Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 15
Pityriasis Lichenoides Et Varioliformis Acuta and Lymphomatoid Papulosis Type F: A Case Report of Two Entities in One Patient
Authors Pomsoong C ![]() , Suchonwanit P
, Suchonwanit P ![]() , Chanprapaph K
, Chanprapaph K ![]() , Rattanakaemakorn P
, Rattanakaemakorn P ![]() , Rutnin S
, Rutnin S ![]()
Received 22 June 2022
Accepted for publication 19 August 2022
Published 30 August 2022 Volume 2022:15 Pages 1759—1765
DOI https://doi.org/10.2147/CCID.S379577
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Cherrin Pomsoong, Poonkiat Suchonwanit, Kumutnart Chanprapaph, Ploysyne Rattanakaemakorn, Suthinee Rutnin
Division of Dermatology, Department of Medicine, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Correspondence: Suthinee Rutnin, Division of Dermatology, Department of Medicine, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, 270 Rama VI Road, Ratchathewi, Bangkok, Thailand, 10400, Tel +66-2-2011141, Fax +66-2-201-1211, Email [email protected]
Abstract: Pityriasis lichenoides et varioliformis acuta (PLEVA) and lymphomatoid papulosis (LyP) are uncommon inflammatory skin disorders that occasionally share clinicopathological features. Differentiating between the two entities remains problematic, and a definitive diagnosis usually requires multi-step investigations, which is an enormous challenge to physicians. We hereby report a rare case of a 22-year-old female patient diagnosed with PLEVA who later developed LyP type F, a new histological variant of LyP. Our report highlights that long-term follow-up is essential to determine associated hematologic malignancies, particularly in cases with recalcitrant or progressive cutaneous lesions of PLEVA and/or LyP.
Keywords: chronic inflammatory skin disease, CD30 positive lymphoproliferative disorders, immunohistochemistry, mycosis fungoides, pityriasis lichenoides, cutaneous lymphoma
Introduction
Pityriasis lichenoides et varioliformis acuta (PLEVA) and lymphomatoid papulosis (LyP) are uncommon skin disorders categorized as different entities. The former is considered a chronic recurrent inflammatory skin disease while the latter refers to a primary CD30+ cutaneous lymphoproliferative disorder. Clinically, PLEVA presents with acute to subacute cutaneous eruption of multiple erythematous papules that rapidly progress into polymorphic lesions at various stages of evolution, whereas LyP is characterized by recurrent crops of papulonecrotic lesions. Both disorders predominantly appear on the trunk and extremities and sometimes share similar clinical manifestations, leading to delayed or misdiagnosis.1–3 Therefore, the correlation between clinical features, histopathology, and immunohistochemistry is essential to distinguish these two conditions. Herein, we report a rare co-occurrence of PLEVA and LyP type F developing in the same patient.
Case Report
A 22-year-old female presented with a recurrent pruritic rash on the trunk and all extremities for two months. She had no systemic symptoms and denied family history of the same condition. Dermatological examination revealed multiple discrete erythematous papules with crusted lesions on the trunk and extremities, and some healed with varioliform scars (Figure 1). Other systems were unremarkable. Skin biopsy taken from a lesion on the left hand demonstrated dense superficial and deep perivascular and interface dermatitis of lymphocytes admixed with some extravasated erythrocytes. Scatter and confluent necrotic keratinocytes, parakeratosis, and exocytosis of lymphocytes into the epidermis were noted (Figure 2). Lymphocytes were stained positive for CD3, but negative for CD 20, demonstrating that they were T cells. Further immunohistochemistry revealed predominate CD8-positive cytotoxic T cells over CD4-positive T cells and negative for CD 30 staining. (Figure 3). The clinicopathological findings were consistent with PLEVA. She had been treated with topical corticosteroids, doxycycline 200 mg daily, and narrow-band ultraviolet B (NB-UVB) phototherapy two times/week. After 30 sessions of NB-UVB, the treatment was stopped during the Coronavirus disease pandemic with partially clinical improvement. Methotrexate 10 mg/week had been prescribed for 1 month; however, the treatment was switched to cyclosporine 2.5 mg/kg/day due to leukopenia. After 4 months of treatment, cyclosporine was discontinued because of gastrointestinal discomfort. However, skin lesions gradually improved and were totally cleared within 1 year.
 |
Figure 1 Clinical characteristics of pityriasis lichenoides et varioliformis acuta. Multiple discrete erythematous papules with crusted lesions on the (A) trunk and (B and C) extremities, some healed with varioliform scar. |
 |
Figure 2 Histopathology of pityriasis lichenoides et varioliformis acuta. (A) Dense superficial and deep perivascular infiltrate and interface dermatitis (H&E 40X), (B) consisting of lymphocytes admixed with extravasated erythrocytes (H&E 100X). (C) Scatter and confluent necrotic keratinocytes, parakeratosis, and exocytosis of lymphocytes into the epidermis (H&E 400X). Abbreviation: H&E, hematoxylin and eosin. |
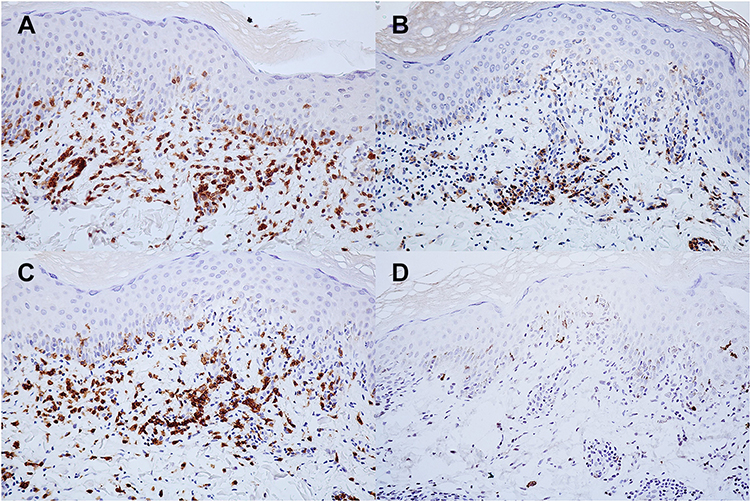 |
Figure 3 Immunohistochemistry of pityriasis lichenoides et varioliformis acuta. Positive (A) CD3, (B) CD4, and (C) CD8 but negative (D) CD30 staining of lymphocytes. (x40 original magnification). |
Three months following the remission, the patient developed a new waxing and waning rash with no systemic symptoms. Dermatological examination revealed multiple discrete erythematous-to-brownish papules with some central necrotic crusts on the trunk and extremities (Figure 4). There was no lymphadenopathy and hepatosplenomegaly. Laboratory investigations, including complete blood count, lactate dehydrogenase level, liver and renal function tests were within normal limits. Skin biopsy obtained from the right arm revealed perifollicular infiltrate of large atypical mononuclear cells with abundant cytoplasm and prominent nuclei admixed with lymphocytes, few neutrophils, and eosinophils with folliculotropism (Figure 5). Immunohistochemistry demonstrated large atypical CD30+ cell infiltrate within the hair follicle and CD3+, CD4+, and CD8+ small lymphocytes in perivascular areas. (Figure 6). Regarding clinicopathological correlation, the definitive diagnosis was LyP type F. Acitretin 20 mg/day and NB-UVB 2–3 times/week were given, and partial remission was observed after nine consecutive months of treatments.
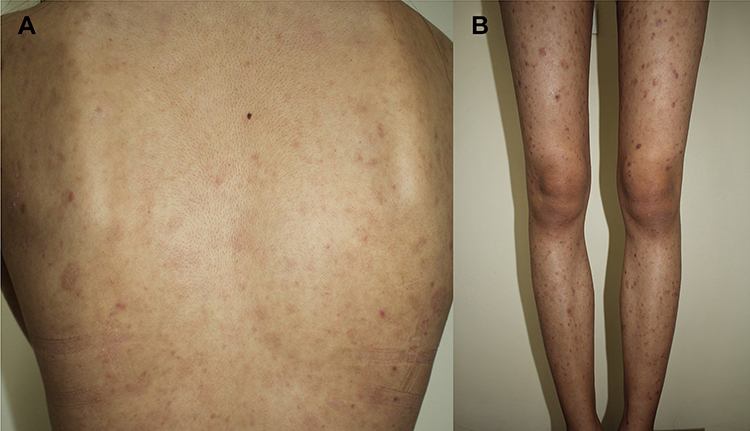 |
Figure 4 Clinical characteristics of lymphomatoid papulosis. Multiple discrete erythematous-to-brownish papules with some central necrotic crusts on the (A) trunk and (B) extremities. |
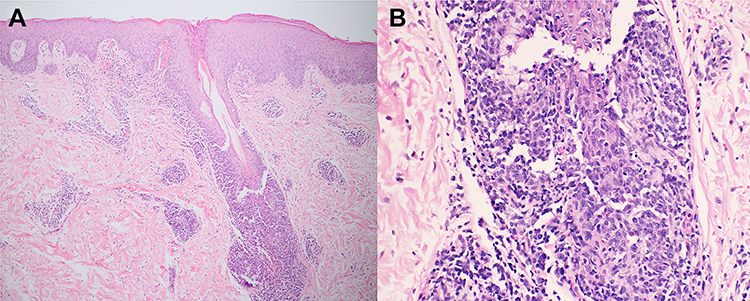 |
Figure 5 Histopathology of lymphomatoid papulosis type F. (A) Superficial and deep perivascular and perifollicular infiltrate with epidermal hyperplasia and focal hyperkeratosis (H&E 100X). (B) Perifollicular infiltrate of atypical large mononuclear cells with folliculotropism (H&E 400X). Abbreviation: H&E, hematoxylin and eosin. |
 |
Figure 6 Immunohistochemistry of lymphomatoid papulosis type F. Positive (A) CD3, (B) CD4, and (C) CD8 staining of small lymphocytes in perivascular area. (D) Perifollicular and follicular infiltrate of positive CD30 large atypical mononuclear cells (x40 original magnification). |
Discussion
PLEVA, one of pityriasis lichenoides variants, is an uncommon inflammatory skin disease, which may potentially be malignant. It generally presents with an acute-to-subacute eruption of multiple erythematous papules with hemorrhagic necrosis and crusting and often heals with atrophic varioliform scars. The lesions are frequently self-healing; however, recalcitrance may occur.1 To confirm the diagnosis, histopathologic examination is required. PLEVA is characterized by interface dermatitis with prominent lymphocytic infiltration and epidermal involvement.4 The exact pathogenesis remains unclear; however, immune dysregulation against medications/infectious agents or an evolution to cutaneous T-cell dyscrasia is an accepted hypothesis.1,4 Despite lacking standard treatment, topical corticosteroids are commonly prescribed as the first-line therapy. Phototherapy, especially NB-UVB, and antibiotics including tetracyclines and erythromycin are also recommended. Low-dose methotrexate and other systemic immunosuppressants are indicated for severe or recalcitrant PLEVA.5
LyP is a rare dermatological condition and classified in primary CD30+ cutaneous lymphoproliferative disorders.6 LyP typically presents with recurrent crops of disseminated papulonodular eruptions, which spontaneously regress within weeks to months.7,8 Healing lesions appear as transient post-inflammatory hyperpigmented/hypopigmented macules and occasionally form atrophic varioliform scars. LyP is categorized into five subtypes (A to E) according to histologic characteristics, with type A being the classic and most common type (>75%).2
LyP type F or follicular LyP is another rare histologic subtype, although not officially classified in WHO classification. It was firstly introduced by Pierard et al in 1980 and mainly manifests with follicular involvement.9 The prevalence of LyP type F is approximately 5.8–10% of all LyP cases.10 Histologically, it is characterized by perifollicular infiltrate with variable degrees of folliculotropism of CD30+ medium to large atypical lymphocytes. The less common features are follicular epithelial hyperplasia, ruptured hair follicle, follicular mucinosis, and intrafollicular pustules.10 The overlapping clinicopathological features between PLEVA, LyP type F, and mycosis fungoides (MF) may exist.11,12 Therefore, clinicopathological correlations and further immunohistochemistry are crucial to distinguish among the three conditions (Table 1).4,10,13–15
 |
Table 1 The Clinicopathological Features and Immunohistochemistry Between Pityriasis Lichenoides Et Varioliformis Acuta, Lymphomatoid Papulosis, and Mycosis Fungoides |
Since LyP is a recurring condition, and its curative therapy is not available, methotrexate and/or phototherapy, including NB-UVB and psoralen ultraviolet-A are the first-line option for cases with disseminated or recurrent scarring lesions on cosmetically sensitive areas. Methotrexate usually reaches disease control after 3–4 weeks, but a relapse rate of up to 40% may occur after its discontinuation.2 Recently, a novel targeted therapy, brentuximab (anti-CD30), demonstrated benefit for severe or recalcitrant LyP.16 Although various therapeutic managements are established, relapse still occurs. Whether LyP has a 10-year disease-specific survival rate of almost 100%, secondary hematologic malignancies including MF, anaplastic large cell lymphoma, and Hodgkin’s lymphoma have been reported in up to 20%. The malignancies may be preceded by, associated with, or followed by LyP.17
We presented a unique case of recalcitrant PLEVA for one year, and developed LyP after 3 months of remission, demonstrating two diseases in one patient. The association between PLEVA and LyP has been proposed but remains debatable.11,18 However, clinicopathological and immunohistochemical evidence in the past decade has suggested PLEVA and LyP as distinct disorders.4,19,20 Currently, there are limited reports of PLEVA and LyP arising in the same patient.21,22 Sidiropoulou et al reported the coexistence of LyP type A, PLEVA, and MF over a 15-year period in one case. Different alterations in host immunity leading to discrete clinical expressions were remarked for these three separate entities.21 Another case presented with prolonged LyP type B for 11 years followed by PLEVA, reflecting the difference in host immune response to antigenic stimulus.22
In conclusion, we reported a patient with PLEVA followed by LyP type F, a rare co-existence. This case underlined the importance of diagnostic confirmation with the clinicopathological and immunohistochemical distinction between these two entities. It is also essential to recognize histologic characteristics of LyP type F to halt misdiagnosis. Additionally, long-term follow-up and re-biopsy should be considered in progressive or recalcitrant cases in order to give a precise diagnosis, provide proper management, and evaluate for associated secondary hematologic malignancies.
Abbreviations
LyP, lymphomatoid papulosis; MF, mycosis fungoides; NB-UVB, narrow-band ultraviolet B; PLEVA, pityriasis lichenoides et varioliformis acuta.
Ethics Approval and Informed Consent
The patient provided written informed consent for the case details and accompanying images to be published. Institutional approval was not required to publish the case details.
Funding
The authors received no financial support for this research.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Khachemoune A, Blyumin ML. Pityriasis lichenoides: pathophysiology, classification, and treatment. Am J Clin Dermatol. 2007;8(1):29–36.
2. Martinez-Cabriales SA, Walsh S, Sade S, Shear NH. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34(1):59–73.
3. Zheng Y, Jia J, Tian Q, et al. Lymphomatoid papulosis misdiagnosed as pityriasis lichenoides et varioliformis acuta: two case reports and a literature review. Exp Ther Med. 2014;8(6):1927–1933.
4. Bowers S, Warshaw EM. Pityriasis lichenoides and its subtypes. J Am Acad Dermatol. 2006;55(4):557–572.
5. Bellinato F, Maurelli M, Gisondi P, Girolomoni G. A systematic review of treatments for pityriasis lichenoides. J Eur Acad Dermatol Venereol. 2019;33(11):2039–2049.
6. Wang HH, Lach L, Kadin ME. Epidemiology of lymphomatoid papulosis. Cancer. 1992;70(12):2951–2957.
7. Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118(15):4024–4035.
8. Kempf W, Kerl K, Mitteldorf C. Cutaneous CD30-positive T-cell lymphoproliferative disorders-clinical and histopathologic features, differential diagnosis, and treatment. Semin Cutan Med Surg. 2018;37(1):24–29.
9. Pierard GE, Ackerman AB, Lapiere CM. Follicular lymphomatoid papulosis. Am J Dermatopathol. 1980;2(2):173–180.
10. Kempf W, Kazakov DV, Baumgartner HP, Kutzner H. Follicular lymphomatoid papulosis revisited: a study of 11 cases, with new histopathological findings. J Am Acad Dermatol. 2013;68(5):809–816.
11. Vonderheid EC, Kadin ME, Telang GH. Commentary about papular mycosis fungoides, lymphomatoid papulosis and lymphomatoid pityriasis lichenoides: more similarities than differences. J Cutan Pathol. 2016;43(4):303–312.
12. Borra T, Custrin A, Saggini A, et al. Pityriasis Lichenoides, Atypical Pityriasis Lichenoides, and Related Conditions: a Study of 66 Cases. Am J Surg Pathol. 2018;42(8):1101–1112.
13. Kodama K, Fink-Puches R, Massone C, Kerl H, Cerroni L. Papular mycosis fungoides: a new clinical variant of early mycosis fungoides. J Am Acad Dermatol. 2005;52(4):694–698.
14. Mitteldorf C, Stadler R, Sander CA, Kempf W. Folliculotropic mycosis fungoides. J Dtsch Dermatol Ges. 2018;16(5):543–557.
15. Ross NA, Truong H, Keller MS, Mulholland JK, Lee JB, Sahu J. Follicular Lymphomatoid Papulosis: an Eosinophilic-Rich Follicular Subtype Masquerading as Folliculitis Clinically and Histologically. Am J Dermatopathol. 2016;38(1):e1–10.
16. Lewis DJ, Talpur R, Huen AO, Tetzlaff MT, Duvic M. Brentuximab Vedotin for Patients With Refractory Lymphomatoid Papulosis: an Analysis of Phase 2 Results. JAMA Dermatol. 2017;153(12):1302–1306.
17. Wieser I, Oh CW, Talpur R, Duvic M. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74(1):59–67.
18. Black MM. Lymphomatoid papulosis and pityriasis lichenoides: are they related? Br J Dermatol. 1982;106(6):717–721.
19. Willemze R, Scheffer E. Clinical and histologic differentiation between lymphomatoid papulosis and pityriasis lichenoides. J Am Acad Dermatol. 1985;13(3):418–428.
20. Jang KA, Choi JC, Choi JH. Expression of cutaneous lymphocyte-associated antigen and TIA-1 by lymphocytes in pityriasis lichenoides et varioliformis acuta and lymphomatoid papulosis: immunohistochemical study. J Cutan Pathol. 2001;28(9):453–459.
21. Sidiropoulou P, Nikolaou V, Marinos L, et al. A case of lymphomatoid papulosis, pityriasis lichenoides acuta, and mycosis fungoides coexistence. Australas J Dermatol. 2019;60(2):e154–e156.
22. Vonderheid EC, Kadin ME, Gocke CD. Lymphomatoid papulosis followed by pityriasis lichenoides: a common pathogenesis? Am J Dermatopathol. 2011;33(8):835–840.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Folliculotropic Mycosis Fungoides: Current Guidance and Experience from Clinical Practice
Roccuzzo G, Mastorino L, Gallo G, Fava P, Ribero S, Quaglino P
Clinical, Cosmetic and Investigational Dermatology 2022, 15:1899-1907
Published Date: 13 September 2022