Back to Journals » OncoTargets and Therapy » Volume 10
PI3K/AKT/mTOR pathway promotes progestin resistance in endometrial cancer cells by inhibition of autophagy
Authors Liu H, Zhang L, Zhang X, Cui Z
Received 27 August 2015
Accepted for publication 5 February 2016
Published 6 June 2017 Volume 2017:10 Pages 2865—2871
DOI https://doi.org/10.2147/OTT.S95267
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Jianmin Xu
Hua Liu,1,2 Liqin Zhang,2 Xuyan Zhang,2 Zhumei Cui1
1Department of Gynecology, Affiliated Hospital of Qingdao Medical College, Qingdao University, Qingdao, 2Department of Gynecology, Affiliated Hospital of Weifang Medical University, Weifang, Shandong, People’s Republic of China
Abstract: Endometrial cancer (EC) is now one of the most common malignant tumors in young women. In all, 90% of young patients with EC have a high expression of progesterone receptor, can be treated with progestin, and have very good prognosis. However, some of the young EC patients are resistant to progestin, the mechanism of which is unclear. To illuminate the mechanism by which endometrial cells acquire progestin resistance, we treated Ishikawa cells by slowly increasing dosage of progestin and established a progestin-resistant cell subline. We show here that progesterone resistant cells acquire increased proliferation rate and interestingly decreased autophagy. To uncover the mechanism by which cells increase proliferation and bypass autophagy, we found higher activation of phosphatidylinositol 3-kinase/AKT/mTOR signaling pathway was necessary to this malignant acquirement by RNAi technique. Further, we elucidated that activation of mTOR was sufficient and necessary for progestin resistance. RAD001, an inhibitor of mTOR, decreased phosphorylation of mTOR and inhibited proliferation of progestin-resistant cancer cells by promoting autophagy. Thus, our results indicated that mTOR can be a target to treat the progestin-resistant EC.
Keywords: progesterone receptor, RAD001, proliferation, Ishikawa, phosphorylation
Introduction
Progestins, such as medroxyprogesterone acetate (MPA), were commonly used in young early endometrial adenocarcinoma patients as conservative endocrine treatment.1–3 But according to clinical data, >30% of young endometrial cancer (EC) patients did not respond to progestin because of de novo or acquired progestin resistance during progestin treatment, despite the different drugs or regimens used.4–7 The mechanism by which cancer cells acquire resistance is still unclear.
Phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR pathway is central to the control of cell transcription, translation, migration, metabolism, proliferation, and survival.8,9 Aberrant hyperactivation of PI3K/AKT/mTOR pathway is one of the most common tumor-related signaling pathways that can be detected in a variety of tumor types, such as breast cancer, colorectal cancer, endometrial carcinoma, lung cancer, and glioblastoma, mainly including PIK3CA mutation, PTEN loss, and AKT1 mutation. In addition, KRAS mutation and BRAF mutation from the MAPK pathway can also result in PI3K/AKT/mTOR pathway activation.10–15 A previous study showed that progestin might activate PI3K/AKT signaling pathway independent of progesterone receptor (PR) in breast cancer.16
Autophagy is a catabolic degradation process in which cellular proteins and organelles are engulfed by double-membrane autophagosomes and degraded in lysosomes. Autophagy has emerged as a critical pathway in tumor development and cancer therapy.17 Previous studies showed that several cytokines and sex steroids can regulate autophagy in different cancers.18–20
Here we show that activation of the PI3K/AKT pathway would result in activation of mTOR and promote cell proliferation by inhibiting autophagy. Inhibition of PI3K/AKT/mTOR pathway could reverse the progestin resistance in EC.
Materials and methods
Plasmid construction and transient transfection
The complete mTOR coding sequence was amplified by polymerase chain reaction (PCR) using primers 5′-GAT CGC TAG CAT GCT TGG AAC CGG ACC-3′ (forward) and 5′-GAT CGC GGC CGC TTA CCA GAA AGG GCA CCA GCC-3′ (reverse). The PCR product was purified and cloned into pcDNA3.1 vector using NheI/NotI. The selected clone was fully sequenced in order to verify that no mutations were introduced by PCR. The mTOR RNAi sequence is 5′-AAG CCA TCC AGA TTG ATA CCT-3′. The AKT1 RNAi sequence is 5′-AAG CAC CGC GTG ACC ATG AAC-3′. Scramble sequence is 5′-CAA GAT GAA GAG CAC CAA A-3′. For transfection, the day before transfection, cells were seeded in six-well plates and grown overnight until the cells had reached 90%–95% confluence. Transient transfections were performed using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol.
Cell culture
Human endometrial carcinoma cell line, Ishikawa cells (European Collection of Cell Cultures; Porton Down, Salisbury, Wiltshire, UK), were grown in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific). Cells were maintained at 37°C under an atmosphere of 5% CO2. All research experiments were approved by the Department of Gynecology, Affiliated Hospital of Qingdao Medical College, Qingdao University Ethics Committee.
Establishment of progestin-resistant EC cell lines
Induction of progestin-resistant EC cell line was described previously.21 Briefly, Ishikawa cells were maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum, and MPA (Sigma-Aldrich Co., St Louis, MO, USA) was added for 6 months. The concentration of MPA was increased twofold every month until it reached 10 μM. The medium containing MPA was changed every 3 days. Cells were passaged when the surviving cells grew to 90% confluent. This method produced a subline refractory to the growth-suppressive effects of MPA, which was considered to consist of progestin-resistant Ishikawa cells, and was thereafter maintained in 10 μM MPA.
Western blot
Western blot analysis was performed as previously described.22 Cells were lysed in hot 1× sodium dodecyl sulfate sample buffer, and 30 μg samples were loaded into each lane of 4%–12% polyacrylamide gels. The proteins were separated by electrophoresis, and the proteins in the gels were transferred onto nitrocellulose membranes (Pall; Port Washington, NY, USA). The membrane was blocked with 5% nonfat milk for 1 hour at room temperature and then incubated with antibodies against PR (3176; Cell Signaling Technology, Danvers, MA, USA), pAKT (Ser473; 4058; Cell Signaling Technology), AKT (4685; Cell Signaling Technology), phosphor-mTOR (Ser2448; 5536; Cell Signaling Technology), mTOR (2972; Cell Signaling Technology), LC3B (2775; Cell Signaling Technology), ATG3 (3415; Cell Signaling Technology), ATG5 (8540; Cell Signaling Technology), KRAS (ab137739; Abcam, Cambridge, UK); PTEN (ab170941; Abcam), EIG121 (ab156275; Abcam), P62 (ab91526; Abcam), Beclin-1 (ab114071; Abcam), and β-actin (sc-47778; Santa Cruz Biotechnology Inc., Dallas, TX, USA) for 16 hours at 4°C. Then, the specific horseradish peroxidase-conjugated rabbit anti-mouse or rabbit anti-mouse IgG was added to the membrane and incubated for 1 hour at room temperature. Detection by the chemiluminescence reaction was carried out using the enhanced chemiluminescence (ECL) kit (Pierce; Rockford, IL, USA).
Cell proliferation assay
Cells were seeded in 24-well plates at low density (2×104 cells) and allowed to attach overnight. Then cells were cultured for indicated times. Twenty microliters of 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT; 5 mg/mL; Sigma-Aldrich Co.) was added into each well at 0 hours, 24 hours, 48 hours, 72 hours, and 96 hours, and the cells were incubated for a further 4 hours. The absorbance was recorded at 570 nm with a 96-well plate reader after the addition of dimethyl sulfoxide.
Statistical analysis
Data were analyzed by using SPSS statistical package (Version 16; SPSS Inc., Chicago, IL, USA). Independent two group’s analyses used Student’s t-test. P<0.05 was considered statistically significant.
Results
Progestin-resistant cells show increased cell growth and decreased autophagy
The progestin-resistant cell (PRC) subline was established by long-term MPA treatment.12 We then characterized the PRCs by examining cell growth and cell viability. The MTT results showed that MPA significantly inhibited the proliferation of parental but not PRCs (Figure 1A). Previous studies21 showed lower expression of PR in PRCs. We also found lower expression of PR in our system (Figure 1B). We showed that MPA promoted autophagy of parental cells, but not in PRCs, by Western blot for autophagy markers (Figure 1C). This result indicates that continuous progestin treatment could lead to PR downregulation and activation of other pathways to promote cell growth and inhibit cell autophagy in EC cells.
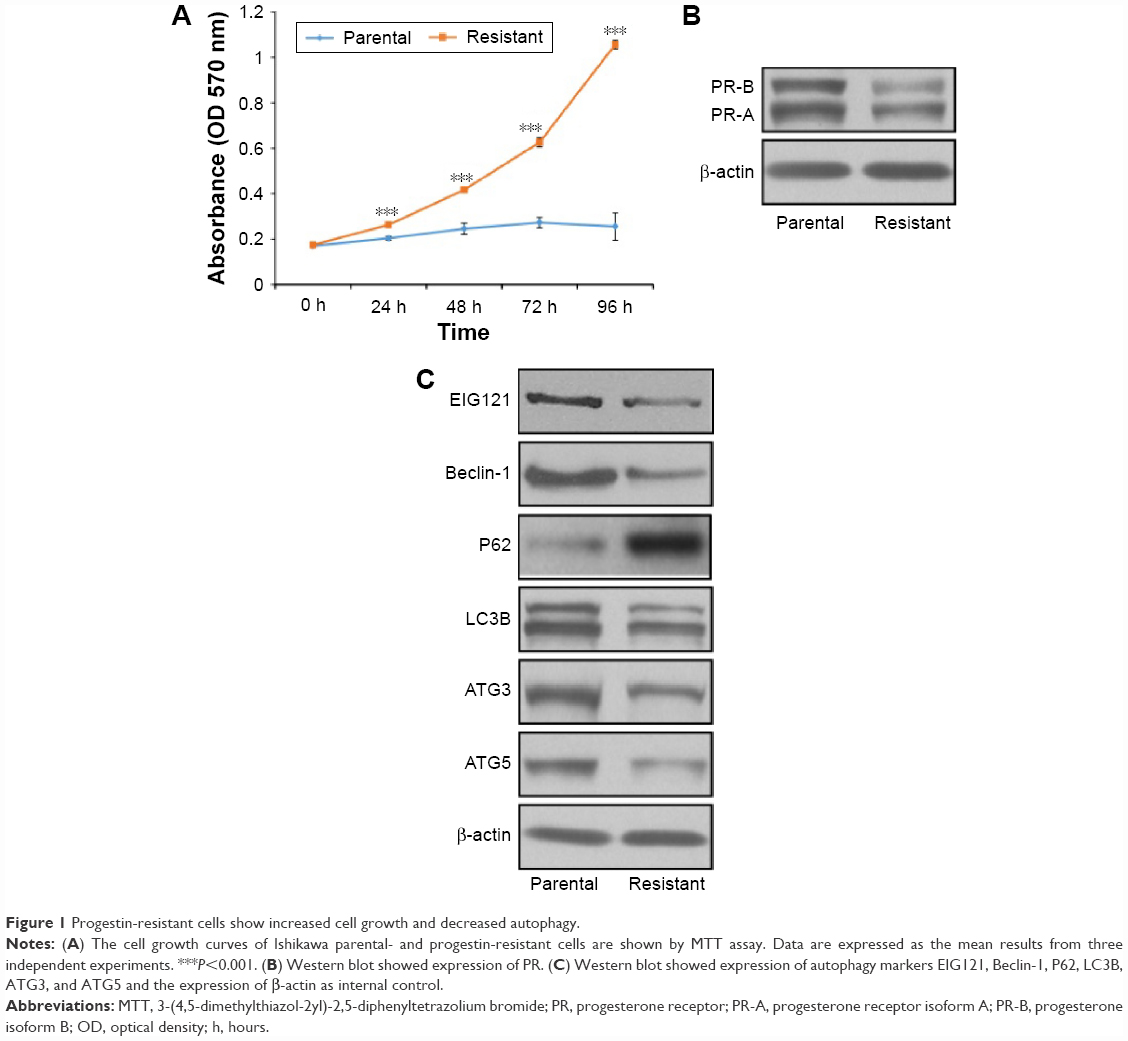 | Figure 1 Progestin-resistant cells show increased cell growth and decreased autophagy. |
Progestin activates the PI3K/AKT/mTOR pathway in progestin-resistant EC cells
Abnormal activation of PI3K/AKT pathway is one of the most common tumor-related signaling abnormalities that regulated cell proliferation and viability in a variety of tumors.12–14 To illustrate if PI3K/AKT pathway was activated by progestin, we examined the phosphorylation of AKT1 on Ser473 in response to MPA treatment in progestin-resistant EC cells. MPA treatment resulted in reduced phosphorylation of AKT1 in parental Ishikawa cells, but interestingly, the phosphorylation of AKT1 increased in the PRCs (Figure 2A). We also showed that the expression of PTEN is decreased, whereas the expression of KRAS is increased in the PRCs, which is consistent with a previous study.23
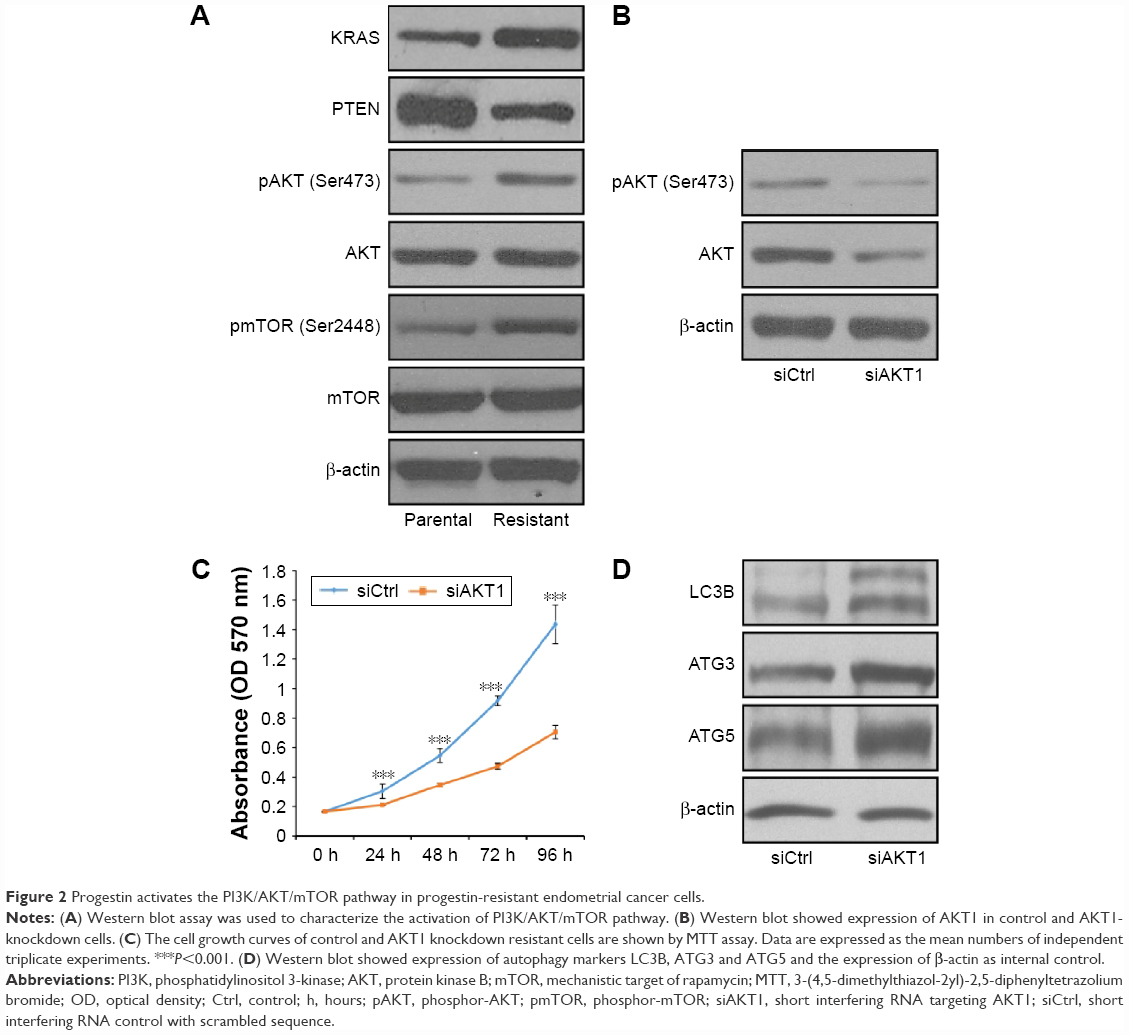 | Figure 2 Progestin activates the PI3K/AKT/mTOR pathway in progestin-resistant endometrial cancer cells. |
It is well known that the PI3K/AKT/mTOR pathway plays a critical role in cell growth and viability; hence, we examined the activation status of mTOR. The result showed that the phosphorylation of mTOR on Ser 2448 is increased in PRCs compared with progestin-sensitive cells (Figure 2A).
PI3K/AKT/mTOR signaling pathway promotes cell growth in EC cells by diminishing autophagy
To address whether PI3K/AKT activation is necessary for the progestin resistance, we reduced AKT1 expression in PRCs using siRNA. The siRNA successfully decreased AKT1 expression and phosphorylation of AKT1 in the cells (Figure 2B). We showed decreased cell growth (Figure 2C) and increased autophagy in AKT1-knockdown cells (Figure 2D).
To address whether mTOR is involved in the progestin resistance, we then decreased mTOR expression in PRCs using siRNA (Figure 3A). The expression of PR showed no significance after knockdown of mTOR in PRCs (Figure 3A), which showed that PR might not be involved. Upon elimination of mTOR, cell growth decreased (Figure 3B) and expression of autophagy markers increased dramatically (Figure 3C). Further, we wondered whether mTOR activity is sufficient to promote progestin resistance in Ishikawa cells; hence, we rescued the expression of mTOR with mutation at the siRNA-recognized site in PRCs. We then found restored phosphorylation of mTOR (Figure 3D). MTT assay showed restored proliferation of PRCs (Figure 3E); Western blot against autophagy markers showed decreased expression of LC3B, ATG3, and ATG5 (Figure 3F).
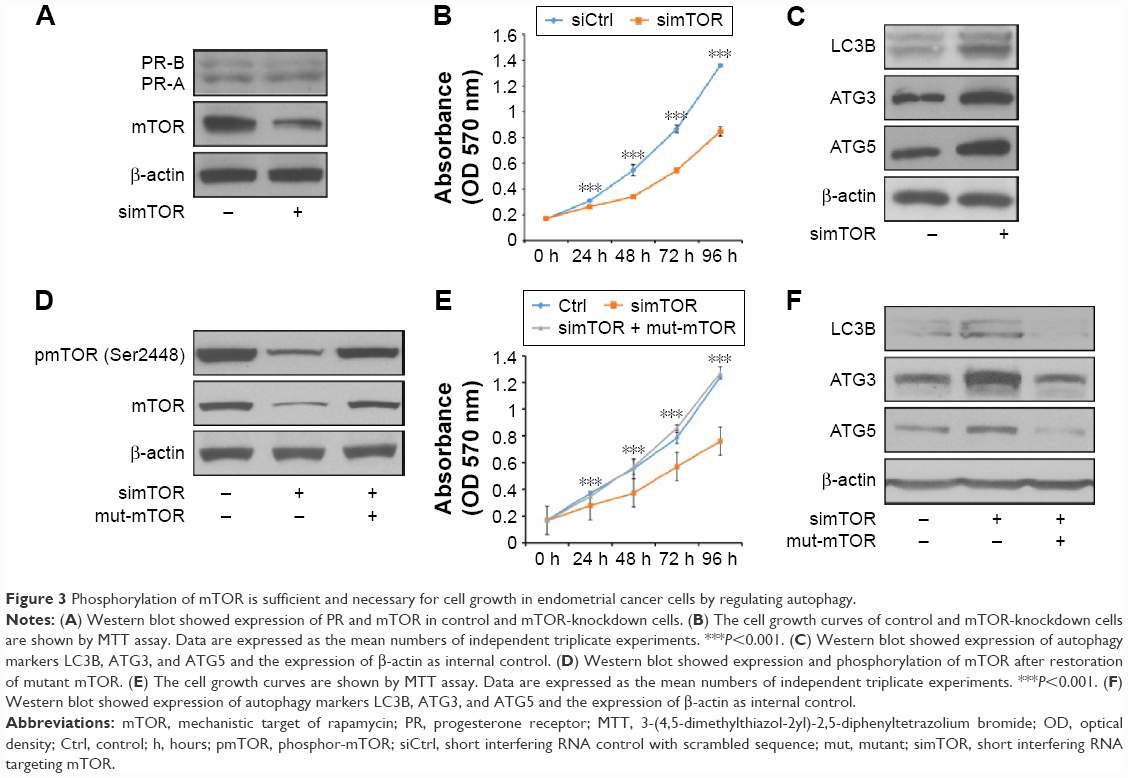 | Figure 3 Phosphorylation of mTOR is sufficient and necessary for cell growth in endometrial cancer cells by regulating autophagy. |
These results confirmed that progestin activated PI3K/AKT/mTOR pathway, and PI3K/AKT/mTOR pathway is both sufficient and necessary for the progestin resistance of EC cells.
RAD001 inhibits proliferation in progestin-resistant EC cells by inducing autophagy
RAD001 (Everolimus) is an inhibitor of mTOR and was approved for the treatment of advanced renal cell carcinoma, advanced breast cancer, and pancreatic neuroendocrine tumors.24,25 To assess whether RAD001 can inhibit cell growth and promote autophagy in EC cells, we treated progestin-resistant Ishikawa cells with RAD001. The phosphorylation of mTOR by Western blot analysis is shown in Figure 4A. RAD001 significantly inhibited phosphorylation of mTOR, and the expression of LC3B, ATG3, and ATG5 was all increased after RAD001 treatment (Figure 4A). MTT results showed decreased cell growth after RAD001 treatment (Figure 4B). These results demonstrate that RAD001 treatment could inhibit proliferation of progestin-resistant EC cells by inducing autophagy.
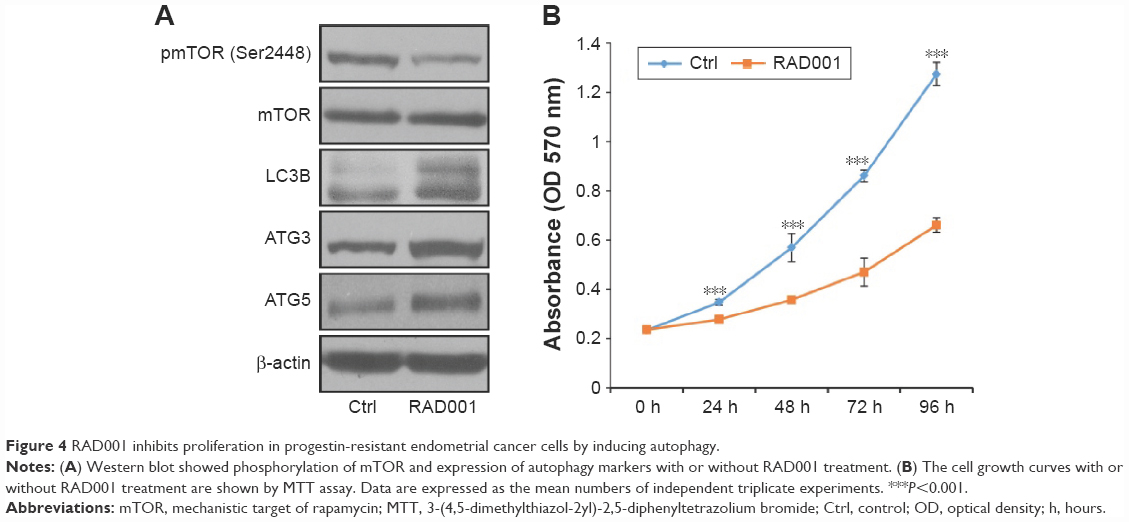 | Figure 4 RAD001 inhibits proliferation in progestin-resistant endometrial cancer cells by inducing autophagy. |
Discussion
Progestin plays a very critical role in EC therapy by inhibiting cell proliferation and inducing autophagy by binding of the nuclear receptor PR. But clinically, MPA treatment stimulated the proliferation of PRCs with low expression of PR. These studies indicate that the proliferation promotion ability of progestin on PRCs might take advantage of a nontranscriptional pathway other than the PR pathway.
It has been shown that AKT hyperactivation could induce endocrine resistance in breast cancer and non-small-cell lung cancer.10–15 We also showed AKT hyperphosphorylation in PRCs with MPA treatment (Figures 2 and 3). But AKT phosphorylation was not due to PR mediation because the PRCs showed very low or no expression of PR (Figure 1B). Knockdown of AKT in MPA-treated PRCs inhibited AKT phosphorylation and inhibited cell growth. Interestingly, inhibition of activity of AKT induced autophagy. Further, we studied the detailed mechanism by which PI3K/AKT regulates progestin resistance, and we found that mTOR signaling pathway is critical to promote cell growth and inhibit autophagy in progestin-resistant Ishikawa cells. According to these results, we proposed that PRCs that do not have or have low PR expression possibly have other hyperactivated signaling and blockade of PI3K/AKT/mTOR pathway could recover progestin resistance in EC. We also showed that combination treatment of progestin and mTOR inhibitor RAD001 can resensitize PRCs to progestin treatment.
To summarize, here we suggest that long-term progestin treatment causes progestin resistance by activating PI3K/AKT/mTOR signaling pathway. In addition, hyperactivation of PI3K/AKT/mTOR signaling pathway inhibits autophagy of PRCs. Blockade of PI3K/AKT/mTOR pathway promotes autophagy and sensitizes PRCs to progestin.
Acknowledgment
This work was supported by the Science and Technology Development Project of Weifang City, People’s Republic of China (2015wf051).
Disclosure
The authors report no conflicts of interest in this work.
References
Prat J, Gallardo A, Cuatrecasas M, Catasus L. Endometrial carcinoma: pathology and genetics. Pathology. 2007;39(1):72–87. | ||
Benshushan A. Endometrial adenocarcinoma in young patients: evaluation and fertility-preserving treatment. Eur J Obstet Gynecol Reprod Biol. 2004;117(2):132–137. | ||
Minaguchi T, Nakagawa S, Takazawa Y, et al. Combined phospho-AKT and PTEN expressions associated with post-treatment hysterectomy after conservative progestin therapy in complex atypical hyperplasia and stage Ia, G1 adenocarcinoma of the endometrium. Cancer Lett. 2007;248(1):112–122. | ||
Hahn HS, Yoon SG, Hong JS, et al. Conservative treatment with progestin and pregnancy outcomes in endometrial cancer. Int J Gynecol Cancer. 2009;19(6):1068–1073. | ||
Hoekstra AV, Kim JJ, Keh P, Schink JC. Absence of progesterone receptors in a failed case of fertility-sparing treatment in early endometrial cancer: a case report. J Reprod Med. 2008;53(11):869–873. | ||
Kim JJ, Chapman-Davis E. Role of progesterone in endometrial cancer. Semin Reprod Med. 2010;28(1):81–90. | ||
Wang S, Pudney J, Song J, Mor G, Schwartz PE, Zheng W. Mechanisms involved in the evolution of progestin resistance in human endometrial hyperplasia-precursor of endometrial cancer. Gynecol Oncol. 2005;88(2):108–117. | ||
Saitoh M, Ohmichi M, Takahashi K, et al. Medroxyprogesterone acetate induces cell proliferation through up-regulation of cyclin D1 expression via phosphatidylinositol 3-kinase/AKT/nuclear factor-kappaB cascade in human breast cancer cells. Endocrinology. 2005;146(11):4917–4925. | ||
Cui X, Zhang P, Deng W, et al. Insulin-like growth factor-I inhibits progesterone receptor expression in breast cancer cells via the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin pathway: progesterone receptor as a potential indicator of growth factor activity in breast cancer. Mol Endocrinol. 2003;17(4):575–588. | ||
Fujiwara M, Izuishi K, Sano T, et al. Modulating effect of the PI 3-kinase inhibitor LY294002 on cisplatin in human pancreatic cancer cells. J Exp Clin Cancer Res. 2008;27:76. | ||
Simoncini T, Genazzani AR. Non-genomic actions of sex steroid hormones. Eur J Endocrinol. 2003;148(3):281–292. | ||
Hubbard PA, Moody CL, Murali R. Allosteric modulation of Ras and the PI3K/AKT/mTOR pathway: emerging therapeutic opportunities. Front Physiol. 2014;5:478. | ||
Chia S, Gandhi S, Joy AA, et al. Novel agents and associated toxicities of inhibitors of the pI3k/Akt/mtor pathway for the treatment of breast cancer. Curr Oncol. 2015;22(1):33–48. | ||
Mabuchi S, Kuroda H, Takahashi R, Sasano T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol Oncol. 2015;37(1):173–179. | ||
Beca F, Andre R, Martins DS, Bilhim T, Martins D, Schmitt F. p-mTOR expression is associated with better prognosis in luminal breast carcinoma. J Clin Pathol. 2014;67(11):961–967. | ||
De Amicis F, Guido C, Santoro M, et al. A novel functional interplay between progesterone receptor-B and PTEN, via AKT, modulates autophagy in breast cancer cells. J Cell Mol Med. 2014;18(11):2252–2265. | ||
Zhao S, Li G, Yang L, Li L, Li H. Response-specific progestin resistance in a newly characterized Ishikawa human endometrial cancer subcell line resulting from long-term exposure to medroxyprogesterone acetate. Oncol Lett. 2013;5(1):139–144. | ||
Ballare C, Vallejo G, Vicent GP, Saragueta P, Beato M. Progesterone signaling in breast and cendometrium. J Steroid Biochem Mol Biol. 2006;102(1–5):2–10. | ||
Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. | ||
Sobolewska A, Gajewska M, Zarzyńska J, Gajkowska B, Motyl T. IGF-I, EGF, and sex steroids regulate autophagy in bovine mammary epithelial cells via the mTOR pathway. Eur J Cell Biol. 2009;88(2):117–130. | ||
Choi S, Shin H, Song H, Lim HJ. Suppression of autophagic activation in the mouse uterus by estrogen and progesterone. J Endocrinol. 2014;221(1):39–50. | ||
Kim J, Kim TY, Cho KS, Kim HN, Koh JY. Autophagy activation and neuroprotection by progesterone in the G93A-SOD1 transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2013;59:80–85. | ||
Janzen DM, Rosales MA, Paik DY, et al. Progesterone receptor signaling in the microenvironment of endometrial cancer influences its response to hormonal therapy. Cancer Res. 2013;73:4697. | ||
Davis ID, Long A, Yip S, et al. EVERSUN: a phase 2 trial of alternating sunitinib and everolimus as first-line therapy for advanced renal cell carcinoma. Ann Oncol. 2015;26(6):1118–1123. | ||
Wolin EM. Long-term everolimus treatment of patients with pancreatic neuroendocrine tumors. Chemotherapy. 2015;60(3):143–150. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.