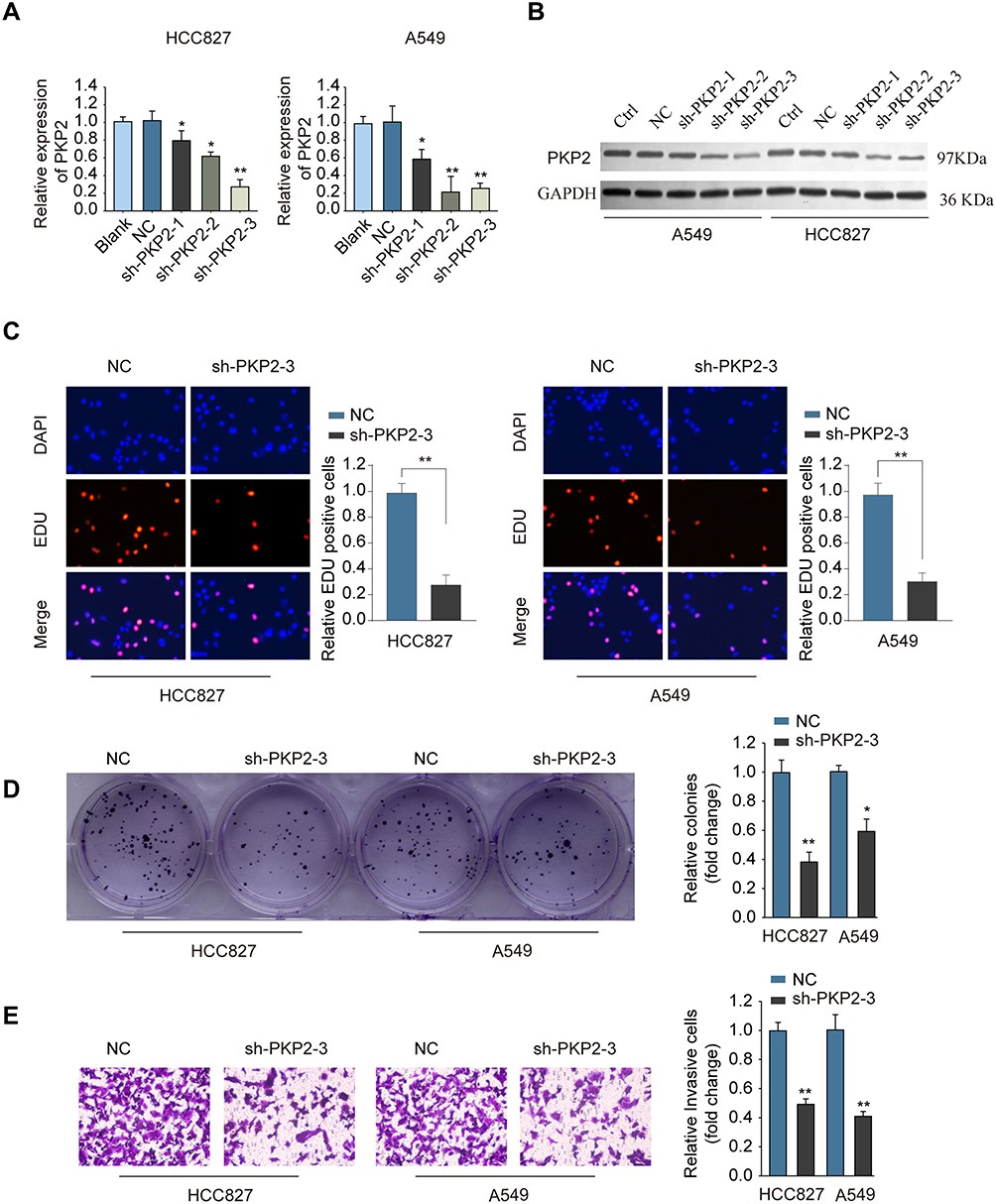 |
Figure 4 Knockdown of PKP2 suppresses cell proliferation and invasion of lung cancer cells. (A and B) HCC827 or A549 cells were transfected with negative control (NC), or shRNA targeting PKP2 (sh-PKP2-1/2/3), or left untreated (Blank). The mRNA (A) or protein (B) expression of PKP2 was analyzed 48 hours later. HCC827 or A549 cells were transfected with NC or sh-PKP2-3. (C) EdU incorporation assay was performed to analyze cell proliferation. (D) Colony formation assay was performed to assess cell growth. (E) Transwell assay was performed to assess cell invasion. *P < 0.05, **P < 0.01. |
Knockdown of PKP2 Inhibits Xenograft Lung Tumor Development in vivo
Xenograft tumor model was established by subcutaneous injection of A549 cells stably knocking down PKP2 (sh-PKP2) or control cells (MOCK) to further investigate the function of PKP2 in vivo. Knockdown of PKP2 significantly delayed the tumor development in vivo, with significant smaller tumors in sh-PKP2 group (Figure 5A and B). The tumor weights from PKP2 knockdown group were markedly lower than those from MOCK group (Figure 5C). In addition, IHC staining of proliferation marker Ki-67 and PKP2 showed that knockdown of PKP2 significantly inhibited lung cancer cell proliferation in vivo, with a much lower Ki-67 expression (Figure 5D). Taken together, these results indicate that knockdown of PKP2 inhibits xenograft lung tumor development in vivo.
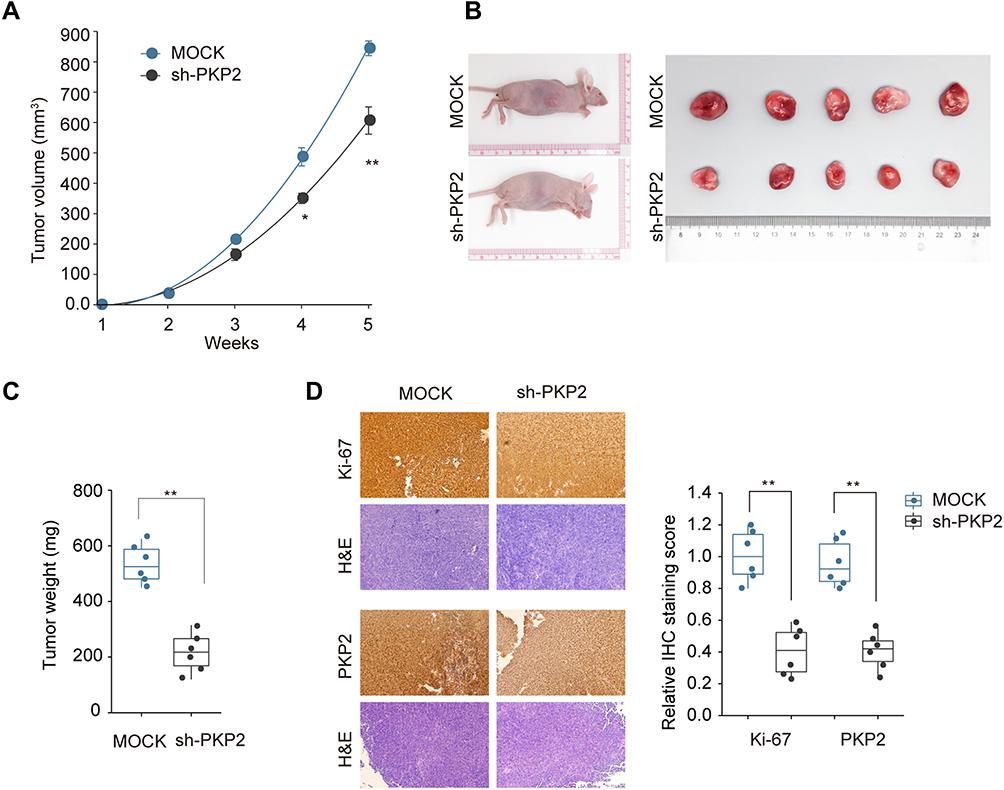 |
Figure 5 Knockdown of PKP2 inhibits xenograft lung tumor development in vivo. A549 cells were stably transfected with sh-PKP2 or MOCK, and then subcutaneously injected into nude mice. (A and B) The tumor growth was monitored for 5 weeks and xenograft tumors from different groups were photographed after dissecting the tumor at the end of experiment. (C) Tumor weights were analyzed. (D) IHC staining was performed to examine the Ki-67 and PKP2 expression in tumor tissue sections. *P < 0.05, **P < 0.01. |
PKP2 Promotes Lung Cancer Development via Enhancing EMT and Focal Adhesion
To further explore the mechanism of PKP2 regulation in lung cancer development, we performed bioinformatics analysis using the Molecular Signatures Database (MsigDB) Hallmark and found that high expression of PKP2 associated with enhanced EMT (Figure 6A). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis found that there was a positive correlation between upregulated PKP2 expression and focal adhesion (Figure 6B). In addition, we revealed that PKP2 expression was positively correlated with the expression of proliferation marker Ki-67 or PCNA in LUAD TCGA cohort (Figure 6C and D). Gene Set Enrichment Analysis (GSEA) also confirmed that the signature gene sets of EMT and focal adhesion were enriched in PKP2 high-expression LUAD (Figure 6E and F).
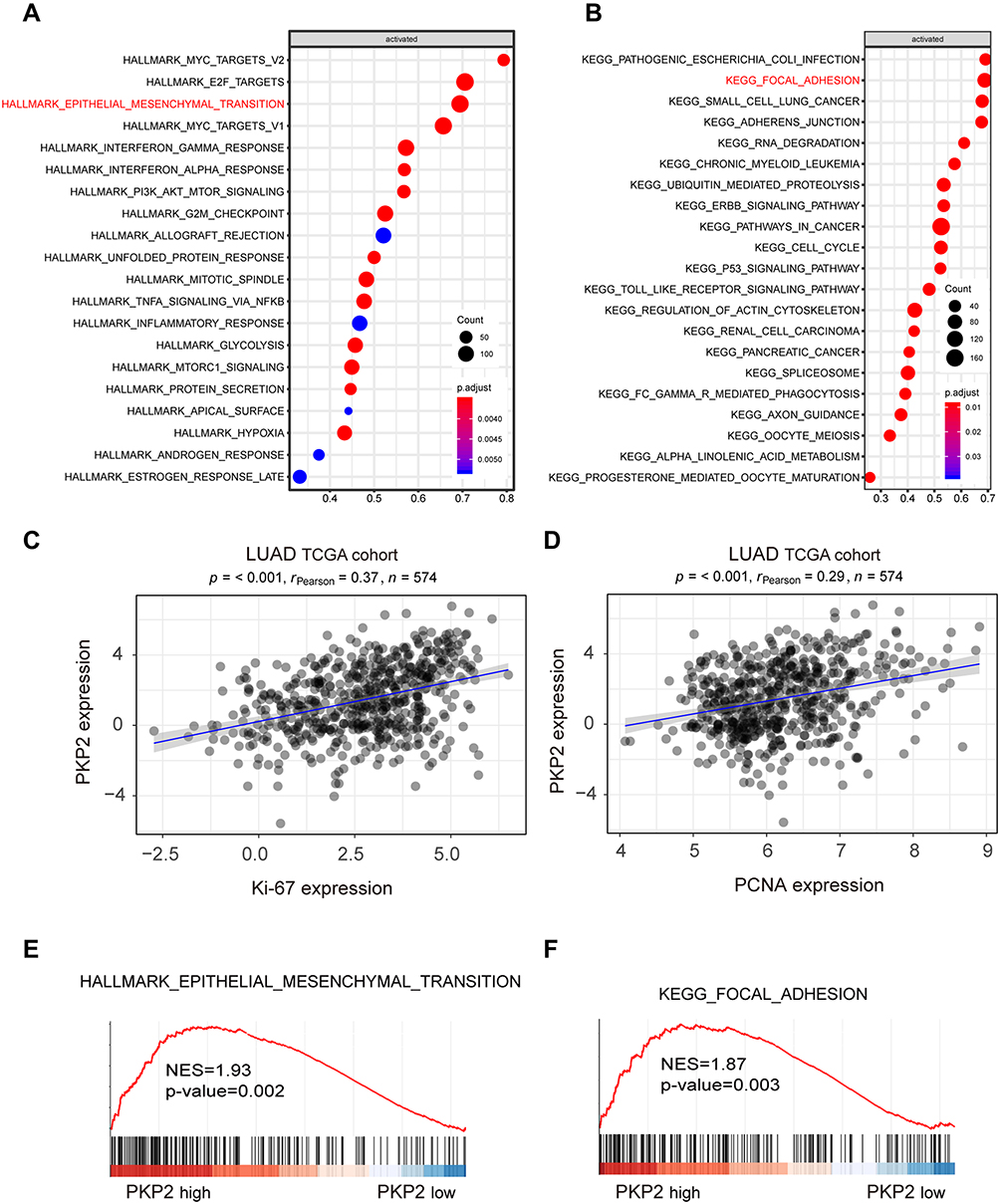 |
Figure 6 Bioinformatics analysis suggests high expression of PKP2 correlates with enhanced EMT and focal adhesion. (A) Bioinformatics analysis using the Molecular Signatures Database (MsigDB) Hallmark revealed the signaling pathways activated with high expression of PKP2 in LUAD TCGA cohort. (B) Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed the signaling pathways activated with high expression of PKP2 in LUAD TCGA cohort. (C and D) Pearson correlation analysis the relationship between PKP2 expression and the expression of proliferation marker Ki-67 or PCNA in LUAD TCGA cohort. (E and F) Gene Set Enrichment Analysis (GSEA) to assess the enrichment of signature gene sets of focal adhesion and EMT in PKP2 high-expression LUAD. |
To verify the bioinformatics analysis results, we performed IHC staining of EMT markers (E-cadherin, vimentin, and N-cadherin) and focal adhesion markers (BMP4, ICAM1, and VCAM1) using xenograft tumor sections. The results showed that knockdown of PKP2 suppressed EMT, with enhanced E-cadherin expression and decreased Vimentin/N-cadherin expression (Figure 7A). Silencing PKP2 also inhibited the focal adhesion and suppressed the expression of BMP4, ICAM1 and VCAM1 (Figure 7A). Consistently, we demonstrated that overexpression of PKP2 promoted the expression of EMT and focal adhesion markers while knockdown of PKP2 showed the opposite effects in HCC827 or A549 cells in vitro (Figure 7B). In summary, these findings suggest that PKP2 promotes lung cancer development via enhancing EMT and focal adhesion.
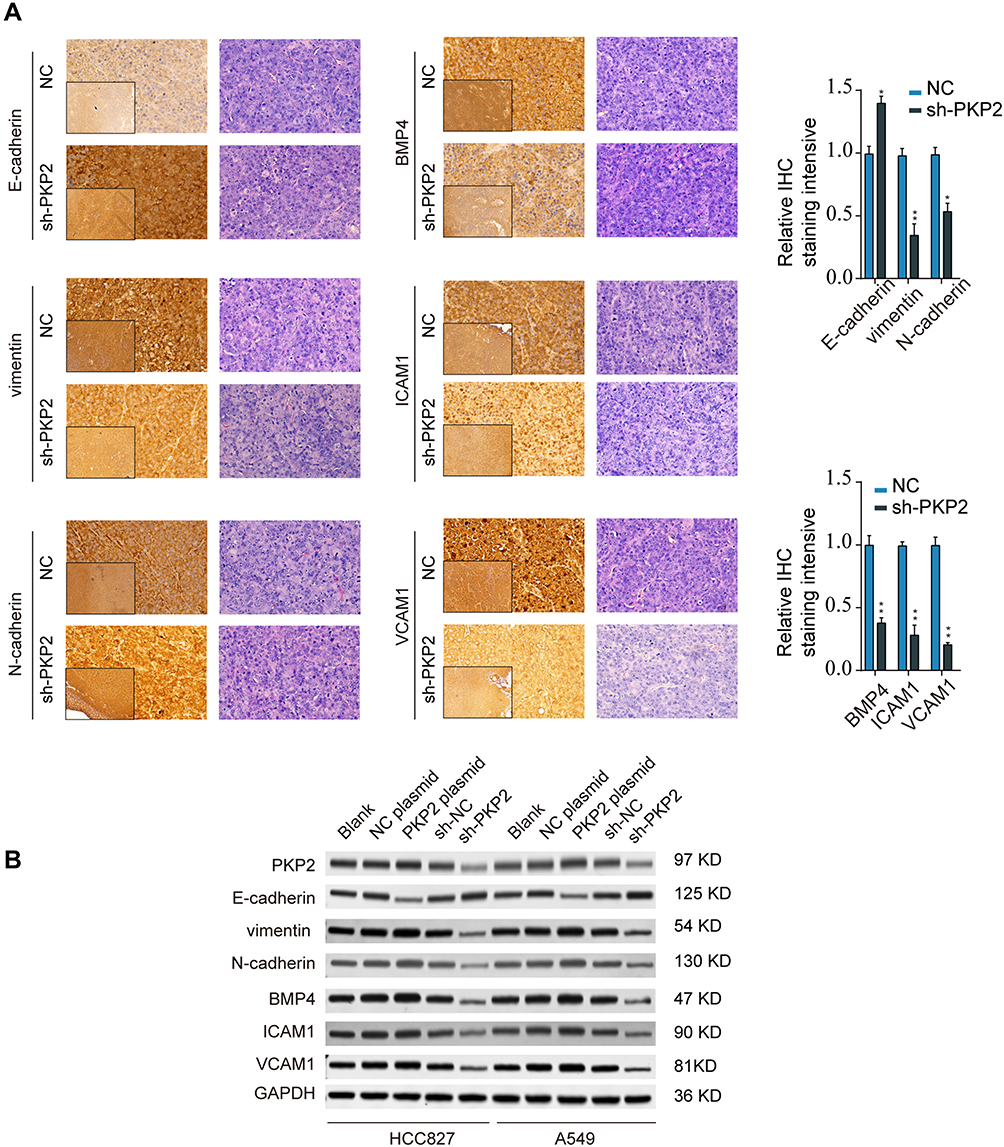 |
Figure 7 Knockdown of PKP2 suppressed EMT and focal adhesion in LUAD. (A) IHC staining of EMT markers (E-cadherin, Vimentin, and N-cadherin) and focal adhesion markers (BMP4, ICAM1, and VCAM1) was performed using xenograft tumor sections from NC or sh-PKP2 groups. (B) HCC827 or A549 cells were transfected with sh-NC, sh-PKP2 or left untreated. The protein expression of EMT and focal adhesion markers was analyzed by Western blot. *P < 0.05, **P < 0.01. |
Discussion
The oncogenic function of PKP2 has been investigated in various malignancies including glioma, colon cancer, bladder cancer, and lung cancer.12,15–17 Here we showed that high expression of PKP2 was found in LUAD tissues and lung cancer cells. Knockdown of PKP2 suppressed lung cancer cell growth and metastasis both in vitro and in vivo. We also demonstrated that PKP2 regulated LUAD development via modulating EMT and focal adhesion. Together, our study suggests that PKP2 serves as a prognostic biomarker to predict the outcome of LUAD patients and could be utilized as a novel therapeutic target for LUAD.
PKP2 has been studied as structural components of desmosomes and serves as multifunctional scaffolds for adhesion and signaling.9 The dysregulated PKP2 expression has been found in cancer pathogenesis.10,13 PKP2 activates ligand-dependent and independent EGFR dimerization, and activation leads to tumor progression.13,18 Immunohistochemical staining demonstrated that enhanced PKP2 expression was observed in 85.7% of metastatic human primary oropharyngeal tumors.19 Hao et al reported that PKP2 was highly expressed in lung cancer and promoted lung cancer cell proliferation and migration via activating EGFR signaling.17 Consistently, we also detected the upregulated expression of PKP2 in LUAD tissues and lung cancer cells. High expression of PKP2 in LUAD was confirmed in TCGA LUAD cohort. Both the TCGA LUAD cohort and CHCMU LUAD cohort suggested that overexpression of TCGA was associated with poor prognosis. The oncogenic function of PKP2 has been validated both in vitro and in vivo. Knockdown of PKP2 suppressed cell proliferation and invasion, while silencing PKP2 inhibited xenograft lung tumor growth in vivo.
Bioinformatics analysis was performed to investigate the signaling pathway involved in PKP2 regulation and we found high PKP2 expression enhanced EMT and focal adhesion. Consistent with our report, Koetsier et al demonstrated that PKP2 regulated cell migration via modulating focal adhesion dynamics and the protein expression of integrins.20 Given that PKP2 functions as an oncogene in multiple tumors, it is intriguing to explore the regulation of PKP2. In normal and colon cancer-related fibroblasts, Wnt/beta-catenin could induce the expression of PKP2.16 The expression of PKP2 was also regulated post-transcriptionally by miR-184 via modulating CpG hypermethylation.21 Nevertheless, how PKP2 expression is regulated in LUAD needs further investigation.
Conclusions
In summary, our findings demonstrate that PKP2 functions as an oncogene in LUAD development and progression via enhancing EMT and focal adhesion. High expression of PKP2 predicts poor outcome of LUAD patients. PKP2 could be utilized as a diagnostic biomarker and therapeutic target for LUAD treatment.
Acknowledgment
This work was supported by the Key Research and Development Project of Liaoning Province of China (2019JH8/10300079).
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bade BC, Dela Cruz CS. Lung cancer 2020: epidemiology, etiology, and prevention. Clin Chest Med. 2020;41(1):1–24. doi:10.1016/j.ccm.2019.10.001
2. Yang D, Liu Y, Bai C, Wang X, Powell CA. Epidemiology of lung cancer and lung cancer screening programs in China and the United States. Cancer Lett. 2020;468:82–87. doi:10.1016/j.canlet.2019.10.009
3. Wang C, Su H, Yang L, Huang K. Integrative analysis for lung adenocarcinoma predicts morphological features associated with genetic variations. Pac Symp Biocomput. 2017;22:82–93. doi:10.1142/9789813207813_0009
4. Yavorski JM, Blanck G. Smoking correlates with increased cytoskeletal protein-related coding region mutations in the lung and head and neck datasets of the cancer genome atlas. Physiol Rep. 2016;4(24):e13045. doi:10.14814/phy2.13045
5. Unni AM, Harbourne B, Oh MH, et al. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. Elife. 2018;7. doi:10.7554/eLife.33718
6. Lemjabbar-Alaoui H, Hassan OU, Yang YW, Buchanan P. Lung cancer: biology and treatment options. Biochim Biophys Acta. 2015;1856(2):189–210. doi:10.1016/j.bbcan.2015.08.002
7. Uramoto H, Tanaka F. Recurrence after surgery in patients with NSCLC. Transl Lung Cancer Res. 2014;3(4):242–249. doi:10.3978/j.issn.2218-6751.2013.12.05
8. Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69(5):363–385. doi:10.3322/caac.21565
9. Bass-Zubek AE, Godsel LM, Delmar M, Green KJ. Plakophilins: multifunctional scaffolds for adhesion and signaling. Curr Opin Cell Biol. 2009;21(5):708–716. doi:10.1016/j.ceb.2009.07.002
10. Rickelt S. Plakophilin-2: a cell-cell adhesion plaque molecule of selective and fundamental importance in cardiac functions and tumor cell growth. Cell Tissue Res. 2012;348(2):281–294. doi:10.1007/s00441-011-1314-3
11. Grossmann KS, Grund C, Huelsken J, et al. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol. 2004;167(1):149–160. doi:10.1083/jcb.200402096
12. Takahashi H, Nakatsuji H, Takahashi M, et al. Up-regulation of plakophilin-2 and Down-regulation of plakophilin-3 are correlated with invasiveness in bladder cancer. Urology. 2012;79(1):240e241–248. doi:10.1016/j.urology.2011.08.049
13. Arimoto K, Burkart C, Yan M, Ran D, Weng S, Zhang DE. Plakophilin-2 promotes tumor development by enhancing ligand-dependent and -independent epidermal growth factor receptor dimerization and activation. Mol Cell Biol. 2014;34(20):3843–3854. doi:10.1128/MCB.00758-14
14. Arimoto KI, Weng S, Zhang DE. Plakophilin-2 induced EGFR phosphorylation: a focus on the intracellular activators of EGFR. Receptors Clin Investig. 2014;2(1):e485. doi:10.14800/rci.485
15. Zhang D, Qian Y, Liu X, Yu H, Zhao N, Wu Z. Up-regulation of plakophilin-2 is correlated with the progression of glioma. Neuropathology. 2017;37(3):207–216. doi:10.1111/neup.12363
16. Niell N, Larriba MJ, Ferrer-Mayorga G, et al. The human PKP2/plakophilin-2 gene is induced by Wnt/beta-catenin in normal and colon cancer-associated fibroblasts. Int J Cancer. 2018;142(4):792–804. doi:10.1002/ijc.31104
17. Hao XL, Tian Z, Han F, Chen JP, Gao LY, Liu JY. Plakophilin-2 accelerates cell proliferation and migration through activating EGFR signaling in lung adenocarcinoma. Pathol Res Pract. 2019;215(7):152438. doi:10.1016/j.prp.2019.152438
18. Kazlauskas A. Plakophilin-2 promotes activation of epidermal growth factor receptor. Mol Cell Biol. 2014;34(20):3778–3779. doi:10.1128/MCB.00968-14
19. Papagerakis S, Shabana AH, Depondt J, Gehanno P, Forest N. Immunohistochemical localization of plakophilins (PKP1, PKP2, PKP3, and p0071) in primary oropharyngeal tumors: correlation with clinical parameters. Hum Pathol. 2003;34(6):565–572. doi:10.1016/S0046-8177(03)00174-6
20. Koetsier JL, Amargo EV, Todorovic V, Green KJ, Godsel LM. Plakophilin 2 affects cell migration by modulating focal adhesion dynamics and integrin protein expression. J Invest Dermatol. 2014;134(1):112–122. doi:10.1038/jid.2013.266
21. Gurha P, Chen X, Lombardi R, Willerson JT, Marian AJ. Knockdown of plakophilin 2 downregulates miR-184 through CpG hypermethylation and suppression of the E2F1 pathway and leads to enhanced adipogenesis in vitro. Circ Res. 2016;119(6):731–750. doi:10.1161/CIRCRESAHA.116.308422
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.