Back to Journals » International Journal of Nanomedicine » Volume 11
Pigment epithelial-derived factor gene loaded novel COOH-PEG-PLGA-COOH nanoparticles promoted tumor suppression by systemic administration
Authors Yu T, Xu B, He L, Xia S, Chen Y, Zeng J, Liu Y, Li S, Tan X, Ren K, Yao S, Song X
Received 26 September 2015
Accepted for publication 30 November 2015
Published 25 February 2016 Volume 2016:11 Pages 743—759
DOI https://doi.org/10.2147/IJN.S97223
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Ting Yu,1,* Bei Xu,1,* Lili He,2 Shan Xia,3 Yan Chen,1 Jun Zeng,1 Yongmei Liu,1 Shuangzhi Li,1 Xiaoyue Tan,4 Ke Ren,1 Shaohua Yao,1 Xiangrong Song1
1State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, and Collaborative Innovation Center for Biotherapy, 2College of Chemistry and Environment Protection Engineering, Southwest University for Nationalities, 3Central Laboratory, Science Education Department, Chengdu Normal University, Chengdu, Sichuan, 4Department of Pathology/Collaborative Innovation Center of Biotherapy, Medical School of Nankai University, Tianjin, People’s Republic of China
*These authors contributed equally to this work
Abstract: Anti-angiogenesis has been proposed as an effective therapeutic strategy for cancer treatment. Pigment epithelium-derived factor (PEDF) is one of the most powerful endogenous anti-angiogenic reagents discovered to date and PEDF gene therapy has been recognized as a promising treatment option for various tumors. There is an urgent need to develop a safe and valid vector for its systemic delivery. Herein, a novel gene delivery system based on the newly synthesized copolymer COOH-PEG-PLGA-COOH (CPPC) was developed in this study, which was probably capable of overcoming the disadvantages of viral vectors and cationic lipids/polymers-based nonviral carriers. PEDF gene loaded CPPC nanoparticles (D-NPs) were fabricated by a modified double-emulsion water-in-oil-in-water (W/O/W) solvent evaporation method. D-NPs with uniform spherical shape had relatively high drug loading (~1.6%), probably because the introduced carboxyl group in poly (D,L-lactide-co-glycolide) terminal enhanced the interaction of copolymer with the PEDF gene complexes. An excellent in vitro antitumor effect was found in both C26 and A549 cells treated by D-NPs, in which PEDF levels were dramatically elevated due to the successful transfection of PEDF gene. D-NPs also showed a strong inhibitory effect on proliferation of human umbilical vein endothelial cells in vitro and inhibited the tumor-induced angiogenesis in vivo by an alginate-encapsulated tumor cell assay. Further in vivo antitumor investigation, carried out in a C26 subcutaneous tumor model by intravenous injection, demonstrated that D-NPs could achieve a significant antitumor activity with sharply reduced microvessel density and significantly promoted tumor cell apoptosis. Additionally, the in vitro hemolysis analysis and in vivo serological and biochemical analysis revealed that D-NPs had no obvious toxicity. All the data indicated that the novel CPPC nanoparticles were ideal vectors for the systemic delivery of PEDF gene and might be widely used as systemic gene vectors.
Keywords: pigment epithelium-derived factor gene, nanoparticles based on PLGA derivative, gene delivery, systemic delivery, tumor
Introduction
Angiogenesis is a physiological process for formation of new blood vessels from preexisting ones and has played a critical role in the development, growth, and metastatic potential of tumors.1 Anti-angiogenic therapy is now considered to be one of the most promising approaches for cancer treatment.2–5 Pigment epithelium-derived factor (PEDF), as a member of the serine protease inhibitor (serpin) superfamily, has been reported to be one of the most potent inhibitors of angiogenesis.6–8 PEDF is a 50 kDa secreted glycoprotein that belongs to a group of proteins having a common three-dimensional structure.7 Compared with the direct protein administration, PEDF gene might be cheaper and achieve higher antitumor efficacy due to easier access to tumor tissue.
Gene therapy, transferring therapeutic genes to tumor cells using diverse delivery vehicles, has been developed as a potential treatment modality for cancers.9–11 Viral vectors and nonviral vectors have been developed for this purpose. Viral vectors can mediate gene transfer with high efficiency, but the disadvantages such as immunogenic/inflammatory responses, low loading capacity, difficult large-scale manufacturing, and quality control limit their applications.12 Consequently, more attention has been paid on nonviral gene vectors due to their low toxicity, relatively easy production, and great versatility.13 Among nonviral vectors, biodegradable and biocompatible polymer-based nanoparticles have shown their advantages over other carriers by their increased stability and their controlled-release efficacy.14,15 Nanoparticles made from poly (D,L-lactide-co-glycolide) (PLGA) have been the focus of extensive researches in gene delivery due to their capability of overcoming the disadvantages of polyethylenimine- or cationic lipids/polymers-based gene carriers, such as cytotoxicity induced by excess positive charge and aggregation on the cell surface.16 PLGA, which has been approved by the US Food and Drug Administration for certain human clinical uses,17 has many advantages including nontoxicity, biodegradable and biocompatible, controlled- and sustained-release efficacy,17,18 long-term stability, and rapid evasion of the endolysosomal pathway.6,19 Thus, PLGA has been widely used to design nanoparticles as the promising gene delivery system in many studies.6,17,20
In our previous study, PLGA nanoparticles loaded with PEDF gene were successfully prepared and had shown great therapeutic efficacy in a C26 xenograft tumor model.6 However, the mice were treated by intratumoral injection, which was invasive and could not cover tumors adequately and treat small metastatic tumors efficiently.21 Thus, this administration route was not used widely like intravenous injection in routine clinical practice.22 Bare PLGA nanoparticles administered intravenously are rapidly removed from circulation in the blood due to opsonization.23 Therefore, versatile strategies are being adopted to overcome the limitations of bare PLGA nanoparticles. Polyethylene glycol (PEG) is a biocompatible, strongly hydrophilic, and nonhazardous polymer. It is known to shield nanoparticles from the immune surveillance and improve the enhanced permeability and retention effect.17,24,25 PEG-functionalized PLGA nanoparticles have been considerably more effective against systemic clearance than similar particles without PEG.26 Hence, a novel COOH-PEG-PLGA-COOH (CPPC), one of PEG-PLGA copolymers, was designed to develop nanoparticles encapsulating PEDF gene (D-NPs) in this study. The carboxyl group in PEG terminus could be used for functional modification such as the conjugation of targeting ligands, and it was hypothesized that the one carboxyl group in PLGA terminus would enhance the interaction of copolymer with gene and thereby improve the drug loading.
Taking into account the above information, CPPC was first synthesized and characterized. D-NPs were subsequently developed to investigate the pharmaceutical properties, in vitro and in vivo antitumor efficacy. The safety of D-NPs was also assessed synchronously.
Materials and methods
Materials
PLGA (molecular weight [MW] =15 kDa; lactic acid:glycolic acid =75:25) was purchased from Jinan Daigang Biomaterial Co., Ltd. (Jinan, People’s Republic of China). PEDF plasmid pAAV2-PEDF (PEDF gene) and null plasmid pAAV2 were constructed according to our previous report.6 PEG (MW =2 kDa), polyvinyl alcohol (PVA, MW =30–70 kDa; high density: 80%), 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT), Hoechst 33258, and poly-L-lysine (PLL, MW =15–30 kDa) were procured from Sigma-Aldrich (St Louis, MO, USA). All other reagents were of analytical grade and were used as supplied.
Cell culture and animals
Murine colon adenocarcinoma cells (C26) and adenocarcinomic human alveolar basal epithelial cells (A549) were purchased from American Type Culture Collection (Manassas, VA, USA) and cultured in Roswell Park Memorial Institute 1640 medium with 10% calf serum and 10% fetal bovine serum, respectively. Primary human umbilical vein endothelial cells (HUVECs) were isolated from human umbilical cord veins by a standard procedure,27 and were grown in endothelial basal medium-2 medium with SingleQuots (Lonza, Allendale, NJ, USA) containing vascular endothelial growth factor and other growth factors. All above cells were maintained at 37°C in a humidified incubator containing 5% CO2. This research followed the principles of the Declaration of Helsinki. All samples were obtained after patients provided informed consent. Both this study and the consent were approved by the Institutional Ethics Committee of West China Hospital of Sichuan University.
Balb/c mice (18±2 g) were used for in vivo tests and purchased from the Laboratory Animal Center of Sichuan University (Sichuan University, Chengdu, Sichuan, People’s Republic of China). The mice were housed and maintained under specific pathogen-free conditions in facilities. All studies were approved and supervised by the State Key Laboratory of Biotherapy Animal Care and Use Committee (Sichuan University).
Synthesis of CPPC
COOH-PEG-COOH (CPC) was synthesized according to the scheme shown in Figure 1A. The mixture containing PEG, 4-dimethylaminopyridine (DMAP), and succinic anhydride (1:0.1:2, molar ratio) in dichloromethane solution was stirred over 24 hours at room temperature (RT). Thereafter, the solvent was removed using rotary evaporator and then the crude product CPC was purified using recrystallization in isopropyl alcohol and collected with vacuum filtration. CPPC was then synthesized by a standard 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride/DMAP-mediated chemistry. CPC, DMAP, and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.6:0.12:3, molar ratio) were dissolved in anhydrous dichloromethane and stirred for 2 hours at RT to activate the carboxylic groups of CPC. A solution of PLGA in anhydrous dichloromethane was then added dropwise into the reaction mixture. After being continuously stirred for 48 hours, the resultant solution was dried, dissolved in dimethyl sulfoxide, and dialyzed (Regenerated Cellulose Membrane Dialysis Tubing, MW cutoff 7 kDa) against deionized (DI) water for 3 days to remove excess unreacted CPC and by-products. The conjugate CPPC was then lyophilized and stored at RT until further use. The obtained polymer was confirmed by proton nuclear magnetic resonance (1H-NMR).
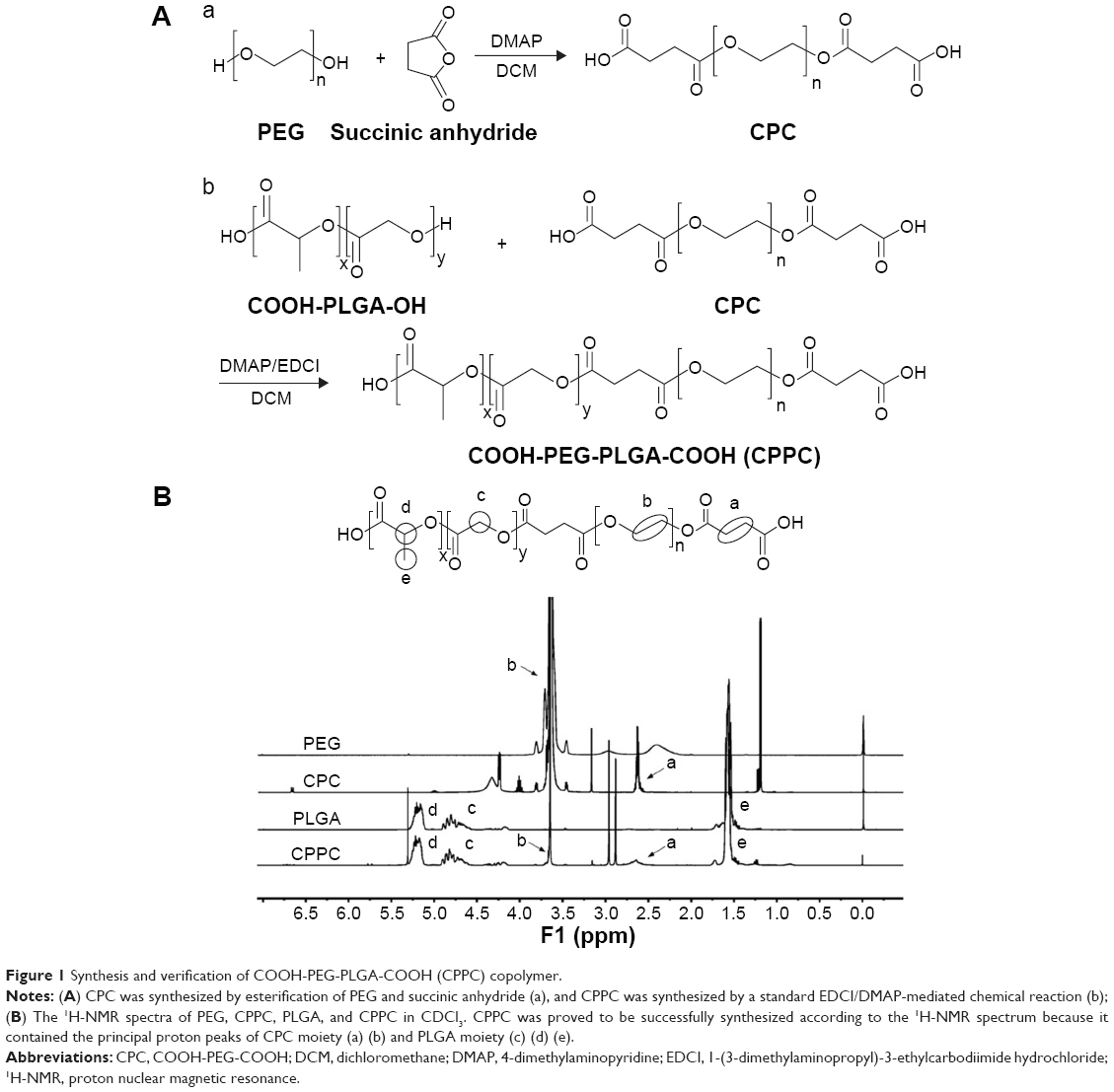 | Figure 1 Synthesis and verification of COOH-PEG-PLGA-COOH (CPPC) copolymer. |
Preparation of D-NPs
D-NPs were prepared by a modified double-emulsion (W/O/W) solvent evaporation method. First, PEDF gene was incubated with equivalent PLL to form the inner aqueous phase. Then the organic phase (dichloromethane/acetone, 4/1, v/v) containing 10 mg CPPC was emulsified with the inner aqueous phase by probe sonication at 20 W for 20 seconds in ice bath to produce the primary W/O emulsion. Six mL 1% PVA solution was added to the primary emulsion and the mixture was further sonicated at 40 W for 40 seconds in ice bath to obtain the multiple emulsion (W/O/W). Finally, the resulting emulsion was treated under vacuum to remove the organic solvent at 37°C. The obtained colloidal solution was then centrifuged (13,300× g) at 4°C for 40 minutes and was washed three times with Milli-Q water to remove PVA and unencapsulated PEDF gene. The sediment was redispersed to obtain D-NPs. Null plasmid loaded PEG-PLGA nanoparticles (Dv-NPs) were also prepared using a similar procedure. Both nanoparticles were stored at 4°C before use.
Characterization of D-NPs
Entrapment efficiency and drug loading
During the preparation of D-NPs, the supernatant obtained by centrifuging the colloidal solution was stored to determine the entrapment efficiency (EE%) and drug loading (DL%) of PEDF gene. The supernatant was first incubated with 0.15 μg/mL Hoechst 33258 for a few minutes in the dark. Then the fluorescence intensity of the mixture was measured by LS55 Luminescence Spectrometer (PerkinElmer Inc., Waltham, MA, USA) at an excitation wavelength of 358 nm and an emission wavelength of 457 nm, which was further used to calculate the amount of PEDF gene unentrapped into D-NPs by a one-point external standard method. EE% and DL% of PEDF gene were determined as follows:
|
|
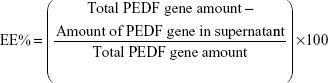
|
|

Particle size and zeta potential
The mean particle size was measured by dynamic laser scattering using a Zetasizer (Zetasizer Nano-ZS 90; Malvern Instruments Ltd., Malvern, UK) at 25°C. The zeta potential was automatically calculated from the electrophoretic mobility using the same instrument. All results were the mean of three different samples and all the data were expressed as the mean ± standard deviation.
Appearance
The morphology of the D-NPs was examined by transmission electronic microscopy (TEM). Before analysis, the prepared nanoparticles were diluted with DI water, and then placed on a copper electron microscopy grid. The sample was negatively stained with a 2% (w/v) phosphotungstic acid solution and dried at RT. Then the TEM image was obtained.
Stability
D-NPs were stored at 4°C for 1 month to investigate the preliminary stability. The changes in particle size, zeta potential, and EE% of PEDF gene were examined by the methods described in the “Entrapment efficiency and drug loading’’ section.
In vitro release
The release profiles of PEDF gene from D-NPs were investigated in different phosphate-buffered saline at pH 5.5, 6.8, and 7.4, respectively. The nanoparticles were first dispersed in phosphate-buffered saline and then divided into nine tubes, which were shaken at 37°C with a gentle rate of 100× g. At a definite time interval, one tube was taken out and treated with centrifugation. The amount of the PEDF gene released in the supernatant was calculated by the same method as described in the “Entrapment efficiency and drug loading” section. Experiments were performed in triplicate.
Hemolysis
The hemolysis test was performed to determine whether the D-NPs were safe for intravenous administration. Briefly, the red blood cells of healthy rabbit were collected by centrifuging the diluted whole blood in normal saline (NS) at 1,000× g for 5 minutes. The supernatant was removed and the pellet was washed with NS at least three times. Finally, the red blood cells pellet was resuspended in NS to obtain a standard 2% erythrocyte dispersion. For the hemolysis experiment, 2% erythrocyte dispersion (2.5 mL) was treated with D-NPs, which had been preliminarily diluted with 2 mL NS. DI water and NS were employed as the positive and negative control, respectively. All the samples were centrifuged after incubation at 37°C for 3 hours. The absorbance (A) of the obtained supernatant was monitored at 545 nm by ultraviolet-visible spectrophotometer (Perkin-Elmer Lambda 35; PerkinElmer Inc.). The percentage of the sample-induced hemolysis was calculated as follows:
|
|
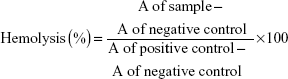
In vitro experiments
Growth inhibition of tumor cells and HUVECs
Growth inhibition assay of D-NPs was performed on C26, A549, and HUVECs by contrast with Dv-NPs. Cells were plated in 96-well plates at a density of 3×103 cells/well and cultured in a humidified atmosphere of 5% CO2 in air at 37°C for 24 hours. Then, cells were exposed to different concentrations of D-NPs or Dv-NPs for 48 hours. The cell growth inhibition or cell viability was subsequently assessed using the modified MTT assay as previously described.28 In brief, 20 μL of MTT stock solution (5 mg/mL in saline) was added to each well. After incubation for 4 hours at 37°C, the culture medium was removed by aspiration and then 150 μL of dimethyl sulfoxide was added to dissolve formazan crystals by shaking at RT. The solubilized formazan product was quantified using an enzyme-linked immunosorbent assay (ELISA) plate reader (Thermo Scientific Multiskan MK3; Thermo Fisher Scientific, Waltham, MA, USA) at the wavelength of 570 nm. The absorbance value (optical density (OD) 570) of each well was proportional to the number of viable cells. Untreated cells were used as controls. The inhibition rate and the cell viability were calculated by the following formulae, respectively:
|
|

|
|
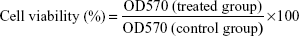
The cytotoxicity test of CPPC was conducted on all the three cell lines by the same method. All data were expressed as the mean ± standard deviation.
HUVEC tube-formation assay
The HUVEC tube-formation assay was carried out as previously described.8 Briefly, 50 μL of Matrigel (BD Biosciences, Bedford, MA, USA) was pipetted into the prechilled 96-well plate on ice and the plate was incubated at 37°C for 30 minutes; 100 μL of trypsinized HUVECs (3×104 cells/well) with 8 μg/mL D-NPs or Dv-NPs were then seeded to Matrigel-coated wells. Untreated cells were used as control. After 6 hours incubation at 37°C, images of capillary-like structure were obtained using a light microscope (Olympus, Tokyo, Japan). Tubular structures were quantified by manually counting the numbers of connected cells in randomly selected fields at 200× magnification and the total tube number in the control group was designated as 100%.
Western blot and ELISA analysis
C26 and A549 cells were plated in 96-well plates at a density of 3×103 cells/well and cultured in a humidified atmosphere of 5% CO2 in air at 37°C for 24 hours. Then both cell lines were treated with D-NPs and Dv-NPs, respectively. Cells without treatment were used as control. After incubation for another 48 hours, cells and media were collected, respectively. The former were lysed with radioimmunoprecipitation assay solution (Beyotime, Jiangsu, People’s Republic of China), and the supernatants obtained through centrifugation were subjected to Western blot analysis. The protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. The membrane was blocked with 5% skimmed milk. Then, the expressed PEDF protein was probed with a primary anti-PEDF antibody (1:1,000; R&D Systems, Boston, MA, USA) and a horseradish peroxidase-conjugated secondary antibody (1:5,000; ZSGB-BIO, Beijing, People’s Republic of China). The binds were observed using an enhanced chemiluminescence detection kit (Pierce, Rockford, IL, USA). A membrane was tested for β-actin to confirm equal loading.
Meanwhile, the concentrations of the secreted PEDF in media were measured using a sandwich ELISA kit (R&D Systems) following the manufacturer’s instructions. Briefly, the collected media were added to the wells which had been precoated with a monoclonal antibody specific for PEDF. After a series of reactions, absorbance of the suspension was measured at the wavelength of 450 nm with an ELISA reader (Thermo Scientific Multiskan MK3). A standard curve was plotted and the protein concentration in each group was interpolated from the standard curve.
Alginate-encapsulated tumor cell assay
Alginate-encapsulated tumor cell assay was performed to evaluate the anti-angiogenesis activity of D-NPs in vivo. Briefly, alginate beads entrapping 5×104 C26 cells/bead were prepared and subcutaneously implanted into both dorsal sides of mice. The mice were injected intravenously every 2 days with NS (control), D-NPs (PEDF gene at dose of 250 μg/kg), and Dv-NPs for 12 days. At the end of the experiment, 0.1 mL of 2% fluorescein isothiocyanate-dextran solution was injected into mice intravenously. Alginate beads were photographed and removed within 20 minutes after being exposed surgically. The beads were transferred to tubes containing 2 mL NS, vortexed, and centrifuged. The fluorescence of the supernatant was measured with a fluorescence spectrophotometer (Thermo Scientific Varioskan Flash) by excitation at 492 nm and emission at 515 nm as described.29
In vivo therapeutic experiment
Animal experiment
Antitumor activity of D-NPs was investigated in subcutaneous C26 tumor-bearing mice model. Briefly, mice were injected subcutaneously on the right flank with 100 μL of cell suspension containing 5×105 viable C26 cells. When the tumor reached ~50 mm3 in volume, mice were randomized into three groups (five mice per group) and were respectively treated with NS (control), D-NPs, and Dv-NPs (PEDF gene at dose of 250 μg/kg) via tail vein every 2 days for 2 weeks. In tumor growth inhibition assay, mice were weighed and tumor sizes were measured every 2 days. Tumor volumes were calculated by the equation: volume =0.5× L × W2, where L and W represented the length and the width of the tumors, respectively. After the mice were sacrificed, tumors in each group were immediately harvested and weighed. To further investigate the antitumor activity of D-NPs, survival times of the mice were also evaluated (ten mice per group).
Serological and biochemical analysis
Blood samples were collected from the mice for serological and biochemical analysis before they were sacrificed on day 14. Serum was obtained by centrifugation at 13,300× g for 10 minutes and was used for biochemical analysis with an automatic analyzer (Hitachi High-Technologies Corp., Minato-ku, Tokyo, Japan) immediately.
CD31 immunohistochemistry
Anti-angiogenesis activity of D-NPs was determined by immunohistochemistry analysis of neovascularization in tumor tissue. After fixation with cold acetone for 15 minutes, the frozen sections of tumors were incubated with anti-CD31 antibody (1:200; Abcam, Cambridge, MA, USA) for the identification of endothelial cells. The vessels were revealed with streptavidin-peroxidase followed by chromogenic substrate diaminobenzidine (ZSGB-BIO). Then the sections were counterstained with hematoxylin. Immunostaining images were observed under a light microscope (Olympus) and microvessel density (MVD) was determined according to the previously reported method.30
Terminal deoxynucleotidyl transferase-mediated nick-end labeling assay
To explore the role of D-NPs on apoptosis of tumor cells, terminal deoxynucleotidyl transferase-mediated nick-end labeling (TUNEL) staining was performed on paraffin sections using an in situ cell death detection kit (Beyotime). TUNEL-positive nuclei with dark green fluorescence were monitored using a fluorescence microscope (Olympus). Four equal-sized fields were randomly chosen and analyzed. The apoptotic index was defined as follows: apoptotic index (%) = apoptotic cells/total tumor cells ×100. The samples in NS and Dv-NPs groups were recorded and processed by the same procedure.
Hematoxylin and eosin staining
The heart, liver, spleen, lung, and kidney were immediately collected after the mice were sacrificed and fixed in a 4% paraformaldehyde solution overnight. Then, the organs were embedded in paraffin, sectioned, and processed for hematoxylin and eosin (H&E) staining. The images for the H&E staining were acquired on a light microscope (Olympus).
Statistical analysis
The statistical analysis was performed using the Statistical Product and Service Solutions software (SPSS V19.0, IBM Corp., New York, USA). Data were analyzed by one-way analysis of variance. Survival curves were generated based on the Kaplan–Meier method and statistical significance was determined by Mann–Whitney U-tests. P<0.05 and 0.01 were considered indications of statistical difference and statistically significant difference, respectively.
Results
Synthesis of CPPC copolymer
The CPPC copolymer was successfully prepared using the synthetic route as shown in Figure 1A. CPC obtained in the first reaction step provided active carboxylic terminal to further synthesize CPPC in the second reaction step. The principal peaks related to CPC moiety (b) and PLGA moiety (c), (d), (e) in the 1H-NMR spectrum of the final product indicated successful synthesis of CPPC (Figure 1B). The degree of substitution (0.7) of the CPC moiety to PLGA was calculated by comparing the integrations of shift “b” (3.64 ppm; methylene hydrogen of the PEG) and shift “d” (5.21 ppm; methine hydrogen of the lactide units in PLGA).
Characterization of D-NPs
D-NPs were successfully produced by the W/O/W solvent evaporation method (Figure 2A). The colloidal solution presented slightly blue opalescence (Figure 2B). The dynamic laser scattering results showed that D-NPs had an average particle size of 176.9±1.3 nm (Figure 2C) and a narrow distribution with a small polydispersity index of 0.085±0.01. TEM image further revealed that D-NPs were spherical and homogeneous in aqueous solution, which was in good accordance with the narrow particle size distribution (Figure 2D). Besides, D-NPs, with a negative surface charge of −23.04±1.03 mV, exhibited a high entrapment efficiency (89.5%±2.6%) and drug loading (1.6%±0.3%). Moreover, the stability study showed that D-NPs remained stable without change of size, zeta potential, and EE% for at least 2 weeks (Figure 2E and F).
 | Figure 2 D-NPs development and the relative pharmaceutical characteristics. |
In vitro PEDF gene release
As presented in Figure 3, D-NPs exhibited a sustained release without a burst-release effect in all the release media (pH 5.5, 6.8, and 7.4) simulating various physiological conditions and showed a typical pH-dependent release behavior. The cumulative PEDF gene release within 48 hours at pH 5.5 was ~100%, while only about 80% and 55% of PEDF gene released at pH 6.8 and pH 7.4, respectively.
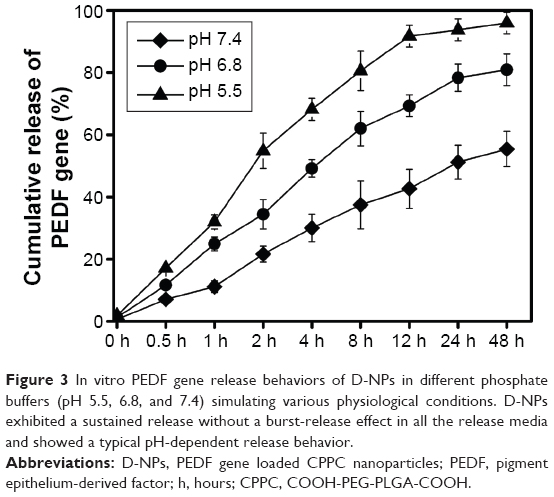 | Figure 3 In vitro PEDF gene release behaviors of D-NPs in different phosphate buffers (pH 5.5, 6.8, and 7.4) simulating various physiological conditions. D-NPs exhibited a sustained release without a burst-release effect in all the release media and showed a typical pH-dependent release behavior. |
Growth inhibition of tumor cells and HUVECs
As shown in Figure 4A and B, D-NPs presented concentration-dependent cell growth inhibition activities on C26 and A549 cells, while no inhibition effects of Dv-NPs on both cell lines were found (P<0.01). D-NPs also significantly inhibited the proliferation of HUVECs in a dose-dependent manner (P<0.01, vs Dv-NPs) (Figure 4C). In addition, CPPC was found to be nontoxic to all the three investigated cell lines even if its concentration was up to 2 mg/mL (Figure 4D).
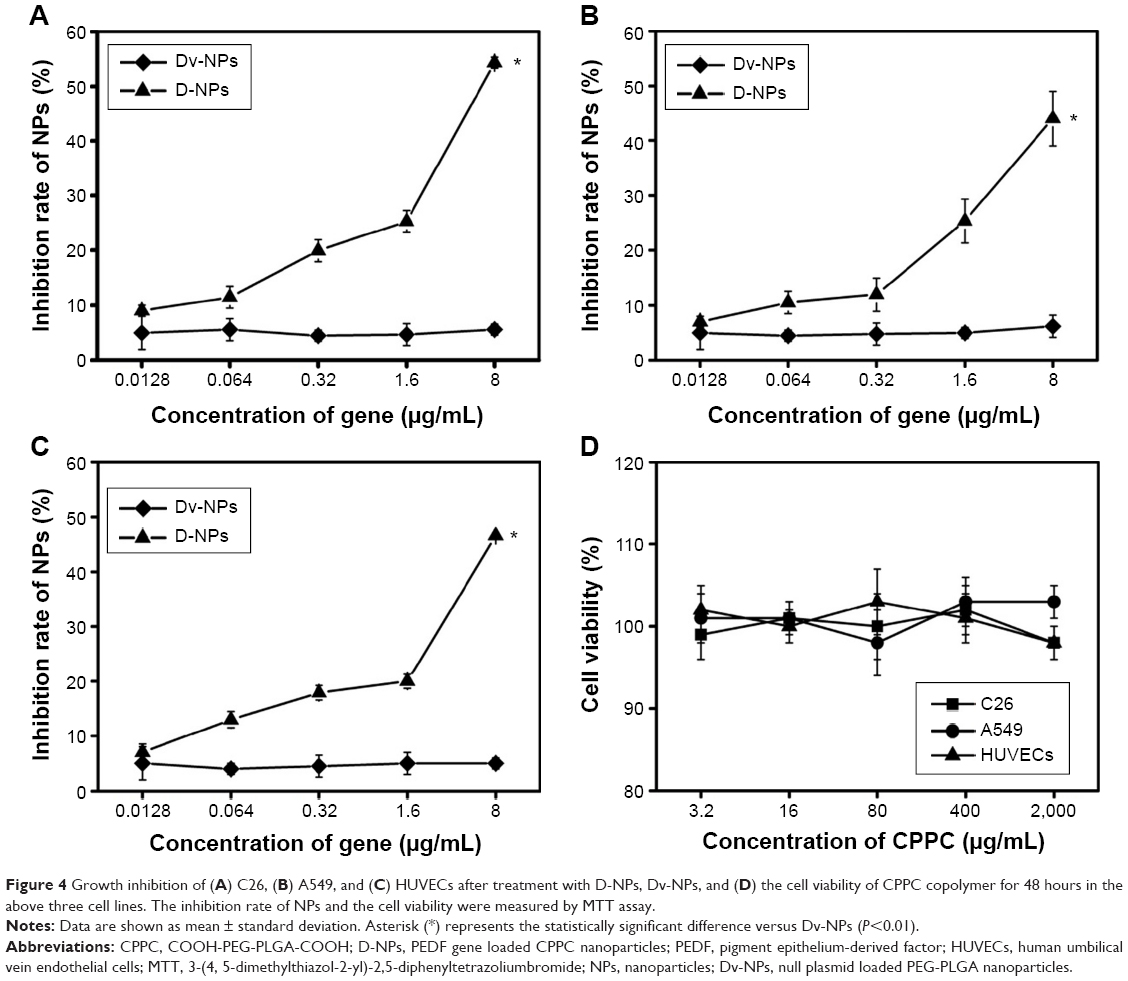 | Figure 4 Growth inhibition of (A) C26, (B) A549, and (C) HUVECs after treatment with D-NPs, Dv-NPs, and (D) the cell viability of CPPC copolymer for 48 hours in the above three cell lines. The inhibition rate of NPs and the cell viability were measured by MTT assay. |
The functional inhibition of HUVECs
As presented in Figure 5A, HUVECs formed capillary-like structure on the surface of Matrigel within 6 hours in control and Dv-NPs groups, while D-NPs dramatically disrupted the tube formation. The tube formation rates of Dv-NPs and D-NPs were 96.9% and 60.1%, respectively (Figure 5B).
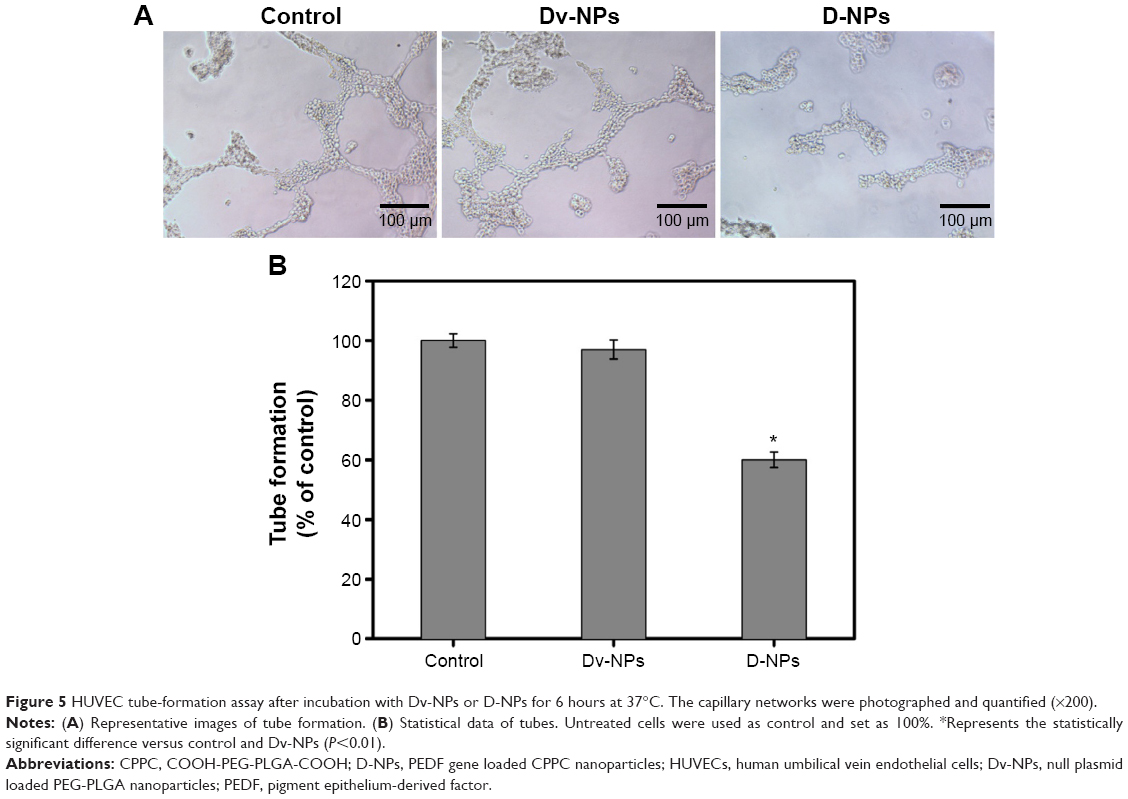 | Figure 5 HUVEC tube-formation assay after incubation with Dv-NPs or D-NPs for 6 hours at 37°C. The capillary networks were photographed and quantified (×200). |
In vitro expression of PEDF
The Western blot analysis showed that C26 cells treated with D-NPs expressed PEDF, but no PEDF was found in C26 cells treated by Dv-NPs or untreated cells. Meanwhile, A549 cells treated with D-NPs also had higher PEDF expression than the cells untreated or treated by Dv-NPs (Figure 6A). In addition, the ELISA analysis displayed that the D-NPs-treated C26 and A549 cells secreted PEDF in the culture media at a higher concentration (178 and 186 ng/mL, respectively) compared with the untreated or Dv-NPs-treated cells (P<0.01) (Figure 6B). These results confirmed that PEDF indeed expressed in C26 and A549 tumor cells and successfully secreted into the extracellular environment.
 | Figure 6 In vitro expression of PEDF in C26 and A549 cells treated by D-NPs and Dv-NPs for 48 hours, respectively. |
Alginate-encapsulated tumor cell assay
As presented in Figure 7A, new blood vessels in alginate beads from D-NPs-treated mice were remarkably sparse, as compared with the NS- and Dv-NPs-treated ones. Besides, the fluorescein isothiocyanate-dextran uptake in D-NPs group was only 1.14±0.16 μg/bead, which was much lower than that in NS (2.25±0.24 μg/bead) (P<0.01) and Dv-NPs (2.16±0.20 μg/bead) groups (P<0.01) (Figure 7B). These data demonstrated that CPPC-mediated PEDF gene transfer and expression was responsible for the inhibition of tumor angiogenesis.
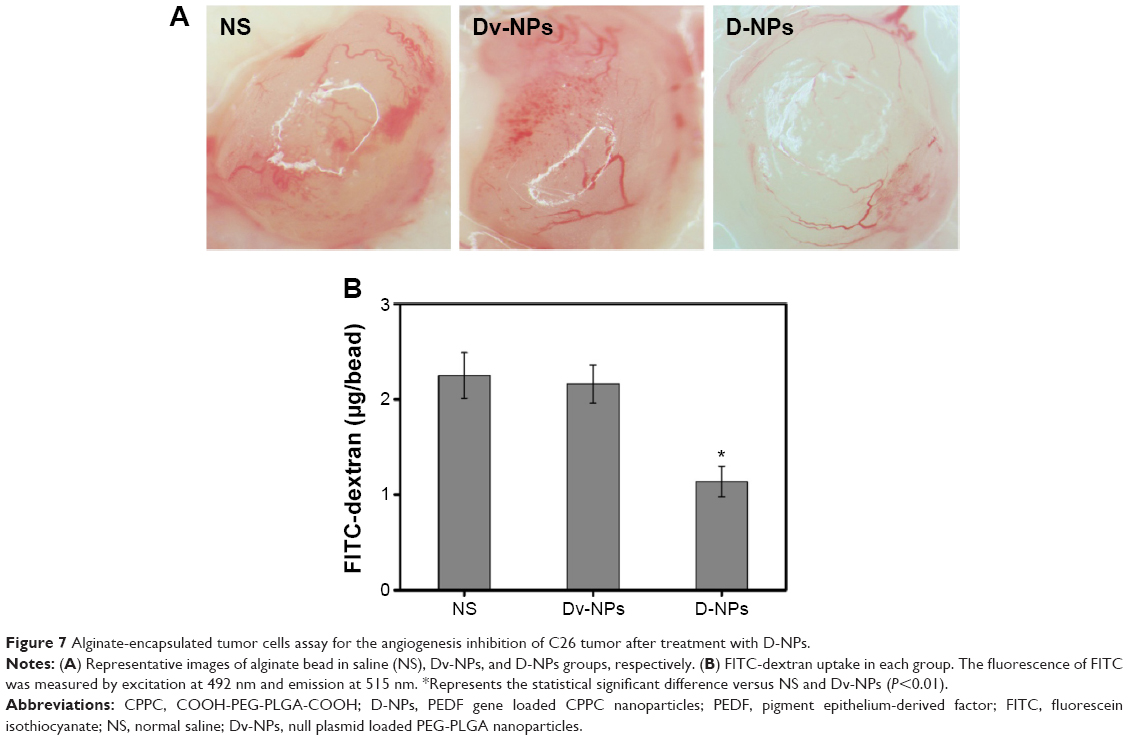 | Figure 7 Alginate-encapsulated tumor cells assay for the angiogenesis inhibition of C26 tumor after treatment with D-NPs. |
In vivo antitumor activity
The ability of D-NPs to inhibit the growth of the C26 colon carcinoma by intravenous administration was further evaluated in a xenograft mouse model. As shown in Figure 8A and B, D-NPs exhibited significantly suppressed efficacy on tumors (P<0.01, vs NS and Dv-NPs). The average weight of tumors in D-NPs group (1.49 g) was much lower compared with that in the NS (3.48 g) (P<0.01) and Dv-NPs (3.27 g) groups (P<0.01) (Figure 8C). In addition, a slight increase in the body weight of the mice was found during the treatment (Figure 8D). NS- and Dv-NPs-treated groups had higher body weight than D-NPs-treated group. D-NPs were efficient in prolonging survival time of C26 tumor-bearing mice (Figure 8E). The median survival of mice was 55 days in D-NPs group but only 42 days in Dv-NPs group.
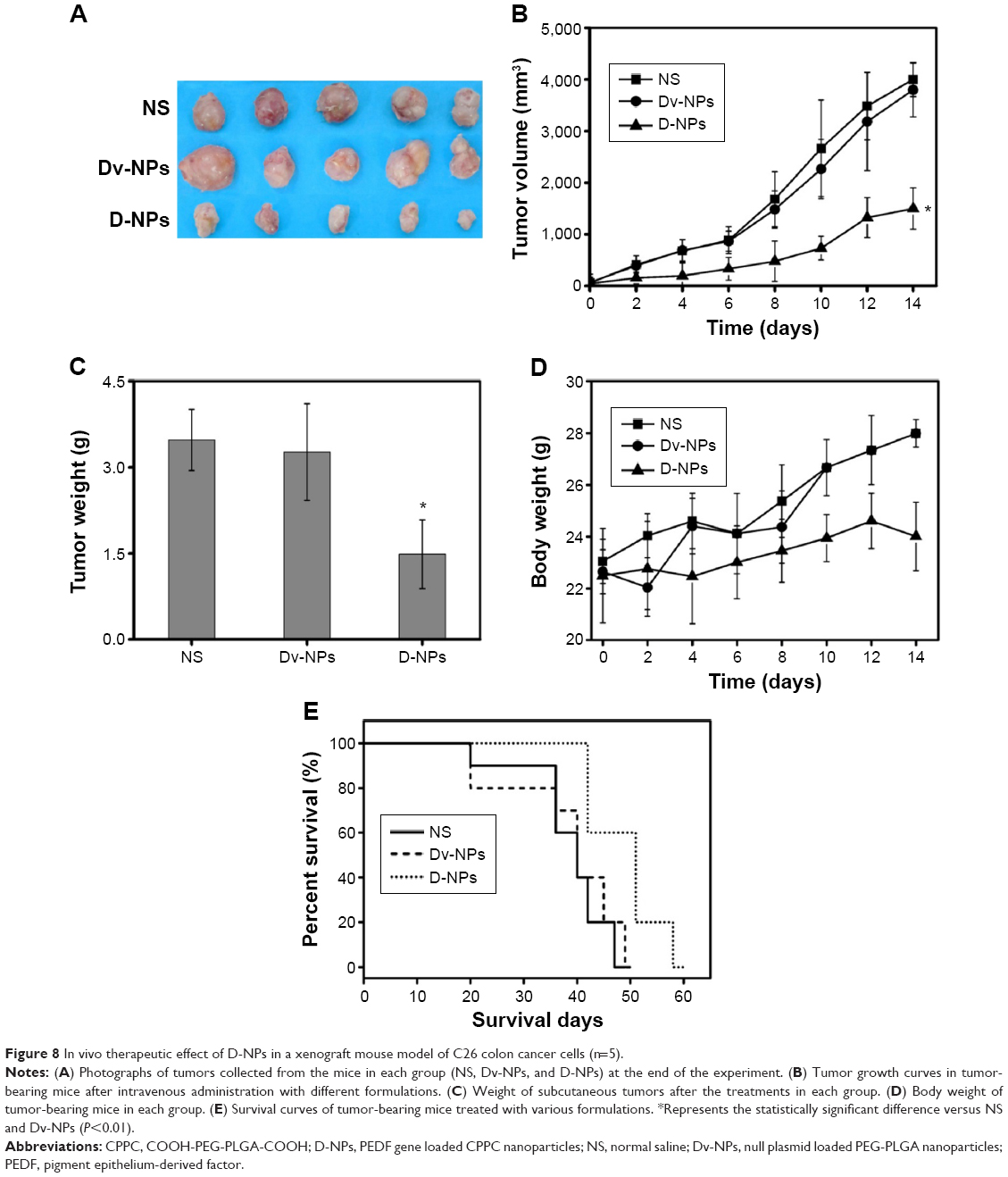 | Figure 8 In vivo therapeutic effect of D-NPs in a xenograft mouse model of C26 colon cancer cells (n=5). |
Effect of D-NPs on tumor angiogenesis and apoptosis
The potential mechanism underlying the efficacy of D-NPs-based therapy was investigated by CD31 staining and TUNEL assay. As seen in Figure 9A, significantly fewer immunoreactive microvessels were observed in tumor tissue after D-NPs treatment. D-NPs significantly decreased the MVD (14.57%±3.05%) compared with that of NS (49.5%±4.03%) (P<0.01) and Dv-NPs (47.6%±2.10%) (P<0.01) (Figure 9B).
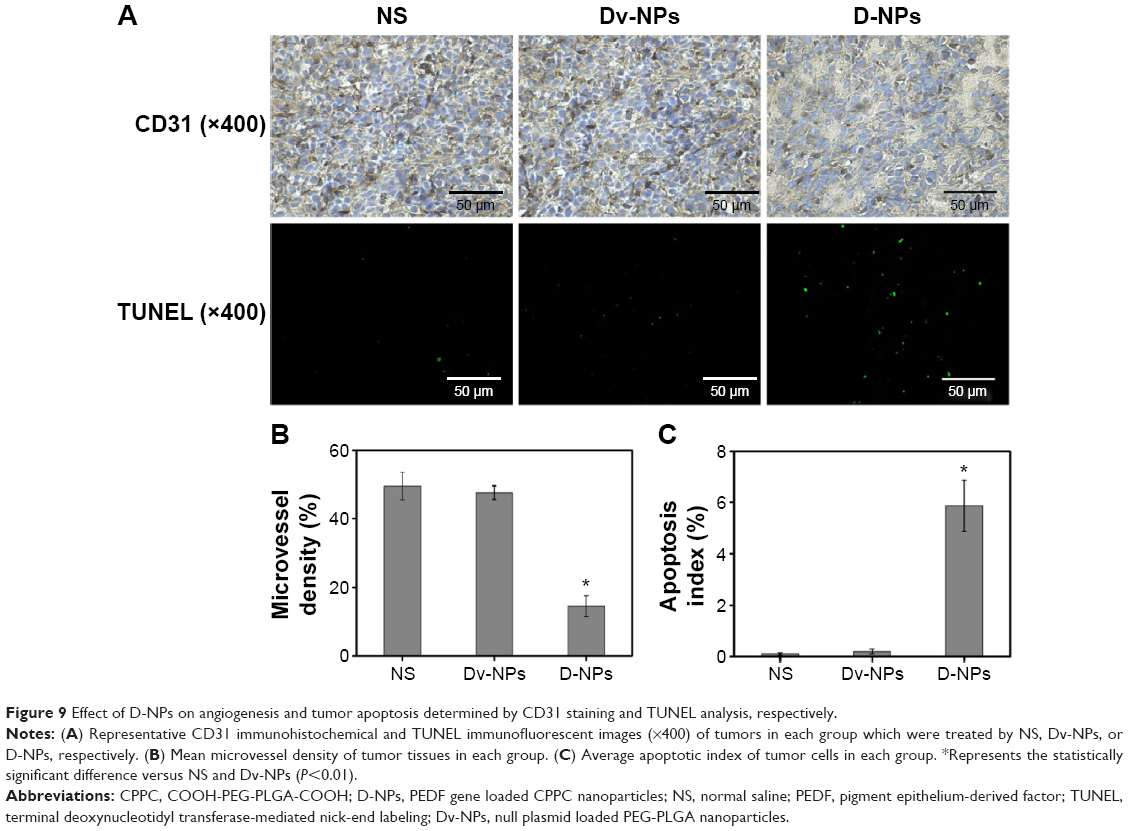 | Figure 9 Effect of D-NPs on angiogenesis and tumor apoptosis determined by CD31 staining and TUNEL analysis, respectively. |
The apoptosis of tumor cells was examined by immunofluorescent TUNEL assay in this study. As shown in Figure 9A, more apoptotic tumor cells were found in D-NPs group. Apoptotic index in D-NPs-treated group was 5.87%±2%, which was much higher than NS (0.1%±0.05%) (P<0.01) and Dv-NPs (0.2%±0.1%) (P<0.01) treated groups (Figure 9C).
Preliminary safety evaluation of D-NPs
The hemolysis analysis was performed to evaluate the blood compatibility of D-NPs. No visible hemolytic effect was observed in D-NPs-treated group as shown in Figure 10A. The percentage of the D-NPs-induced hemolysis was below 5% (Figure 10B).
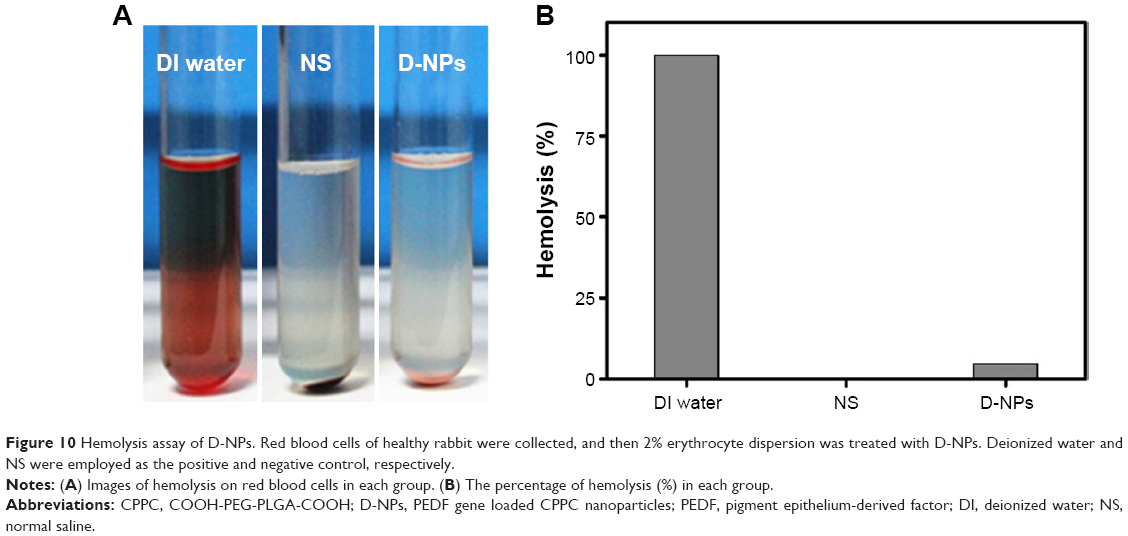 | Figure 10 Hemolysis assay of D-NPs. Red blood cells of healthy rabbit were collected, and then 2% erythrocyte dispersion was treated with D-NPs. Deionized water and NS were employed as the positive and negative control, respectively. |
The in vivo therapeutic experiment showed that a slight increase in the body weight of the mice was found during the treatment (Figure 8D) and no apparent toxicities such as depressed hair quality were observed.
The serological and biochemical analyses were further carried out to study the effects of D-NPs on mice physiology. As presented in Figure 11, D-NPs-treated mice had no obvious side effects compared with the group treated by NS, and meaningfully, several biochemical indicators of these mice including aspartate transaminas, creatine kinase, glucose, and total cholesterol approached the normal ranges.
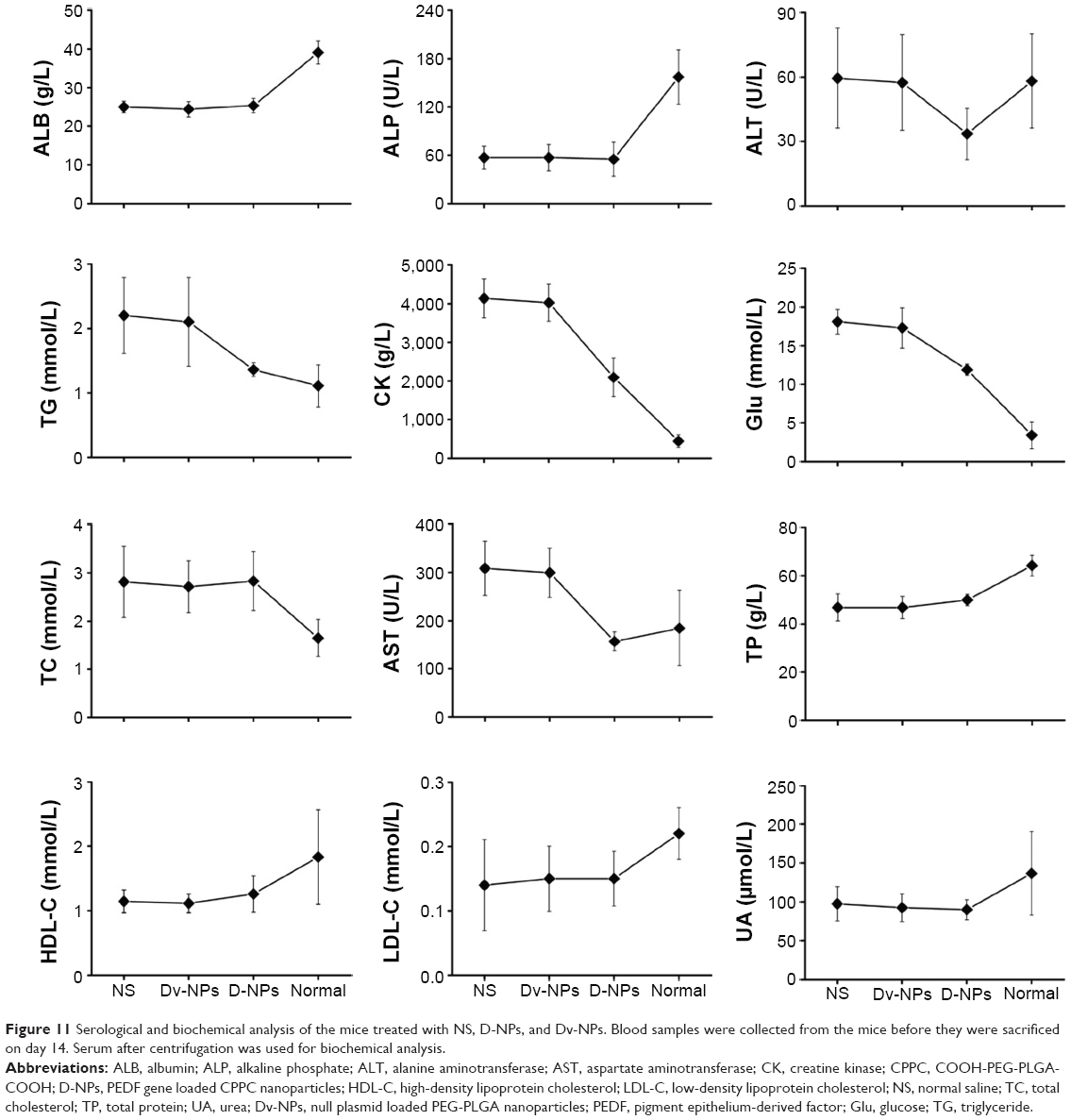 | Figure 11 Serological and biochemical analysis of the mice treated with NS, D-NPs, and Dv-NPs. Blood samples were collected from the mice before they were sacrificed on day 14. Serum after centrifugation was used for biochemical analysis. |
H&E staining was performed to investigate the pathological changes of mice organs. No significantly toxic pathological changes in the heart, liver, spleen, lung, and kidney were detected as shown in Figure 12.
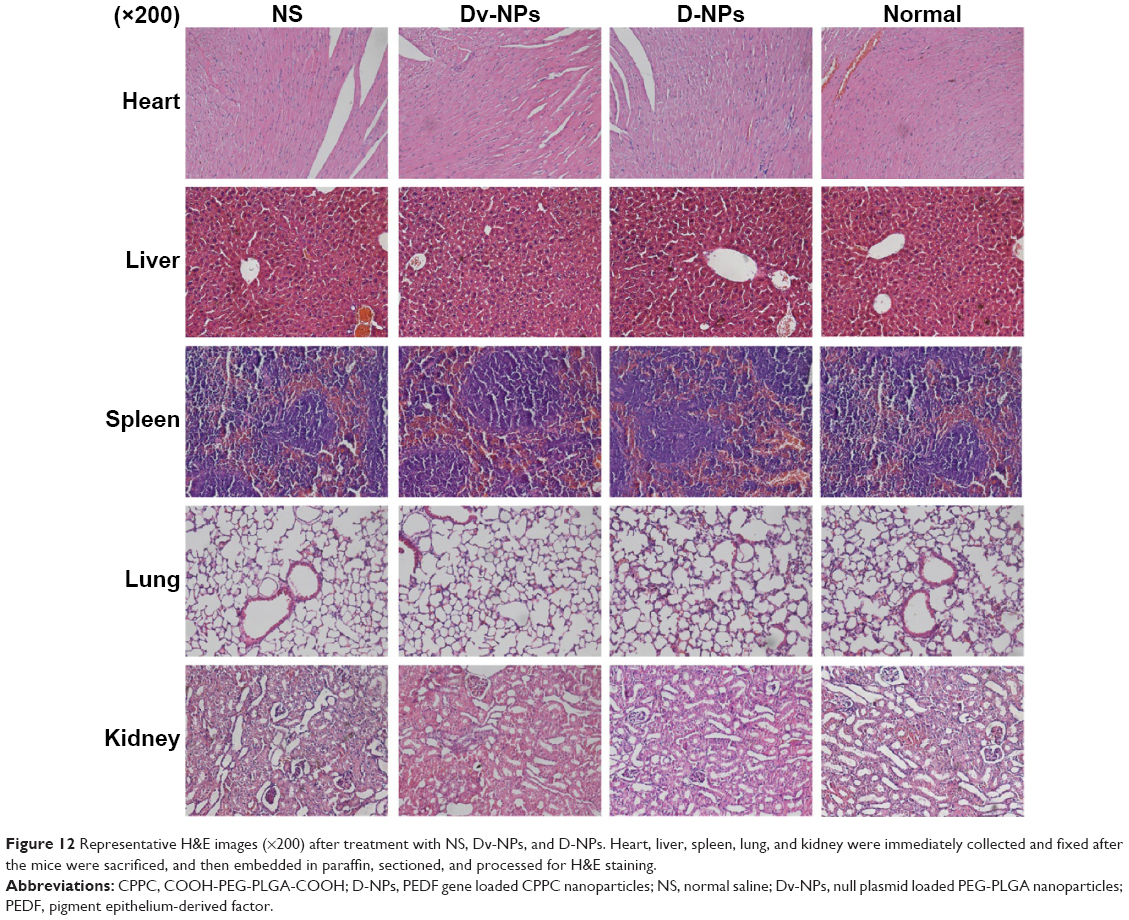 | Figure 12 Representative H&E images (×200) after treatment with NS, Dv-NPs, and D-NPs. Heart, liver, spleen, lung, and kidney were immediately collected and fixed after the mice were sacrificed, and then embedded in paraffin, sectioned, and processed for H&E staining. |
Discussion
PEDF gene is a promising treatment option for various tumors. To develop a safe and valid vector for the systemic delivery of PEDF gene, a novel copolymer CPPC, one of PLGA derivatives, was synthesized for the first time in this study. CPPC, confirmed by 1H-NMR spectrum, was successfully obtained through two simple chemical reaction steps. It might be easily synthesized in large amounts on an industrial scale and controlled in its quality using this simple and inexpensive method.
In the application of PLGA nanoparticles, one of the limitations was their low EE% and DL% of plasmids. For instance, EE% and DL% of p53 plasmid loaded PLGA microparticles were only 40.6% and 0.04%, respectively.31 In another report, DL% of pVITRO2 loaded PLGA nanoparticles was only 0.8%.32 Therefore, to increase the capacity of PEDF plasmid, CPPC, a negatively charged terminal carboxyl-modified PEG-PLGA polymer, was designed and successfully synthesized in our study. According to our previous experiments, the preliminary condensed PLL/PEDF gene complexes exhibited a positive zeta potential of +18.5 mV. Hence, due to the presence of terminal carboxyl groups on the PLGA-COOH polymer, the positive PLL/PEDF gene complexes could be encapsulated into the PLGA polymer core through electrostatic interactions easily and tightly, leading to a high entrapment efficiency (89.5%±2.6%) and drug loading (1.6%±0.3%) of PEDF gene. Besides, D-NPs exhibited a typical pH-dependent release behavior, which was a result of the diffusion of PEDF gene through PLGA matrix as well as the erosion of polymer.33 The controllable release profile of PEDF gene indicated the potential applicability of CPPC nanoparticles as a gene delivery system. Moreover, particles with nanoscale tend to aggregate because of their high surface/volume ratio, but a stable system can be prepared by stereospecific blockade and/or electrostatic interactions of the particles.5 In our work, the presence of the carboxy-modified PEG on the nanoparticle surface resulted in a negative surface charge (ζ=−23.04±1.03 mV), thus the stability of D-NPs might be attributed to both electrostatic repulsions and stereospecific blockade formed by the hydrophilic PEG chain. On the other hand, the terminal carboxyl groups of PEG could be used for functional modification such as the conjugation of targeting ligands, which would enhance the antitumor activities. Moreover, it has been reported that particles with diameter less than 200 nm could easily extravasate to an extravascular tumor site through the leaky blood vessels and accumulate in tumor tissues.34 Therefore, D-NPs which have the average diameter of 177 nm in our work might enhance drug accumulation in tumor sites and increase the antitumor activity, indicating that D-NPs might have applications as a potential formulation for cancer therapy. As a result, the prepared D-NPs showed excellent properties in terms of particle size, EE, DL, stability, and drug release behavior, indicating that CPPC might be a promising gene delivery carrier.
Several investigations concerning the antitumor activity of D-NPs were made in our study. D-NPs showed an increased cytotoxicity on C26 and A549 cells when compared with Dv-NPs and control groups, contributing to the successful PEDF expression which was confirmed by ELISA and Western blot, respectively. It could be inferred that D-NPs could be internalized into C26 and A549 tumor cells and PEDF gene loaded in CPPC nanoparticles could be transferred successfully. In addition, PEDF gene entrapped in PLGA or CPPC nanoparticles has shown its inhibitory effect on the growth of C26 cells/tumors in our previous6 and present studies, whereas it has not been used for the therapy of A549 tumors to the best of our knowledge. It has been reported that PEDF apparently induced apoptosis in A549 cells predominantly via the Fas-L/Fas death signaling pathway.35 Thus, our findings of the PEDF gene/CPPC delivery system might provide a potential gene therapeutic strategy for human lung cancers. Otherwise, D-NPs efficiently inhibited the growth of tumors and prolonged the survival of tumor-bearing mice in a subcutaneous C26 mouse model, which suggested that CPPC nanoparticles could transfer PEDF gene to the C26 cells effectively through the enhanced permeability and retention effect and PEDF gene was successfully expressed.
Tumor neovascularization is a highly complicated process with multiple steps, which plays a vital role in tumor growth and metastasis. Anti-angiogenesis has been a promising therapeutic strategy for clinical therapy of tumors.5 PEDF is a potent inhibitor of angiogenesis and numerous studies have shown its anti-angiogenic effects on tumors. In this study, D-NPs displayed a significant in vitro antiproliferative effect on HUVECs. Besides, results of CD31 staining of tumor tissues and alginate-encapsulated tumor cell assay showed the improved anti-angiogenesis effect of D-NPs. These results indicated that PEDF protein was successfully produced by PEDF gene and was highly bioactive. Moreover, the tube formation of endothelial cells is one of the key steps of angiogenesis.8 It is well established that tube formation of endothelial cells on Matrigel recapitulates some angiogenesis steps, such as cell migration, alignment, formation of tubes, and tube branching and anastomosing with adjacent tubes.36 The capillary morphogenesis of HUVECs on Matrigel was found to be strongly disrupted after treated by D-NPs. Taken together, all the results revealed that D-NPs could block angiogenesis in vitro and in vivo by inhibiting the proliferation and tubular formation of endothelial cells.
PEDF is a multifunctional protein with therapeutic effect on tumors and has the ability to induce tumor cell apoptosis directly.6 TUNEL assay showed a pronounced apoptosis-inducing activity of D-NPs in this work. The reason might be that the PEDF which successfully expressed in the tumor tissue increased the levels of p53 and BAX and concomitantly inhibited BCL-2.7 This observation indicated that the growth inhibitory effect of C26 tumors was also associated with the apoptosis of C26 cells in vivo. Considering these results, D-NPs could successfully delivery PEDF gene into the tumor cells and would be a promising approach for antitumor therapy.
Finally, several experiments in vitro and in vivo were explored to investigate the systemic toxicity or potential side effects of the treatment. Hemolysis analysis, a simple and widely used method to study surfactant-membrane interaction,37 was first performed and showed that D-NPs had good blood compatibility and could be used for intravenous administration. In the in vivo antitumor experiment, no obvious toxicities were observed in the tumor-bearing mice as determined by appearance, body weight, as well as fecal and urinary excretions. Moreover, serological and biochemical analyses were carried out and showed that D-NPs had no side effects on the physiology of the tumor-bearing mice. H&E staining also indicated that no significantly toxic pathological changes in the heart, liver, spleen, lung, and kidney were detected. To draw a definitive conclusion, D-NPs were evaluated as safe formulations for intravenous administration.
Conclusion
The new designed copolymer CPPC was synthesized for the first time. D-NPs were successfully prepared and their anticancer effects in vitro and in vivo were systemically investigated. This novel formulation encapsulating PEDF gene, with high EE% and DL%, good stability, and pH-dependent release behavior, showed great cytotoxic and antiproliferative effect on C26, A549, and HUVEC cells in vitro. Moreover, D-NPs were effective in suppressing tumor growth and prolonging survival in a C26 subcutaneous tumor model. The inhibitory effects of D-NPs on tumors might be associated with the induction of apoptosis, the decrease of MVD, and the inhibition of angiogenesis. More importantly, no obvious toxicities of D-NPs were observed, indicating that D-NPs were safe for systemic administration. Altogether, D-NPs offered an innovative strategy for delivery of PEDF gene by intravenous injection and this delivery system might be a potential therapeutic approach for various cancers.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (No 81302729), the National High Technology Research and Development Program (No 2012AA020803), and the Fundamental Research Funds for the Central Universities (No 2013SCU04A19).
Disclosure
The authors report no conflicts of interest in this work.
References
Keeley EC, Mehrad B, Strieter RM. CXC chemokines in cancer angiogenesis and metastases. Adv Cancer Res. 2010;106:91–111. | ||
Samant RS, Shevde LA. Recent advances in anti-angiogenic therapy of cancer. Oncotarget. 2011;2(3):122–134. | ||
Mahtabifard A, Merritt RE, Yamada RE, Crystal RG, Korst RJ. In vivo gene transfer of pigment epithelium-derived factor inhibits tumor growth in syngeneic murine models of thoracic malignancies. J Thorac Cardiovasc Surg. 2003;126 (1):28–38. | ||
Ding Q, Niu T, Yang Y, Guo Q, Luo F, Qian Z. Preparation of curcumin-loaded poly (ester amine) nanoparticles for the treatment of anti-angiogenesis. J Biomed Nanotechnol. 2014;10(4):632–641. | ||
Gong C, Deng S, Wu Q, et al. Improving anti-angiogenesis and anti-tumor activity of curcumin by biodegradable polymeric micelles. Biomaterials. 2013;34(4):1413–1432. | ||
Cui FY, Song XR, Li ZY, et al. The pigment epithelial-derived factor gene loaded in PLGA nanoparticles for therapy of colon carcinoma. Oncol Rep. 2010;24(3):661–668. | ||
Becerra SP, Notario V. The effects of PEDF on cancer biology: mechanisms of action and therapeutic potential. Nat Rev Cancer. 2013;13(4):258–271. | ||
Wu Q, He S, Wei X, et al. Synergistic antitumor effect of recombinant adeno-associated virus-mediated pigment epithelium-derived factor with hyperthermia on solid tumor. Hum Gene Ther. 2014;25(9):811–823. | ||
He SS, Wu QJ, Gong CY, et al. Enhanced efficacy of combination therapy with adeno-associated virus-delivered pigment epithelium-derived factor and cisplatin in a mouse model of Lewis lung carcinoma. Mol Med Rep. 2014;9(6):2069–2076. | ||
Nowroozi MR, Pisters LL. The current status of gene therapy for prostate cancer. Cancer Control. 1998;5(6):522–531. | ||
Palmer DH, Chen MJ, Kerr DJ. Gene therapy for colorectal cancer. Br Med Bull. 2002;64:201–225. | ||
Itaka K, Kataoka K. Recent development of nonviral gene delivery systems with virus-like structures and mechanisms. Eur J Pharm Biopharm. 2009;71(3):475–483. | ||
Wong SP, Argyros O, Harbottle RP. Vector systems for prenatal gene therapy: principles of non-viral vector design and production. Methods Mol Biol. 2012:891:133–167. | ||
Li SD, Huang L. Non-viral is superior to viral gene delivery. J Control Release. 2007;123(3):181–183. | ||
Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly (D, L-lactide-co-glycolide) and its derivatives. J Control Release. 2008;125(3):193–209. | ||
Liang GF, Zhu YL, Sun B, et al. PLGA-based gene delivering nanoparticle enhance suppression effect of miRNA in HepG2 cells. Nanoscale Res Lett. 2011;6(1):447–455. | ||
Devulapally R, Sekar NM, Sekar TV, et al. Polymer nanoparticles mediated codelivery of antimiR-10b and antimiR-21 for achieving triple negative breast cancer therapy. ACS Nano. 2015;9(3):2290–2302. | ||
Qi X, Song X, Liu P, et al. Antitumor effects of PLGA nanoparticles encapsulating the human PNAS-4 gene combined with cisplatin in ovarian cancer. Oncol Rep. 2011;26(3):703–710. | ||
Stern M, Ulrich K, Geddes DM, Alton EW. Poly (D, L-lactide-co-glycolide)/DNA microspheres to facilitate prolonged transgene expression in airway epithelium in vitro, ex vivo and in vivo. Gene Ther. 2003;10(16):1282–1288. | ||
Zou W, Liu C, Chen Z, Zhang N. Studies on bioadhesive PLGA nanoparticles: a promising gene delivery system for efficient gene therapy to lung cancer. Int J Pharm. 2009;370(1–2):187–195. | ||
Huang HS, Hainfeld JF. Intravenous magnetic nanoparticle cancer hyperthermia. Int J Nanomedicine. 2013;8:2521–2532. | ||
Lammers T, Peschke P, Kühnlein R, et al. Effect of intratumoral injection on the biodistribution, the therapeutic potential of HPMA copolymer-based drug delivery systems. Neoplasia. 2006;8(10):788–795. | ||
Sah H, Thoma LA, Desu HR, Sah E, Wood GC. Concepts and practices used to develop functional PLGA-based nanoparticulate systems. Int J Nanomedicine. 2013;8:747–765. | ||
Ma Q, Li B, Yu Y, et al. Development of a novel biocompatible poly (ethylene glycol)-block-poly (γ-cholesterol-l-glutamate) as hydrophobic drug carrier. Int J Pharm. 2013;445(1–2):88–92. | ||
Devulapally R, Sekar TV, Paulmurugan R. Formulation of anti-miR-21 and 4-hydroxytamoxyfen co-loaded biodegradable polymer nanoparticles and its anti-proliferative effect on breast cancer cells. Mol Pharm. 2015;12(6):2080–2092. | ||
Farokhzad OC, Cheng J, Teply BA, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103(16):6315–6320. | ||
Jaffe EA, Nachman RL, Becker CG, et al. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52(11):2745–2756. | ||
He Z, Yu Y, Zhang Y, et al. Gene delivery with active targeting to ovarian cancer cells mediated by folate receptor α. J Biomed Nanotechnol. 2013;9(5):833–844. | ||
Hoffmann J, Schirner M, Menrad A, Schneider MR. A highly sensitive model for quantification of in vivo tumor angiogenesis induced by alginate-encapsulated tumor cells. Cancer Res. 1997;57(17):3847–3851. | ||
He ZY, Wei XW, Luo M, et al. Folate-linked lipoplexes for short hairpin RNA targeting Claudin-3 delivery in ovarian cancer xenografts. J Control Release. 2013;172(3):679–689. | ||
Shi X, Li C, Gao S, et al. Combination of doxorubicin-based chemotherapy and polyethylenimine/p53 gene therapy for the treatment of lung cancer using porous PLGA microparticles. Colloids Surf B Biointerfaces. 2014;122:498–504. | ||
Yang C, Hu T, Cao H, et al. Facile construction of chloroquine containing PLGA-based pDNA delivery system for efficient tumor and pancreatitis targeting in vitro and in vivo. Mol Pharm. 2015;12(6):2167–2179. | ||
Tahara K, Sakai T, Yamamoto H, Takeuchi H, Kawashima Y. Establishing chitosan coated PLGA nanosphere platform loaded with wide variety of nucleic acid by complexation with cationic compound for gene delivery. Int J Pharm. 2008;354(1):210–216. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Li L, Yao YC, Fang SH, et al. Pigment epithelial-derived factor (PEDF)-triggered lung cancer cell apoptosis relies on p53 protein-driven Fas ligand (Fas-L) up-regulation and Fas protein cell surface translocation. J Biol Chem. 2014;289(44):30785–30799. | ||
Arnaoutova I, George J, Kleinman HK, Benton G. The endothelial cell tube formation assay on basement membrane turns 20: state of the science and the art. Angiogenesis. 2009;12(3):267–274. | ||
Liu J, Jiang Y, Cui Y, Xu C, Ji X, Luan Y. Cytarabine-AOT catanionic vesicle-loaded biodegradable thermosensitive hydrogel as an efficient cytarabine delivery system. Int J Pharm. 2014;473(1):560–571. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.