Back to Journals » Drug Design, Development and Therapy » Volume 13
Physicochemical characterization and phase I study of CMAB008, an infliximab biosimilar produced by a different expression system
Authors An Q ![]() , Zheng Y, Zhao Y, Liu T, Guo H, Zhang D, Qian W, Wang H, Guo Y, Hou S, Li J
, Zheng Y, Zhao Y, Liu T, Guo H, Zhang D, Qian W, Wang H, Guo Y, Hou S, Li J
Received 12 April 2018
Accepted for publication 31 July 2018
Published 12 March 2019 Volume 2019:13 Pages 791—805
DOI https://doi.org/10.2147/DDDT.S170913
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Qing An,1,* Yingxin Zheng,2,3,* Yirong Zhao,2,* Tao Liu,2 Huaizu Guo,2 Dapeng Zhang,2–4 Weizhu Qian,5 Hao Wang,5 Yajun Guo,2,4,6,7 Sheng Hou,2,4 Jing Li2,8
1Nanjing Medical University Affiliated Cancer Hospital, Jiangsu Cancer Hospital, Jiangsu Institute of Cancer Research, Jiangsu, China; 2State Key Laboratory of Antibody Medicine and Targeted Therapy; Shanghai, China; 3Obstetrics and Gynecology Hospital of Fudan University; Shanghai, China; 4School of Pharmacy, Liaocheng University, Liaocheng, China; 5Shanghai Key Laboratory of Cell Engineering, Shanghai, China; 6School of Bioscience and Bioengineering, South China University of Technology, Guangzhou, China; 7Institute of Molecular and Cell Biology, Proteos, Singapore; 8Shanghai Zhangjiang Biotechnology Co., Ltd; Shanghai, China
*These authors contributed equally to this work
Background: Infliximab (Remicade), a chimeric monoclonal antibody against human TNFα, will inevitably face competition from biosimilar products, because of its effectiveness in autoimmune diseases and rapidly increasing market demand. According to guidelines for biosimilar development, the “biosimilar-expression system” may differ from that of the innovator, but more appropriate studies should be carried out to demonstrate the comparability between biosimilar and innovator. CMAB008 is an infliximab biosimilar candidate developed by the State Key Laboratory of Antibody Medicine and Targeted Therapy of China. Infliximab was expressed in SP2/0 cells, while CMAB008 was produced in a CHO-expression system.
Methods: In this study, infliximab and CMAB008 were compared on physicochemical and biological characterizations, including protein content, activity, physiochemical integrity, impurities, additives, and immunogenicity.
Results: The results showed that they were highly similar and comparable, except some differences in glycosylation. As glycosylation profiles can influence immunogenicity and occurrence of allergy or other adverse reactions of antibody therapeutics, primary tolerability and pharmacokinetics of CMAB008 were evaluated. In the phase I clinical trial, plasma concentration of CMAB008 and antidrug antibodies were also measured using ELISA and bridging ELISA, respectively. CMAB008 exhibited favorable clinical tolerability, no adverse events in the 3 mg/kg single-dose group (recommended therapeutic dosage), and no serious adverse events in the multiple-dose group. Also, no injection-site reactions were observed in the experiment.
Conclusion: In summary, CMAB008 might have the potential to be an effective drug compared with infliximab.
Keywords: infliximab, biosimilar, “biobetter”, CMAB008, immunogenicity
Introduction
Rheumatoid arthritis (RA) is a chronic systemic autoimmune disorder principally characterized by destruction and ankylosis of synovial joints, leading to a certain degree of disability and premature mortality.1 TNFα has been identified as a key regulator of abnormal immunoinflammatory responses in RA. The application of TNFα antagonists, including antibodies (infliximab, adalimumab) and Fc-fusion proteins (etanercept), for treatment of rheumatic diseases has significantly improved outcomes of patients.2
However, recombinant monoclonal antibodies (mAbs) represent a class of advanced but relatively expensive medicines. It is necessary to increase investment to develop affordable biosimilar mAbs by both innovator and generic drug companies. Another driving force for the interest in biosimilars is the upcoming patent expiration for marketed protein products. This may improve access to expensive biological agents. The European Medicines Agency has pioneered the regulatory framework for approval of biosimilar products since 2005, and defined a biosimilar as “a medicine which is similar to a biological medicine that has already been authorized”. A regulatory pathway for approval of follow-on biologics has also been established by the US Food and Drug Administration.3 Two biosimilar TNFα-mAb products, with trade names Remsima and Inflectra, were approved for clinical use in the European Union on September 10, 2013.4–6 This approval presented the feasibility of using a biosimilar pathway for mAbs and paved the way for further biosimilar mAb products.
Different from small-molecule generics, which are relatively easy to reproduce with identical quality and properties, biosimilar Abs require much more extensive assessment for comparability, in which the boundaries of criteria are not usually well defined, due to the complex nature of biologics and their manufacturing process.7,8 In addition, because of the complicated structural conformation and complex posttranslational modifications (PTMs), even a well-controlled product may consist of several hundred isoforms with the same amino-acid sequence.9 Also, different modifications generate heterogeneity in biologics. Therefore, it is not possible to produce exact copies of large proteins, especially glycoproteins.10
Demonstration of comparability and similarity between Ab-based biosimilar products and reference products in structure and function must be based on a series of comprehensive comparability studies on protein content, activity, physiochemical integrity, stability, impurities, additives, and immunogenicity. The primary amino-acid sequence should be identical for both. Small differences in the microheterogeneity pattern of the molecule may be acceptable if appropriately justified with regard to its potential effect on safety and pharmacokinetic (PK) and pharmacodynamic properties. Whenever possible, it is better to consider all characteristics, including mechanism of activity, potency, immunogenicity, and identical pharmacokinetics, when the comparability of the biosimilar protein and its corresponding reference drugs are evaluated. The similarity of two products should be demonstrated on a case-by-case basis.5,10
Infliximab is a chimeric mAb against TNFα used to treat autoimmune diseases,11 especially as an effective treatment for RA.12 In Europe and the US, infliximab is approved for use in RA with methotrexate, although in clinical practice it is often used as monotherapy or in combination with other disease-modifying antirheumatic drugs if patients have intolerance of methotrexate.13 In this study, we describe physicochemical and biological characterizations of a biosimilar candidate of infliximab, CMAB008, which was developed by the State Key Laboratory of Antibody Medicine and Targeted Therapy. In addition to evaluation of the primary structure, glycosylation, high-order structure, and bioactivity demonstrating similarity between the two biologics (CMAB008 and infliximab), the primary safety and PK profile of CMAB008 through a single-dose study in healthy volunteers and a multiple-dose study in RA patients were evaluated.
Methods
Samples and materials
The reference anti-TNFα Ab, infliximab (Remicade 100 mg/vial; Janssen), was purchased from the Chinese market and stored according to the manufacturer’s instructions. The proposed biosimilar, CMAB008, was developed by the State Key Laboratory of Antibody Medicine and Targeted Therapy in China. Infliximab was expressed in SP2/0 cells, while CMAB008 was produced in a CHO-expression system. CMAB008 (100 mg/vial) was manufactured in-house according to good manufacturing practice standards. Tetramethylbenzidine (TMB) substrate was obtained from Jingmei Bioengineering (Shenzhen, China); Trypsin, carboxypeptidase B, peptide N-glycosidase (PNGase F), dithiothreitol (DTT), iodoacetamide, GlycoProfile II labeling kit, D-galactosamine, guanidine hydrochloride, NaBH3CN, and ammonium bicarbonate (NH4HCO3) were purchased from Sigma-Aldrich (St Louis, MO, USA). Formic acid, acetonitrile, and solid-phase extraction cartridges were obtained from Waters (Milford, MA, USA).
Size-exclusion chromatography and unreduced SDS-PAGE were used to separate low- and high-molecular mass variants, impurities, and formulation ingredients. Size-exclusion HPLC was performed under nondenaturing conditions with a Waters 2695 Alliance HPLC system on a TSK G3000SWXL column (Tosoh, Tokyo, Japan) with aqueous-buffered mobile phase. Samples were separated on 7 cm-long 12% total concentration of acrylamide plus bis-acrylamide in 100 mL SDS-PAGE gels with a 5% T-stacking gel. In SDS-PAGE, the identity of the test samples was determined by visual comparison of the positions of the major band, with the reference standard applied to the same gel.14
Intact protein mass measurements
Molecular masses of CMAB008 and the reference product were determined with a Waters Synapt G2 quadrupole time-of-flight mass spectrometry (MS) system. N-linked polysaccharides of Abs were deglycosylated by adding PNGase F. Proteins were reduced with DTT. Intact and reduced samples were analyzed by reverse-phase liquid chromatography-MS. Reverse-phase desalting separations of intact samples were performed on a Waters MassPrep micro-desalting column (2.1×5.0 mm) using a gradient (3–9 minutes, 5%–90% B). Reverse-phase separations of reduced samples were performed on a Waters C4 column (2.1×50 mm, 1.7 μm) using a gradient (6–18 minutes, 5%–45% B). The mobile phase B was acetonitrile containing 0.1% formic acid, whereas mobile phase A was water containing 0.1% formic acid. The flow rate was maintained at 0.40 mL/minute and column temperature at 80°C to the desalting column and 40°C to the C4 column, respectively. MS analysis was performed on the Synap G2 system in positive-ion mode. Desolvation gas and source temperatures were set to 350°C and 120°C, respectively. Capillary and cone voltages were set at 3,000 and 45 V, respectively. Transfer-collision energy was set at 6.0 V. The scan range was set to 500–3,000 m/z. Raw protein spectra of intact and reduced samples were deconvoluted before evaluation using the maximum-entropy algorithm in BiopharmaLynx MS software.
Peptide-mapping analysis
Peptide mapping with reverse-phase ultrahigh-performance liquid chromatography (UPLC) MS/MS was performed to identify the primary sequences of the tested products. Protein samples were digested with trypsin. Before digestion, proteins were reduced with DTT and alkylated with iodoacetamide. After digestion, tryptic and alkylated samples were separated on a Symmetry C18 reverse-phase column. Detection was carried out at 214 nm or using MS/MS equipped with electrospray ionization.
N-glycosylation analysis
After denaturation with RapiGest SF and reduction with DDT, N-linked polysaccharides from samples were deglycosylated by adding PNGase F and incubating at 37°C overnight. Released polysaccharides were extracted and labeled with 2-aminobenzamide (2-AB) using a Sigma GlycoProfile II labeling kit. The 2-AB-labeled polysaccharides were subsequently separated by normal-phase chromatography and detected using fluorescence detection (FLD) alone or combined with MS/MS.
Circular dichroism (CD) spectroscopy
CD assessment was performed using a Jasco 715. Cells of quartz glass with optical path lengths of 0.1 cm were used for near-ultraviolet (UV; 340–250 nm) and far-UV (250–190 nm) CD measurement. Protein concentrations used for far-UV and near-UV CD spectra were 0.2 and 1.0 mg/mL, respectively. Scan speed was 50 nm/min. The formulation buffer was measured as a blank and subsequently subtracted. Noise reduction was applied to baseline-corrected protein spectra using the smoothing option of the device software for the spectrometer.
TNFα-neutralization in vitro assay
Assays for the neutralization of TNFα cytotoxicity were used to compare the biological activity of CMAB008 and reference product. TNFα-sensitive L929 cells (1.5×104/well, ATCC CCL1) were seeded in a 96-well culture plate and incubated in 100 μL RPMI 1640/DMEM supplemented with 10% new bovine serum and cultured overnight (18–24 hours) at 37°C, 5% CO2. Serial dilutions of the Abs to be tested for neutralization were prepared in medium containing 20 μg/mL dactinomycin and 4.9 ng/mL rhTNFα (R&D Systems). Consequently, 100 μL prepared serially diluted test samples were added to each well of a 96-well tissue-culture plate in duplicate. After 14–16 hours of incubation at 37°C, 5% CO2, 20 μL MTS/PMS (Promega) solution was added before another 1–4 hours of incubation. Absorbance was determined with a BioTek ELx800 microplate reader at 490 and 630 nm. The following equation established from a four-parameter logistic model was used to calculate EC50: Y=(A−B)/(1+[X/C]D)+B, where X represents the concentration of CMAB008 in the samples, Y absorbance at 450 nm, D the slope of the logit-log plot, C EC50, and A and B maximal and minimal absorbance, respectively. The calculated value of each sample was required to be within the range of the standard curve.
Murine protection model in vivo
A total of 120 Kunming mice were randomly divided into two large groups. Each group was then randomly divided into six small groups of 20 animals each. The large groups were treated first with single intravenous tail injections of 500 μg/mouse CMAB008 or 500 μg/mouse reference product. At 72 hours later, the small groups of CMAB008 or the reference product received intraperitoneal injections of 0, 1.25, 2.5, 5, 10, or 20 μg/mouse rhTNFα (dissolved in 0.1 mL sterile PBS) and 18 mg/mouse D-galactosamine. The study was terminated after 72 hours of treatment. Mortality in each group was recorded. Mice were killed with CO2 asphyxiation. All animals were treated in accordance with the guidelines of the Committee on Animals of the Second Military Medical University. All experiments were conducted in accordance with institutional guidelines and approved by Institutional Animal Care and Use Committee (IACUC) of the Second Military Medical University, and all applicable international, national, and institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Competitive inhibition assay
Culture plates (96 wells) were coated with rhTNFα at 0.1 μg mL in PBS by incubating 100 μL/well for 2 hours at 37°C. Wells were washed three times with PBST (PBS containing 0.1% Tween 20), then blocked with PBS containing 3% BSA. After incubation for 2 hours at 37°C, the plate was again washed three times in PBST. CMAB008 and the reference product were diluted to 10 μg/mL in PBS, then mixed with an equal volume of PBS containing 0.34 μg mL horseradish peroxidase (HRP)-conjugated CMAB008, respectively. The tested samples were prepared by serial dilutions of the aforementioned in PBS containing 0.17 μg mL HRP-conjugated CMAB008. Samples (100 μL/well) were dispensed onto a coated plate and incubated for 1 hour at 37°C. The plate was then washed three times with PBST. Then, a TMB substrate dispensed 100 μL/well and incubation continued for 10 minutes at room temperature. Color development was stopped by the addition of 50 μL 0.5 M H2SO4 to each well. The absorbance of each well was then read at 450 nm with reference at 630 nm. The EC50 value was obtained by a logistics four-parameter curve fit.
Surface plasmon resonance (SPR)
SPR measurements were performed on a Biacore T200 instrument (GE Healthcare). A capture assay was used to allow accurate assessment of Ab affinity. Abs at 20 μg mL were captured for 60 seconds at 10 μL/min on a Series S CM5 chip surface immobilized with ~3,000 RU anti-human IgG (Fc). rhTNFα antigen was flowed over the captured IgG surface using a range from 1 μg/mL to 0.0625 μg/mL in each case. Surfaces were regenerated with 3 M MgCl2 for 180 seconds. Association and dissociation kinetic constants (Ka and Kd) were determined from a best fit of the data using the 1:1 Langmuir global fitting procedure to sensorgrams in the Biacore T200 evaluation software version 1.0.
Study design and subjects
The studies were approved by the State Food and Drug Administration of China (approval 2007L00616). Study 1 was performed at the First Affiliated Hospital of the Third Military Medical University. It was designed as an open-label, dose escalation study in 27 healthy volunteers who were randomly assigned to receive one of three initial doses of CMAB008 (1, 3, or 10 mg/kg) as a 2-hour infusion. Study 2 was also conducted at the First Affiliated Hospital. It was designed as an open-label, single-arm, multiple-dose study in patients with RA. Nine patients were administered CMAB008 intravenously at a dose of 3 mg/kg in 2 hours at 0, 2, 6, 10, and 14 weeks. Considering that liver and renal function and other disease states may affect PK parameters and therapeutic outcomes, we selected healthy volunteers in study 1 and patients strictly according to the eligibility/exclusion criteria. We excluded patients with hepatitis or renal disease or a history of cancer. In study 2, patients eligible for enrollment were aged 18–65 years with active RA.
To assess the safety and primary efficacy of CMAB008, physical examinations, including vital signs and clinical laboratory tests, were performed before receipt of study medication. Biochemical examinations were also monitored at 1, 28, and 56 days after administration in studies 1 and 2 and even after the last administration in study 2. Adverse events (AEs) were monitored during the treatment and follow-up period. The studies were conducted in accordance with the revised Declaration of Helsinki and good clinical practice requirements. All subjects consented to the study after a full explanation of what was involved and signed informed consent forms prior to participation.
Sample collection
In study 1, venous blood samples for analysis of serum concentration were drawn before administration, 1 hour after infusion began, and 0, 1, 2, 4, 8, 12, 24, and 72 hours and 7, 14, 21, 28, 42, and 56 days after the infusion ended. In study 2, blood samples for the analysis were drawn before each infusion and 1, 2, 4, 8, 12, and 24 hours, and 1, 2, 4, 6, 8, and 12 weeks after each infusion. Serum was harvested by means of centrifugation from whole blood and stored at −20°C.
ELISA for CMAB008 levels
Serum concentrations of CMAB008 were determined by ELISA using rhTNFα and HRP-labeled anti-Ig polyclonal Ab. Plates (96 wells) were coated with rhTNFα and then incubated with the serum sample and HRP-labeled anti-Ig polyclonal Ab. The bound HRP conjugate was colored with TMB substrate. CMAB008 serum concentrations in each sample were calculated through a standard curve measured at 450 nm with reference at 630 nm using a plate reader. The following equation established from a four-parameter logistic model was used: Y=(A1−A2)/(1+[X/X0]P)+A2, where X represents the concentration of CMAB008 in the samples, Y absorbance at 450 nm, P the slope of the logit-log plot, X0 EC50, and A1 and A2 maximal and the minimal absorbance, respectively. The calculated value of each sample was required to be within the range of the standard curve.
Immunogenicity
In study 1, serum samples in the three groups were collected to assess antidrug Abs (ADAs) before administration and 14, 28, and 56 days after infusion. Abs against CMAB008 were first measured using an Ab-bridge method. In detail, this was a double-Ab-capture ELISA based on CMAB008 as the capture Ab and the revealing Ab. If ADAs were positive, a high-sensitivity cell-based bioassay was utilized to detect the neutralization Ab. In study 2, drug immunogenicity was assessed through serum samples collected just before each infusion and at 1, 2, 4, 6, 8, and 12 weeks after the last intravenous infusion of CMAB008. Analysis methods were in accordance with study 1.
Statistical analysis
Statistical analysis was performed with SPSS 13.0 software for Windows (SPSS, Chicago, IL, USA). All grouped data are expressed as mean±SD. To test differences between the mortality of mice treated with infliximab and CMAB008, χ2 tests were used. ANOVA was used to test differences among three groups when the data followed a normal distribution, whereas the Kruskal–Wallis test was used when measurement variables were not normally distributed. For differences between two groups, Student’s t-test was employed when data were of normal distribution and the variance homogeneous; otherwise, the Wilcoxon test was used. P<0.05 was considered statistically significant.
Data analysis
Noncompartmental pharmacokinetic parameters were estimated with WinNonlin Professional version 6.1 software (Pharsight). PK parameters in the single-dose study included AUC, AUC from 0 to last time point (AUC0–t), AUC from 0 to infinity (AUC0–∞), maximum concentration (Cmax), time to Cmax (Tmax), t½, clearance (Cl), mean residence time (MRT), and elimination rate constant (K). PK parameters in study two also included steady-state AUC (AUCss), steady-state clearance (Clss), steady-state distribution volume (Vss), steady-state Cmax (Cmaxss), minimum steady-state concentration (Cminss), and average steady-state concentration (Cavg). AUC was calculated by the linear-up/log-down approach. K was determined by linear regression of the terminal points of the log-linear serum concentration–time curve. Cmax in serum, Tmax, Cmaxss, and Cminss were derived directly from observed data.
Results
Primary structure
Infliximab was expressed in SP2/0 cells, while CMAB008 was produced in a CHO-expression system. Confirmation of the primary structure was critical in the verification of the identity of CMAB008 as the reference product, due to its complex molecular structures and PTMs. The samples (CMAB008 and infliximab), determined by size-exclusion HPLC analysis, showed the same retention time and purity of the main peak (Figure 1A). The similarity was also investigated by SDS-PAGE, and the results confirmed that CMAB008 and infliximab bore similar profiles regarding monomer purity (Figure 1B). The intact protein mass profile of CMAB008 was almost identical to that of the reference product, varying only in peak shapes (Figure 1C), and the main peaks of the two mAbs became indistinguishable when they were digested with PNGase F to remove complexity caused by the N-linked polysaccharides at the intact protein level (Figure S1). Reduced intact mass analysis further confirmed the identical heavy chain when the C-terminal lysine was removed with carboxypeptidase B (Figure S2). These data suggest that the primary structures of Infliximab and CMAB008 are identical.
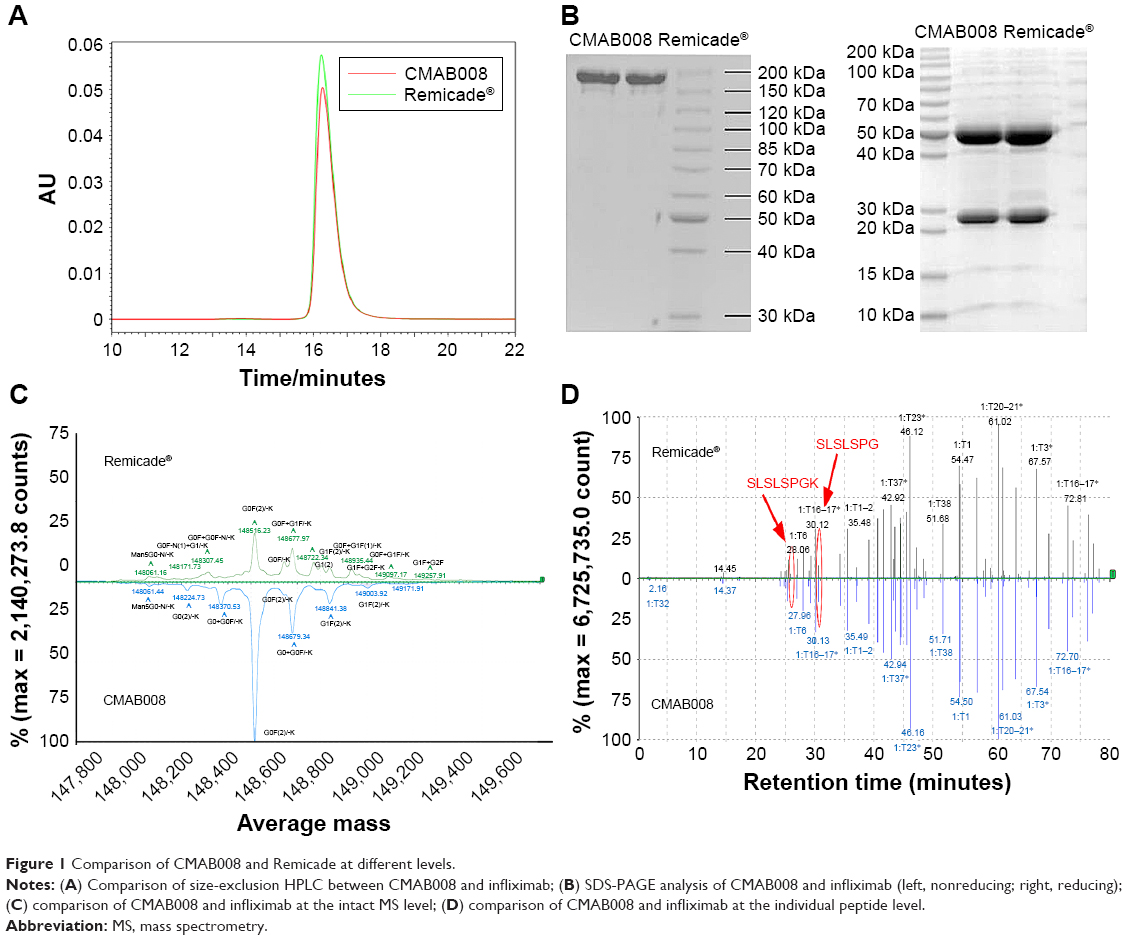 | Figure 1 Comparison of CMAB008 and Remicade at different levels. |
Reverse-phase UPLC-MS/MS peptide maps for the reduced Abs, which resulted in 100% sequence coverage and nearly identical chromatograms, were also used to evaluate the similarity of the test samples. As shown in Figure 1D, peptide mapping of CMAB008 was similar to that of the reference product, despite the difference in the two C-terminal peptide peaks. One peptide sequence of the UPLC-MS/MS peak at RT 25.96 minutes for the reference product was identified as “SLSLSPGK”, and the other at RT 30.56 minutes was confirmed as “SLSLSPG”. Their relative abundance (SLSLSPGK/SLSLSPG) was ~50:50 in the reference product compared to 5:95 in CMAB008. Differences in peptide maps between CMAB008 and the reference product were attributed to C-terminal lysine heterogeneity in the PTM process. This was also accordance with the difference in intact-mass and reduced intact-mass analysis. A 162 Da mass difference was found between the two C-terminal lysine variants.
N-glycosylation profile
Although accounting for only 2%–3% of Ab mass, glycosylation of IgG is essential to the activation of downstream biological mechanisms. Additionally, the precise structure of the attached oligosaccharide can influence biological efficacy.15 The major difference between CMAB008 and the reference product was glycoforms and polysaccharide proportion. The reference product comprised more complex-type polysaccharides with two branches and hybrid-type polysaccharides than CMAB008.
In this study, 2-AB labeled polysaccharides and normal-phase chromatography with FLD-MS/MS detection were used to evaluate differences in N-polysaccharide profiles between CMAB008 and the reference product. As shown in Figure 2, the N-polysaccharide profile of CMAB008 exhibited major differences in glycoforms and polysaccharide proportion compared with the reference product. The reference Ab comprised more complex-type and hybrid-type polysaccharides than CMAB008. The relative abundance of the unfucosylated complex-type polysaccharides, such as 3, 6, 0, 0, 0, 3, 5, 0, 0, 0 and 3, 5, 1, 0, 0, was >5%. The relative abundance of the fucosylated complex-type polysaccharides was 1.58%. In addition, the relatively low-abundance Gal-α1,3-Gal polysaccharide species, such as 4, 6, 1, 0, 0 and 4, 6, 1, 0, 1, were also observed. These nonhuman polysaccharides may induce immunogenicity.16 Compared with 4% of the reference product, the relative abundance of sialic acid-containing polysaccharides of CMAB008 was <1%. Overall, there were ~40 polysaccharide species with relative abundance >0.1% in the reference product, whereas there were about 27 in CMAB008. However, glycoforms with relative abundance >1% in the two samples were highly similar.
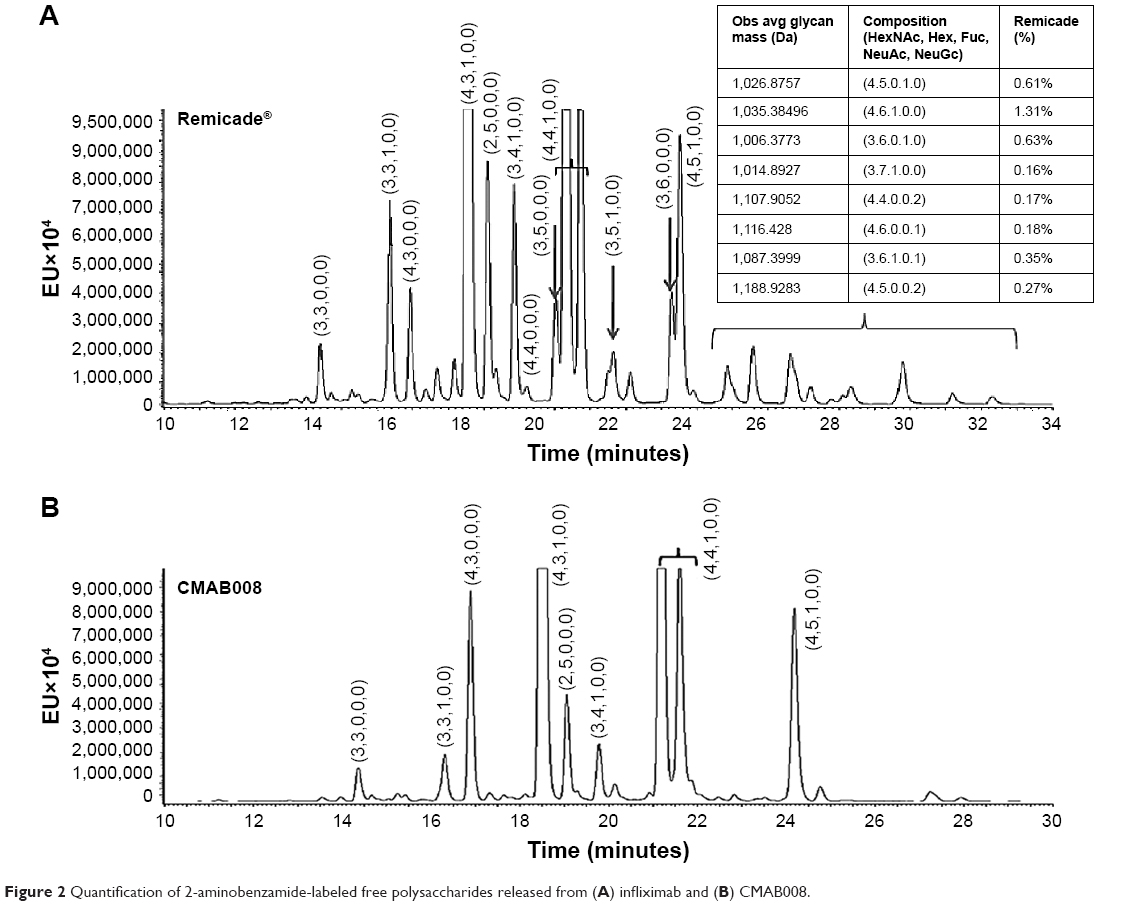 | Figure 2 Quantification of 2-aminobenzamide-labeled free polysaccharides released from (A) infliximab and (B) CMAB008. |
Higher-order structures
CD spectroscopy in the far- and near-UV spectral regions provided insight into the secondary structure (α-helix, β-sheet, random coil) and tertiary structure, respectively. In this study, CD spectroscopy was used to obtain information on the folding state of CMAB008 and the reference product. Three batches for each product were tested. Far-UV and near-UV CD spectra from CMAB008 and the reference product were shown to overlap indistinguishably (Figure 3). These results confirmed that the secondary and tertiary structures of CMAB008 and the reference product (infliximab) were consistent.
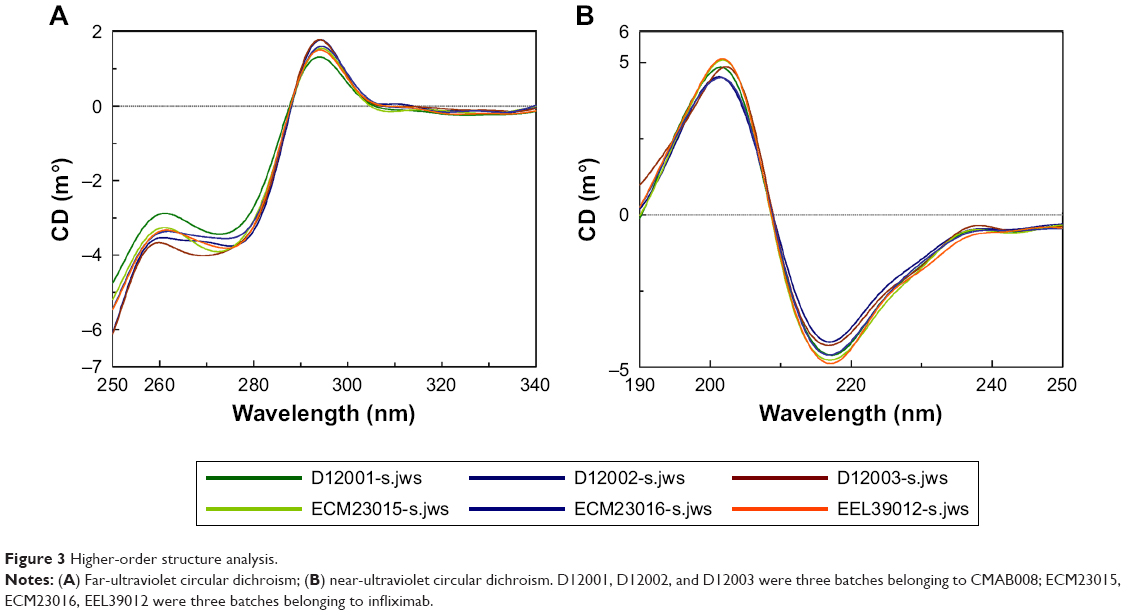 | Figure 3 Higher-order structure analysis. |
Affinity and bioactivity analyses
Affinity analysis can be used to evaluate binding properties of anti-TNFα products with TNFα.17,18 In this study, the proposed biosimilar CMAB008 and the reference product (infliximab) were evaluated for their relative binding affinity for purified rhTNFα by a competition-inhibition assay with solid-phase ELISA. A constant amount of HRP-labeled CMAB008 mixed with varying amounts of unlabeled competitor to TNFα was measured. The results of one experiment for the tested samples are shown in Figure 4A. Relative affinity activity (EC50 ratio of infliximab and CMAB008) was calculated by averaging the results from three such experiments (Table 1). As shown in Table 1, EC50 values of CMAB008 and reference were 0.146 and 0.139 μg/mL (P=0.448, paired Student’s t-test), respectively. Additionally, we also analyzed the two tested Abs by real-time SPR analyses. We measured the binding affinity and kinetics of rhTNFα (analytes) to each Ab coupled as a ligand to a CM5 sensor chip. Figure 4B and C shows the experimental sensorgrams obtained. CMAB008 and the reference product (infliximab) showed binding kinetics similar to TNFα, with binding affinities of 1.91×10−10 M and 1.67×10−10 M, respectively. Therefore, the affinity activities of the two tested Abs were comparable. That was also in accordance with a previously reported affinity constant (Ka) for the reference product (infliximab), which was about 0.18×10−10 M.19
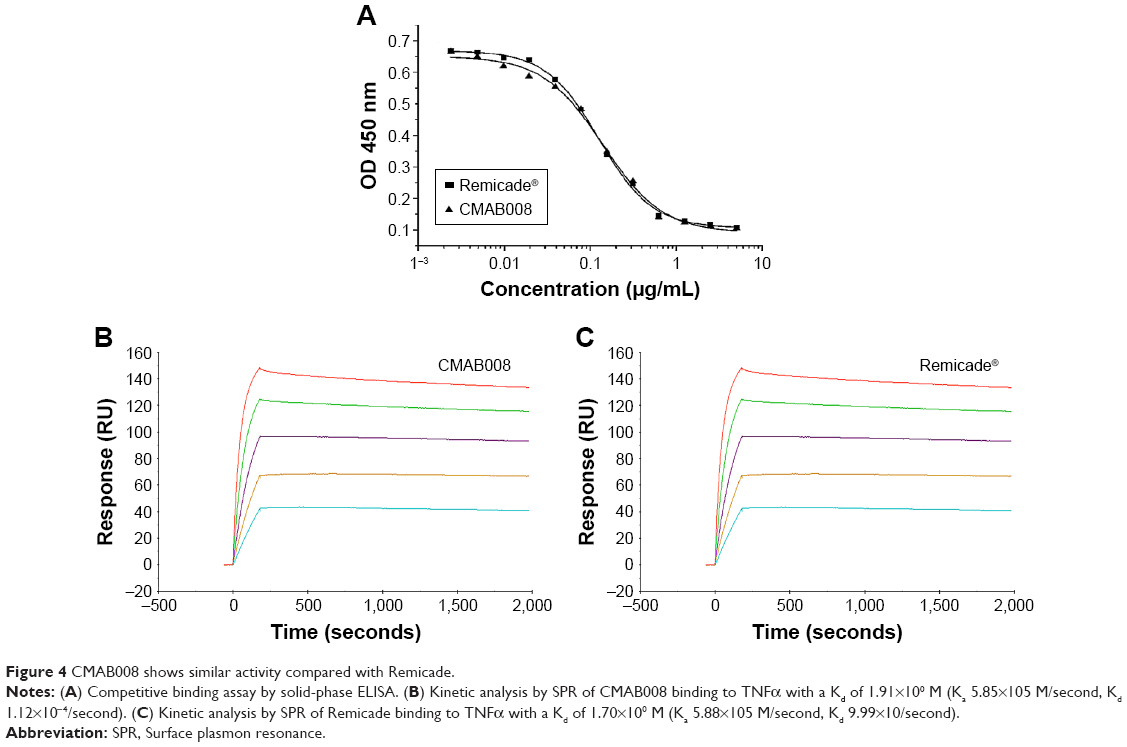 | Figure 4 CMAB008 shows similar activity compared with Remicade. |
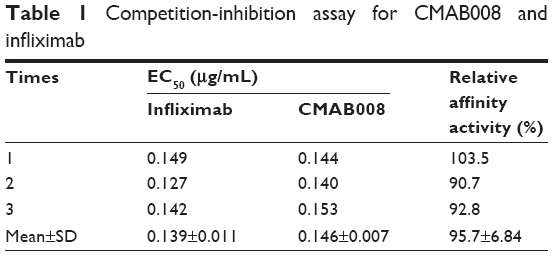 | Table 1 Competition-inhibition assay for CMAB008 and infliximab |
Bioactivity of the tested Abs was also compared by a TNF-sensitive L929 cell cytotoxicity neutralization assay. TNFα exhibits cytotoxicity toward the mouse fibroblast cell line L929. The capacity of Abs to neutralize this TNFα-dependent cytotoxicity was shown in Table 2. CMAB008 and reference EC50 values were 16.81±0.57 ng/mL and 16.74±0.64 ng/mL (P=0.894, paired Student’s t-test), respectively. Relative bioactivity was calculated according to: relative neutralization activity (%)=(EC50 [reference]/EC50 [sample])×100%. The relative neutralization activity of CMAB008 to the reference product was 100.58%±6.71%.
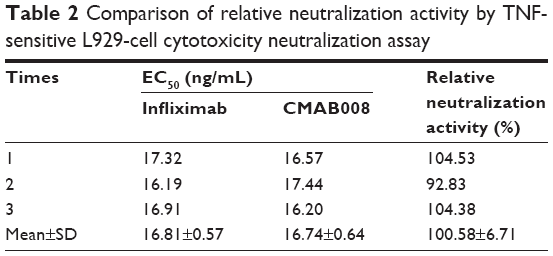 | Table 2 Comparison of relative neutralization activity by TNF-sensitive L929-cell cytotoxicity neutralization assay |
A murine lethality protection model was used to assess the in vivo neutralizing bioactivity of CMAB008 and the reference product. The tested anti-TNFα mAbs effectively prevented mortality induced by rhTNFα (Table 3). The LD50 of rhTNFα for the Kunming mice pretreated with infliximab was 8.56 μg/mouse while for those pretreated with CMAB008, it was 8.71 μg/mouse. Compared with the LD50 of the non-Ab-treated group (0.46 μg/mouse), the LD50 of rhTNFα increased 18.6 and 18.9 times, respectively. No dose mortality between the two groups showed statistically significant differences (χ2, P>0.05). Kaplan–Meier analysis also indicated that mortality induced by rhTNFα was not significantly different between the two groups of products.
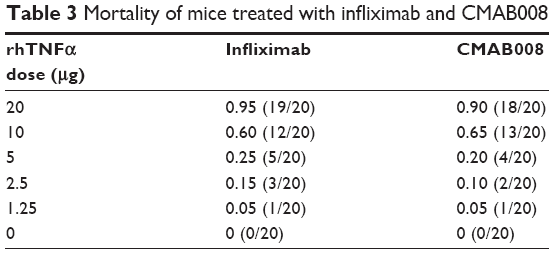 | Table 3 Mortality of mice treated with infliximab and CMAB008 |
Demographics
A total of 38 Chinese men were enrolled into the two studies. In study 1, 27 healthy male subjects were administered CMAB008 intravenously at single doses of 1, 3, or 10 mg/kg. All subjects completed the study and were included in the safety and PK analyses. In study 2, 11 patients with RA received CMAB008 at 3 mg/kg at 0, 2, 6, 10, and 14 weeks. Nine subjects completed the study and were included in PK, pharmacodynamic, and safety assessments. However, two subjects withdrew from the study for personal reasons. The majority of patients were women (nine of 11) in study 2. Demographic characteristics for all study groups are summarized in Table 4. Overall, demographics and baseline characteristics were generally similar among the dose cohorts.
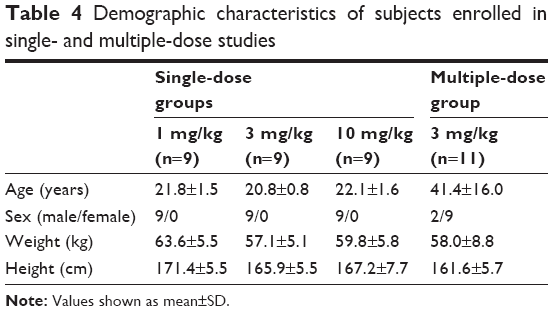 | Table 4 Demographic characteristics of subjects enrolled in single- and multiple-dose studies |
Safety and tolerability
Treatment-related AEs are summarized in Tables 5 and S1 (Remicade included). In study 1, a single intravenous injection of CMAB008 was well tolerated in healthy male Chinese subjects. No serious clinical or laboratory AEs were observed, and no subjects discontinued the study due to an AE. A total of 13 treatment-related AEs were reported by eight (29.6%) of the 27 subjects during the study, including one subject (11.1%) in the 1 mg/kg group and seven subjects (77.8%) in the 10 mg/kg group. Dizziness and muscular pain were the most common AEs associated with the treatment protocol, while other AEs, such as pyrexia, abdominal pain, diarrhea, colds, and transaminase increase, were reported only once. In study 2, one subject (11.1%) experienced nausea, red blood cell decrease, and white blood cell decrease, and Hb decrease occurred in another subject (11.1%). All AEs were evaluated as mild in accordance with the treatment protocol, and patients recovered without medication. Remarkably, in study 1, one subject in the 10 mg/kg group experienced abdominal pain and diarrhea after the first administration, and had cold symptoms and transaminase increase 27 days after CMAB008 injection. The cold symptoms and transaminase increase recovered at 32 and 56 days after administration, respectively.
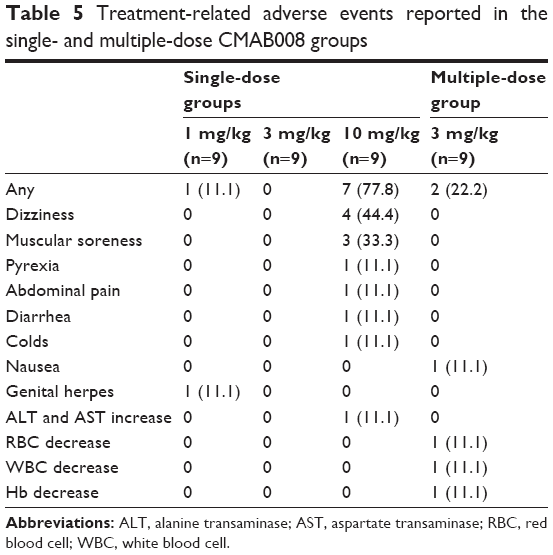 | Table 5 Treatment-related adverse events reported in the single- and multiple-dose CMAB008 groups |
Single-dose pharmacokinetics of CMAB008 in healthy volunteers
No blood samples were missed in study 1. The mean serum concentration–time curves of CMAB008 following doses of 1, 3, or 10 mg/kg are shown in Figure 5. PK parameters were observed by noncompartmental analysis (Table 6). The data indicated that drug concentration reached its peak level at the end of injection. Mean AUC was 2,434±692–41,687±7,599 μg/h·mL across the 1–10 mg/kg doses. Mean Cmax was 22.0±4.7 μg/mL, 51.6±13.0 μg/mL, and 218.9±47.6 μg/mL in the 1, 3, and 10 mg/kg dose groups, respectively. Tmax ranged from 2 to 3 hours among the three groups. Concentrations slowly decreased, with t½ at 116±36, 149±44, and 214±35 hours, respectively. MRT and t½ showed the same tendency. In conclusion, these results indicated that CMAB008 exhibited a nonlinear PK profile over the dose range of 1–10 mg/kg in healthy volunteers. Significant differences were observed in Ke, AUC0−t, AUC0–∞, MRT, t½, and Cmax (P<0.05, ANOVA).
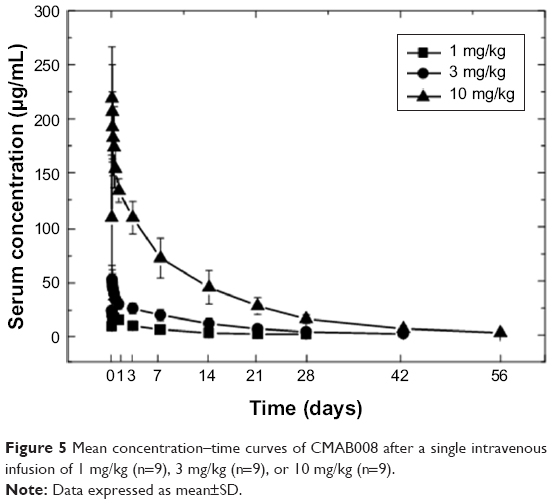 | Figure 5 Mean concentration–time curves of CMAB008 after a single intravenous infusion of 1 mg/kg (n=9), 3 mg/kg (n=9), or 10 mg/kg (n=9). |
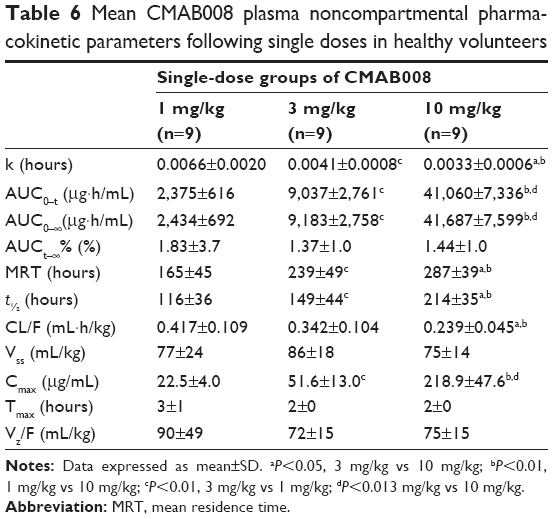 | Table 6 Mean CMAB008 plasma noncompartmental pharmacokinetic parameters following single doses in healthy volunteers |
Multiple-dose pharmacokinetics of CMAB008 in RA patients
Mean serum concentration–time curves of CMAB008 in study 2 following multiple injections of 3 mg/kg are shown in Figure 6. PK parameters are summarized in Table 7. Analysis showed that the serum CMAB008 concentration increased with accumulation of administration times and the steady-state serum concentration level was achieved following the third dose (week 6), based on the similarity of peak concentrations at weeks 2, 6, 10, and 14. Mean Cmaxss after the last dose was 55.4±6.2 mg/L. Mean Cminss was 7.0±1.3 mg/L. Mean steady-state concentration of CMAB008 was 8.7±1.4 mg/L. The mean fluctuation index value was 5.5±0.6. There was no difference between serum concentrations before the 6th and 10th weeks: 6.2±0.9 mg/L and 7.1±1.2 mg/L, respectively. Therefore, no systemic accumulation was found after multiple doses of CMAB008 with 3 mg/kg every 4 weeks in RA patients.
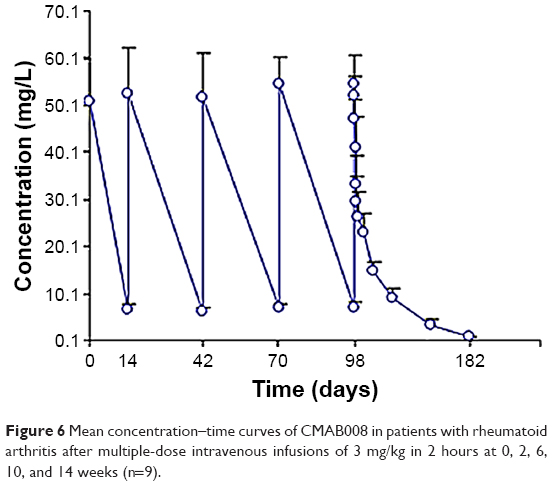 | Figure 6 Mean concentration–time curves of CMAB008 in patients with rheumatoid arthritis after multiple-dose intravenous infusions of 3 mg/kg in 2 hours at 0, 2, 6, 10, and 14 weeks (n=9). |
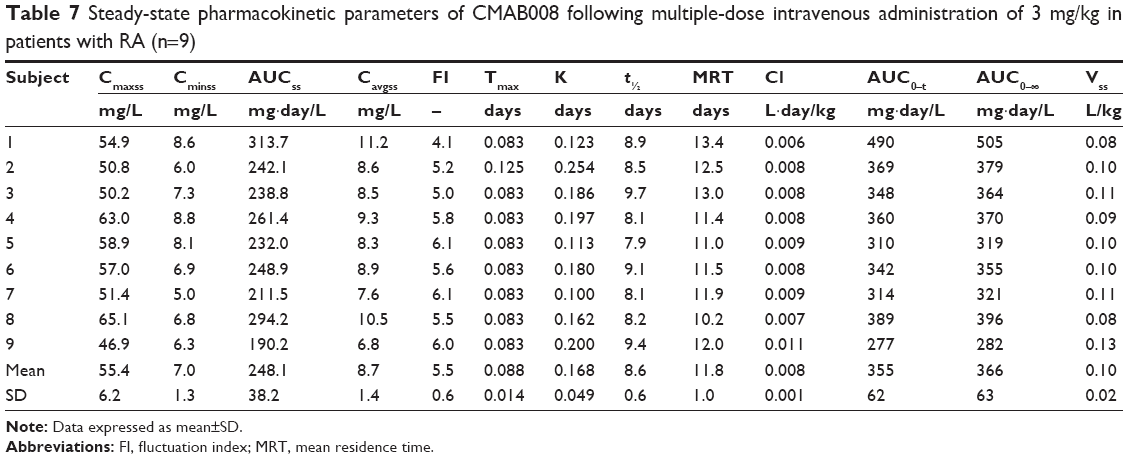 | Table 7 Steady-state pharmacokinetic parameters of CMAB008 following multiple-dose intravenous administration of 3 mg/kg in patients with RA (n=9) |
In study 2, patients with RA receiving CMAB008 3 mg/kg at 0, 2, 6, 10, and 14 weeks achieved peak serum concentrations quickly. Mean Tmax and Cmax were 0.088±0.014 days and 55.4±6.2 mg/L, respectively. However, elimination of CMAB008 was slow: t½ was 8.6±0.6 days. After multiple injections of CMAB008, mean AUC0–t was 355±62 mg/day·L and mean AUC0–∞ 366±63 mg/day·L. In addition, mean Cl and VSS were 0.008±0.001 L/day·kg and 0.10±0.02 L/kg, respectively.
Immunogenicity
In study 1, two of nine subjects in the 1 mg/kg group were found to have anti-CMAB008 Abs, which were demonstrated to be nonneutralizing Abs by immunoassay. No ADAs or neutralizing ADAs were detected from the other subjects, including those in the 3 mg/kg and 10 mg/kg groups. In study 2, only one subject was detected ADA-positive before infusion at 6, 10, and 14 weeks and at 1, 2, and 4 weeks after the last administration of CMAB008, and these were demonstrated to be nonneutralizing Abs in the following studies.
Discussion
RA is the most common inflammatory arthritis, affecting 0.5%–1% of the adult population worldwide. Estimates of RA occurrence suggest that the annual incidence is ~0.2 per 1,000 males and 0.4 per 1,000 females.20 The treatment protocol of RA has traditionally included corticosteroids and conventional disease-modifying antirheumatic drugs.21 Although these therapies provide some benefit, their efficacy has been limited by slow onset of drugs and several AEs. Newer biologically based therapies including molecules that inhibit cytokine activity, such as TNF inhibitors (infliximab), have been effective in moderate–severe RA and delayed disease progression determined radiographically, particularly when combined with methotrexate.22 Owing to the high price of these drugs, companies are commercializing these molecules after expiration of patents on the innovator product. Biosimilar Abs must have the same amino-acid sequences as the original immunoglobulins, but host cells, media, or manufacturing processes can be different, and thus the product may have small physicochemical differences that need to be assessed, due to their potential impact on the biological activity of the Ab. A functional evaluation comparing both products could support a demonstration of biosimilarity and justify clinical testing with the Abs.23
In this study, comparison of the structure, physicochemical characteristics, and potency of CMAB008 (developed by the State Key Laboratory of Antibody Medicine and Targeted Therapy in China) with the reference product (infliximab) showed that they were highly similar and comparable. However, there were still some differences in the glycosylation of the Abs. The different expression systems for CMAB008 (CHO) and Remicade (SP2/0) may be the reason for the differences in the glycosylation pattern. The CHO cell line was chosen to produce the Ab, as it presents efficient posttranslational processing of Abs, while the glycosylation patterns of native and CHO-derived Abs are similar.24
Unlike most mAbs, infliximab was produced in the mouse cell line SP2/0, which may induce some anaphylactic reactions as it expresses the gene for α1,3-galactosyltransferase.25 It is recognized that in some areas of the USA, subjects treated with cetuximab have a high prevalence of hypersensitivity reactions due to galactose-α1,3-galactose, which is present on the Fab portion of the cetuximab heavy chain.26,27 Nevertheless, the mechanism of these anaphylactic reactions has not been defined. For the reference product (infliximab), the relative abundance of unfucosylated complex-type polysaccharides was >5%. Also, the relative abundance of fucosylated complex-type polysaccharides was 1.58%. In addition, the relative abundance of Gal-α1,3-Gal polysaccharide species was >1%. This nonhuman polysaccharide may induce immunogenicity.16 Compared with 4% of the reference product, the relative abundance of sialic acid-containing polysaccharides of CMAB008 was <1%. In total, there were ~40 glycoforms with relative abundance >0.1% in the reference product, while there were about 27 in CMAB008. Sialic acids occupy terminal positions on many mammalian glycoprotein and glycolipid oligosaccharides. Sialylation can significantly influence the safety and efficacy profiles of glycoprotein drugs.28 Higashi et al reported that a sialic acid, N-glycolylneuraminic acid, was also an immunogenic nonhuman polysaccharide.29 As such, it is important to know the sialic acid content of a glycoprotein when assaying its function or efficacy as a pharmaceutical therapeutic.
Noncompartmental analysis provided evidence that CMAB008 had a nonlinear PK profile at doses of 1, 3, and 10 mg/kg. According to our data, as dose levels increased at a ratio of 1:3:10, Cmax and AUC0–∞ increased at a ratio of 1:2.3:9.7 and 1:3.8:17.1, respectively. Cmax increased in a roughly dose-proportional manner, but AUC0–∞ showed more than a dose-proportionate increase. In our single-dose escalation study, almost all PK parameters of the same dose were consistent with those of previous studies of Remicade.30 We thus concluded that the expression system and formulation did not affect the PK parameters of CMAB008. As we know, using Remicade always accompanies some AEs, including infusion-related reactions, infections, lupus-like syndrome, and even lymphoproliferative disorders.31,32
No serious AEs occurred after injection, and no subjects withdrew during the study because of AEs. Some drug-related AEs were observed, but were evaluated as mild in accordance with the study protocol, and subjects returned to normal without medication. In study 2, only one subject was detected to be ADA-positive before 6, 10, and 14 weeks and at 1, 2, and 4 weeks after the last administration of CMAB008, and these were demonstrated to be nonneutralizing Abs in the following studies.
Nearly all characteristics of a biosimilar protein and its corresponding reference product have to be as similar as possible. That means that a biosimilar therapeutic protein cannot be better than the reference product in any aspect: a biosimilar must be as good (or as bad) as the innovator. “Biobetter” Abs are those that target the same validated epitope as a marketed Ab, but have been engineered to have improved properties, eg, optimized glycosylation profiles to enhance effector functions or an engineered Fc domain to increase serum half-life.33 In this respect, CMAB008 may be a biobetter Ab based on the advantage of safety.
Conclusion
The present research shows that CMAB008 was well tolerated, with a similar or comparable profile to Remicade, except for some differences in glycosylation. CMAB008 exhibited favorable clinical tolerability, and might have the potential to be a biobetter based on the advantage of safety.
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology of China (863 projects 2014AA021004), Shanghai Rising-Star Program (16QB1404300), and Shanghai Key Technologies R&D Program of Biological Medicine (15431906100, 16431901200, 16431904700, 16431904100, and 16DZ1910400).
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Levesque MC. Biologic rheumatoid arthritis therapies: do we need more comparative effectiveness data? BioDrugs. 2012;26(2):65–70. | ||
Lin J, Ziring D, Desai S, et al. TNFalpha blockade in human diseases: an overview of efficacy and safety. Clin Immunol. 2008;126(1):13–30. | ||
Kálmán-Szekeres Z, Olajos M, Ganzler K. Analytical aspects of biosimilarity issues of protein drugs. J Pharm Biomed Anal. 2012;69:185–195. | ||
Xie H, Chakraborty A, Ahn J, et al. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. MAbs. 2010;2(4):379–394. | ||
Beck A, Reichert JM. Approval of the first biosimilar antibodies in Europe: a major landmark for the biopharmaceutical industry. MAbs. 2013;5(5):621–623. | ||
Thorpe R, Wadhwa M. Biosimilar monoclonal antibodies approved for use in the EU. GaBI J. 2014;3(1):9–10. | ||
Gottlieb S. Biosimilars: policy, clinical, and regulatory considerations. Am J Health Syst Pharm. 2008;65(14 Suppl 6):S2–S8. | ||
Ahmed I, Kaspar B, Sharma U. Biosimilars: impact of biologic product life cycle and European experience on the regulatory trajectory in the United States. Clin Ther. 2012;34(2):400–419. | ||
Schiestl M, Stangler T, Torella C, et al. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29(4):310–312. | ||
Tan Q, Guo Q, Fang C, et al. Characterization and comparison of commercially available TNF receptor 2-Fc fusion protein products. MAbs. 2012;4(6):761–774. | ||
Maini RN, Feldmann M. How does infliximab work in rheumatoid arthritis? Arthritis Res. 2002;4(Suppl 2):S22–S27. | ||
Rahman MU, Strusberg I, Geusens P, et al. Double-blinded infliximab dose escalation in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66(9):1233–1238. | ||
Zintzaras E, Dahabreh IJ, Giannouli S, Voulgarelis M, Moutsopoulos HM. Infliximab and methotrexate in the treatment of rheumatoid arthritis: a systematic review and meta-analysis of dosage regimens. Clin Ther. 2008;30(11):1939–1955. | ||
Sörgel F, Lerch H, Lauber T. Physicochemical and biologic comparability of a biosimilar granulocyte colony-stimulating factor with its reference product. BioDrugs. 2010;24(6):347–357. | ||
Jefferis R. Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol Sci. 2009;30(7):356–362. | ||
Lammerts van Bueren JJ, Rispens T, Verploegen S, et al. Anti-galactose-α-1,3-galactose IgE from allergic patients does not bind α-galactosylated glycans on intact therapeutic antibody Fc domains. Nat Biotechnol. 2011;29(7):574–576. | ||
Puthoor PR, de Zoeten EF. Pediatric ulcerative colitis: the therapeutic road to infliximab. Biol Ther. 2013;3:1–14. | ||
Scallon B, Cai A, Solowski N, et al. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301(2):418–426. | ||
Knight DM, Trinh H, Le J, et al. Construction and initial characterization of a mouse-human chimeric anti-TNF antibody. Mol Immunol. 1993;30(16):1443–1453. | ||
Voulgari PV, Kaltsonoudis E, Papagoras C, Drosos AA. Adalimumab in the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2012;12(12):1679–1686. | ||
Fries JF, Williams CA, Morfeld D, Singh G, Sibley J. Reduction in long-term disability in patients with rheumatoid arthritis by disease-modifying antirheumatic drug-based treatment strategies. Arthritis Rheum. 1996;39(4):616–622. | ||
Genovese MC, van den Bosch F, Roberson SA, et al. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. 2010;62(4):929–939. | ||
Dorvignit D, Palacios JL, Merino M, et al. Expression and biological characterization of an anti-CD20 biosimilar candidate antibody: a case study. MAbs. 2012;4(4):488–496. | ||
Birch JR, Racher AJ. Antibody production. Adv Drug Deliv Rev. 2006;58(5–6):671–685. | ||
Qian J, Liu T, Yang L, et al. Structural characterization of N-linked oligosaccharides on monoclonal antibody cetuximab by the combination of orthogonal matrix-assisted laser desorption/ionization hybrid quadrupole-quadrupole time-of-flight tandem mass spectrometry and sequential enzymatic digestion. Anal Biochem. 2007;364(1):8–18. | ||
Kawasaki N, Itoh S, Hashii N, et al. The significance of glycosylation analysis in development of biopharmaceuticals. Biol Pharm Bull. 2009;32(5):796–800. | ||
Chung CH, Mirakhur B, Chan E, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. 2008;358(11):1109–1117. | ||
Takeuchi M, Inoue N, Strickland TW, et al. Relationship between sugar chain structure and biological activity of recombinant human erythropoietin produced in Chinese hamster ovary cells. Proc Natl Acad Sci U S A. 1989;86(20):7819–7822. | ||
Higashi H, Naiki M, Matuo S, Okouchi K. Antigen of “serum sickness” type of heterophile antibodies in human sera: indentification as gangliosides with N-glycolylneuraminic acid. Biochem Biophys Res Commun. 1977;79(2):388–395. | ||
Keating GM, Perry CM. Infliximab: an updated review of its use in Crohn’s disease and rheumatoid arthritis. BioDrugs. 2002;16(2):111–148. | ||
Maini RN, Breedveld FC, Kalden JR, et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998;41(9):1552–1563. | ||
Elliott MJ, Maini RN, Feldmann M, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344(8930):1105–1110. | ||
Beck A, Biosimilar BA. Biosimilar, biobetter and next generation therapeutic antibodies. MAbs. 2011;3(2):107–110. |
Supplementary materials
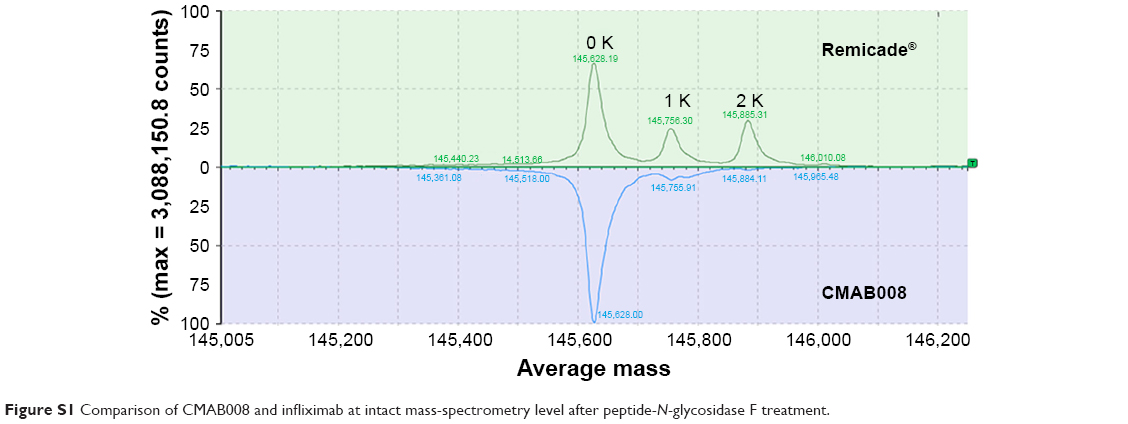 | Figure S1 Comparison of CMAB008 and infliximab at intact mass-spectrometry level after peptide-N-glycosidase F treatment. |
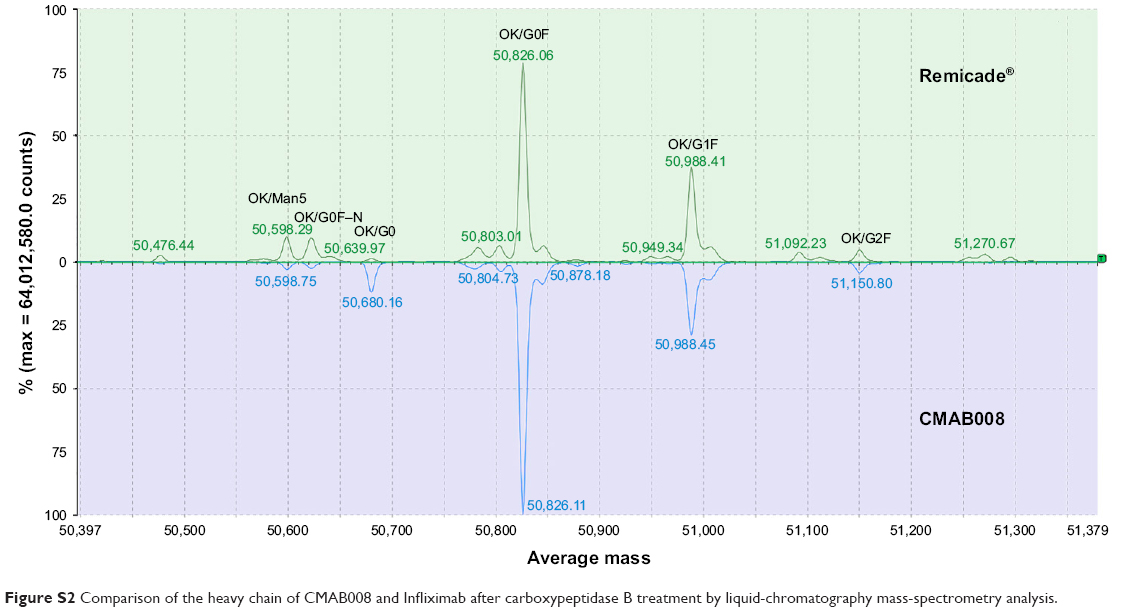 | Figure S2 Comparison of the heavy chain of CMAB008 and Infliximab after carboxypeptidase B treatment by liquid-chromatography mass-spectrometry analysis. |
 | Table S1 AE profiles of CMAB008 and Remicade |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.