Back to Journals » OncoTargets and Therapy » Volume 13
Phosphoproteomics Reveals Key Regulatory Kinases and Modulated Pathways Associated with Ovarian Cancer Tumors
Authors Hu Y, Sun L, Zhang Y, Lang J, Rao J
Received 27 November 2019
Accepted for publication 6 March 2020
Published 29 April 2020 Volume 2020:13 Pages 3595—3605
DOI https://doi.org/10.2147/OTT.S240164
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Yingchao Hu,1 Lejia Sun,2 Yinglan Zhang,3 Jinghe Lang,1 Jun Rao4
1Department of Obstetrics and Gynecology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100730, People’s Republic of China; 2Department of Liver Surgery, Peking Union Medical College (PUMC) Hospital, PUMC & Chinese Academy of Medical Sciences, Beijing 100730, People’s Republic of China; 3Department of Obstetrics and Gynecology, Affiliated Beijing Chaoyang Hospital of Capital Medical University, Beijing 100020, People’s Republic of China; 4Jiangxi Provincial Key Laboratory of Translational Medicine and Oncology, Jiangxi Cancer Hospital, Jiangxi Cancer Center, Nanchang 330029, People’s Republic of China
Correspondence: Jinghe Lang; Jun Rao Email [email protected]; [email protected]
Background: Ovarian cancer (OC) is the seventh most common cancer worldwide for women. However, there are no sufficient diagnostic methods and few treatment options available due to poor understanding of its pathogenic mechanisms.
Methods: To comprehensively analyze the phosphoproteomic characterization for OC, we took advantage of a quantitative global phosphoproteomics method, titanium(IV) immobilized metal affinity chromatography (Ti4+-IMAC) coupled to nanoscale liquid chromatography and quadrupole time-of-flight tandem mass spectrometry (nanoLC/Q-TOF-MS/MS) on ovarian tissue samples obtained from five OC patients and five matched controls.
Results: A total of 722 phosphorylated sites corresponding to 534 proteins were significantly different (fold change ≥ 2, p < 0.01) between OC patients and the controls. Among them, 83 transcription factors mainly consisted of transcription cofactors, zf-C2H2, and chromatin remodeling factors and 29 kinases were included. Further functional analysis suggested significantly biological processes were highly enriched and involved in the pathogenesis of OC, especially fructose and mannose metabolism. Moreover, the regulatory roles of modulated pathways, including MAPK, ErbB, and GnRH signaling pathways were also identified as critical processes involved in OC. The results here highlighted key phosphorylated proteins, particularly kinases, and the corresponding cancer-related metabolic and signal pathways that played important roles in the development of OC. Additionally, the expression levels of two kinases, phosphorylated CDK (T14) and phosphorylated PRKCQ (S695), were validated by Western blot analysis in the other group of ovarian tissue samples.
Conclusion: Altogether, our data not only provided novel insights into the potential biomarkers and therapy options for OC but also extended our knowledge on its pathophysiological mechanism.
Keywords: ovarian cancer, phosphoproteomics, Ti4+-IMAC, kinases, biomarker
Introduction
Ovarian cancer (OC) is the seventh most common cancer worldwide for women.1 Especially in the United States, OC is the leading cause of death among fatal gynecologic malignancies. There were approximately 22,440 new cases of ovarian cancer diagnosed and 14,080 OC deaths in 2018 in the United States.2 Unfortunately, OC is a disease with a poor prognosis, which frequently has no obvious symptoms at its early stages.3 It is usually advanced when it is diagnosed but with low survival rates ranging from 30% to 50%. Nevertheless, risk factors for OC remain largely unknown and there are few promising therapy options for patients, which motivated researchers to characterize the molecular changes associated with OC development and to identify potential biomarkers for diagnostic purposes.
In the last decade, a series of critical discoveries have clearly demonstrated that OC is a highly heterogeneous disease with a wide spectrum of distinct clinical and molecular entities.4 Generally, epithelial ovarian neoplasms can be divided into three major groups: borderline, Type I and Type II tumors. Among them, Type I tumors are genetically stable, carrying a distinct set of frequently mutated genes such as BRAF, KRAS, CTNNB1, and PTEN. On the other hand, Type II tumors are the most common type of OC and comprise high-grade serous (HGS) tumors, primarily responsible for the low survival rate.4 The rapid development of high-throughput omics techniques including genomics, transcriptomics, proteomics, and metabolomics has dramatically extended our understanding on systems biology of OC. The Cancer Genome Atlas (TCGA) performed both genomic and transcriptomic characterization of ovarian high-grade serous carcinoma (HGSC) and identified the critical role of mutations in the TP53, BRCA1 and BRCA2 genes.5 Other key findings were also determined including extensive DNA copy alterations and CCNE1 aberrations in HGSC. Moreover, further analysis of genomic data from the TCGA consortium and the integration of microRNA and mRNA expression profiles associated with HGS were conducted to improve the statistical association with patient outcome and identify candidate microRNA targets.6,7
Besides, proteomics and metabolomics have recently emerged as powerful technologies for the identification of cancer-specific proteins and metabolites, which provides potential biomarkers for early diagnosis of OC and expands the insights of their pathophysiological mechanisms. Proteomics allows detecting both qualitatively and quantitatively thousands of proteins and/or peptides in large clinical studies. Coscia et al employed quantitative proteomics on FFPE tumor samples from 25 chemotherapy-naive patients with advanced-stage HGSOC.8 The results indicated that cancer/testis antigen 45 (CT45) enhances chemo-sensitivity in metastatic OC, which plays a possible role in the DNA damage response. Importantly, the expression of CT45 may improve the treatment efficacy of immunotherapy or platinum-based chemotherapy for patients with advanced-stage OC. Moreover, Tajmul et al used fluorescence-based 2D-DIGE coupled with MALDI/TOF-MS to profile the salivary protein signatures for non-invasive detection of OC.9 Forty-four significantly different expressed proteins (p < 0.05) between cancer patients and healthy controls were determined, three of which including lipocalin-2, indoleamine-2, 3-dioxygenase1 and S100A8 were further validated using Western blotting and ELISA. Their work indicated that salivary proteomics might be exploited as a diagnostic biomarker for the screening of OC after further large-scale validation. Another powerful technology is metabolomics, aiming to simultaneously detection of small molecule metabolites in biological specimens. Likewise, it holds great potential for discovering potential biomarkers for early diagnosis of the disease, and sheds insight into key regulatory metabolic pathways involved in its pathophysiology, thus leading to new therapeutic strategies. For example, Zeleznik et al conducted a prospective analysis of circulating plasma metabolomics and ovarian cancer risk and revealed that an important metabolite, pseudouridine, may be a novel risk factor for OC.10 And meanwhile, TAGs were also determined to play important roles especially on rapidly fatal tumors.
Recently, phosphoproteomics has been an emerging tool in the study of cancer for the discovery of potential biomarkers and therapy targets.11 Protein phosphorylation is considered to be one of the most abundant post-translational modifications (PTMs), acting as a key regulator of a series of subcellular processes including proliferation, apoptosis and tumorigenesis. In order to obtain a global view of protein phosphorylation events, here we employed Ti4+-IMAC coupled with nano-LC-MS/MS technology to comprehensively characterize the phosphoproteomics profiles of OC patients and controls. We aimed to not only find out potential biomarkers and therapy options for OC but also provide novel insights into its pathophysiological mechanism.
Materials and Methods
Subjects
The present study was approved by the local ethics committee of Peking Union Medical College Hospital (PUMCH) and was conducted in accordance with the Declaration of Helsinki, while written informed consent document for each participant was obtained. Totally 20 women were recruited, including 10 patients all with epithelial ovarian cancer (EOC) and 10 matched patients without OC as the controls. Their clinical characteristics were shown in Table S1. All OC patients were defined as serous histological type (type II, G3) without LNM. And meanwhile, their FIGO characterizations were also shown, mostly ranging from stage II to IV. All patients with OC had been diagnosed without any disease of metabolic, kidney, liver and pelvic inflammatory, as well as any other cancers. Additionally, 10 matched patients in the control group had no history of OC and mainly had hysteromyoma instead. All patients had undergone gynecologic surgery and their diagnosis had been validated by histopathologic examinations before sampling.
Sample Preparation and Pretreatment for Phosphoproteomics Analysis
All collected tissue samples were ovarian surface epithelium and immediately stored at −80°C prior to further analysis. 5 µL samples were diluted with lysis buffer containing 8M Urea (Sigma, MO, USA), 1 mM EDTA, 1 mM PMSF, 100 mM Tris-HCL (Sigma, MO, USA, pH 8.5); then, centrifuged at 15000 g for 15 min at 4°C to remove the sediment. The concentration of protein was measured according to the BCA protein assay kit (Bi Yuntian, Shanghai, China). For phosphoproteomics analysis, the extracted proteins were further reduced for 60 min at 35°C with 10 mM dithiothreitol and alkylated with 50 mM iodoacetamide for 40 min at room temperature in the dark. Subsequently, protein digestion was conducted by the filter-aided sample preparation (FASP) protocol with trypsin (Promega, Madison, WI) in 100 mM NH4HCO3 (Sigma, MO, USA).12 The concentration of peptide was determined by NanoDrop 2000 instrument (Thermo Scientific, USA) at an absorbance of A280 nm. Moreover, phosphopeptide enrichment was carried out by Ti4+-IMAC as previously reported.13 Phosphopeptides were finally diluted from the beads by 10% ammonium hydroxide and dried by vacuum centrifugation.
Nano-HPLC-MS/MS Analysis
Afterwards, the peptides were diluted with 10 µL solvent A (water with 0.1% formic acid) and further analyzed using on-line nanospray LC-MS/MS on an Orbitrap Fusion lumos connected to an EASY-nLC system (Thermo Scientific, MA, USA). For LC-MS/MS analysis, 5 µL peptide sample was loaded onto the trap column (Thermo Scientific Acclaim PepMap C18, 100 µm x 2 cm) at a flow rate of 5 µL/min and subsequently separated on the analytical column (Acclaim PepMap C18, 75 µm x 15 cm) using a 120-min non-linear gradient, ranging from 3% solvent B (ACN with 0.1% formic acid) to 32% solvent B. The column flow rate was 300 nL/min. The electrospray voltage of 2.1 kV relative to the inlet of the mass spectrometer was used. The mass spectrometer was operated under data-dependent acquisition mode, and automatically switched between MS and MS/MS mode in 3.5 s cycle. MS1 mass resolution was set as 120 K with m/z 350–1550, MS/MS isolation window was set 1.6 Da, and MS/MS resolution was set as 30 K under HCD mode with a collision energy was 32%. The dynamic exclusion was set as n=1, and the dynamic exclusion time was 30 s, AGC target was 5e4, max injection time was 120 ms.
Data Analysis
MaxQuant (version 1.5.2.8) was employed here for all data processing. The data were searched against a UniProt human proteome fasta file amended to included common contaminants. A decoy database containing reverse sequences was used to estimate false discovery rates and set the false discovery rate at 1%. Default MaxQuant parameters were used with the following adjustments: Phospho (STY) was added as a variable modification. Match between runs was selected. We obtained the high confidence phosphosite with the following threshold: a PTM site localization probability of 0.75, an Andromeda score of 40 and an Andromeda delta score of 6. The intensity of phosphosite was further preprocessed by metaX. Phosphosites with missing values were imputed with the minimum value to further improve the data quality. Intensity of each phosphosite was normalized with variance stabilizing normalization method. The following criteria were adopted for screening differentially expressed phosphosite: fold change ≥ 1.5 or ≤ 0.67 and p value ≤ 0.05. Here, p values were adjusted using Bonferroni corrections. Functional annotations were further performed by BLAST these proteins against the GO (http://www.geneontology.org/), KEGG (http://www.genome.jp/kegg/pathway.html), COG (http://www.ncbi.nlm.nih.gov/COG/) and eggNOG (http://eggnogdb.embl.de/). Meanwhile, we employed WoLF PSORT to predict protein subcellular localization and used AnimalTFDB for transcription factor analysis.
In the GO and KEGG pathway enrichment analysis of differential phosphoproteins, we compared the significant phosphoproteins to all of the identified proteins, as a background, and the hypergeometric test formula used in these analyses was as follows:
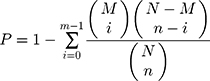
where N is the number of GO/KEGG pathway entries in all identified proteins; n is the number of GO/KEGG pathway entries that represent significant differentially abundant proteins; M is the number of GO/KEGG pathway entries that can be matched to all identified proteins; and m is the number of GO/KEGG pathway entries that can be matched to a significant differentially expressed protein. If the p-value of the hypergeometric test was less than 0.05, the differential protein was significantly enriched in the GO/KEGG pathway entry.
In addition, a multi-omics data analysis tool, OmicsBean (http://www.omicsbean.com), was used in the present study for protein–protein interaction network analysis. P-values < 0.05 were considered to be significantly enriched KEGG pathways.
Western Blot Analysis
For Western blot analysis, extracted proteins were separated by sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) and electroblotted onto polyvinylidene difluoride (PVDF) membranes.14 The antibodies used here were: Rabbit anti-PRKCQ (phosph S695) antibody (Cat. # BS-5584R, Bioss), rabbit anti-CDK2 (phospho T14) antibody (Cat. # ab68265, Abcam), and mouse anti-β-actin antibody (Cat. # GB12001, Servicebio). The final films were photographed and analyzed by using an AlphaEaseFC software (Alpha Innotech). β-actin was used as the loading control.
Results
Phosphoproteomic Profiling of Human Ovarian
To comprehensively analyze the phosphoproteomic characterization for human ovarian, we took advantage of the label-free method for 10 samples from the patients with OC (Table S1, OC1-OC5) and the controls (Table S1, OV1-OV5). A total of 9619 different phosphopeptides (10,934 unique phosphorylation sites) mapped to 3920 proteins were identified across the test 10 samples, which contained 91.5% phosphoserine sites, 8.2% phosphothreonine sites and 0.3% phosphotyrosine sites (Figure 1). Moreover, 83.8% singly phosphorylated phosphopeptides, 14.3% doubly phosphorylated phosphopeptides and 1.9% phosphopeptides phosphorylated at more than three sites were included in the detected 9619 phosphopeptides. In addition, most of the 3920 proteins had a large number of phosphosites. Especially, more than 10 phosphorylated proteins had nearly four thousands of phosphosites to perform various biological functions. The identified 3920 proteins were further annotated according to multiple databases including COG, GO, and KEGG database. The publicly available program WolfPsort was also employed here for subcellular localization prediction. As shown in Table S2, the majority of 3920 proteins distributed predominantly in the nucleus and cytoplasm space. Finally, 2140 transcription factors and 193 kinases were identified among those proteins detected in OC tissues (Tables S3 and S4).
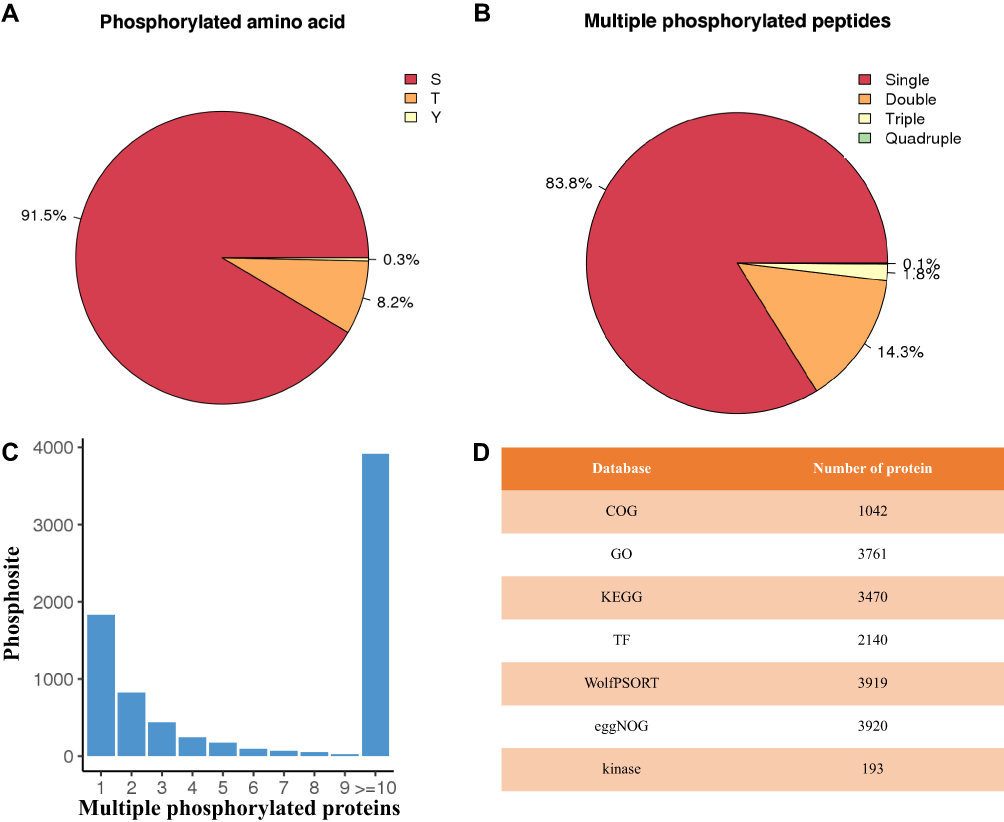 |
Figure 1 Summary of the phosphoproteomic profiling. (A) The percentages of phosphoserines, phosphothreonines and phosphotyrosines. (B) The distribution of phosphopeptides. (C) The number of phosphosite for phosphorylated proteins. (D) The number of phosphorylated protein annotated according to multiple databases including COG, GO, and KEGG database. |
Phosphoproteomic Characterization in Patients with Ovarian Cancer
To overview the relationship between patients with OC and the controls, we employed pearson correlation analysis and principal component analysis (PCA) based on the intensities of each phosphosite. As shown in Figure 2A, the correlation coefficients between patients with OC ranged from 0.65 to 0.82, the average of which seemed to be higher than those between patients with OC and the controls, but lower than those between the controls. On the other hand, PCA score plots revealed that the samples from the patients with OC were tightly clustered together, but to some extent, separated from the controls (Figure 2B). The results here suggested that patients with OC had a significantly distinct phosphoproteomic profiling. Further statistical analysis indicated 722 out of 10,934 phosphorylated sites that corresponded to 534 proteins were significantly different (fold change ≥ 2, p < 0.01) (Figure 3 and Table S5). Of these phosphosites, 233 sites exhibited significantly greater phosphorylation in patients with ovarian cancer, while 489 phosphorylated sites exhibited less phosphorylation in patients with ovarian cancer than the controls.
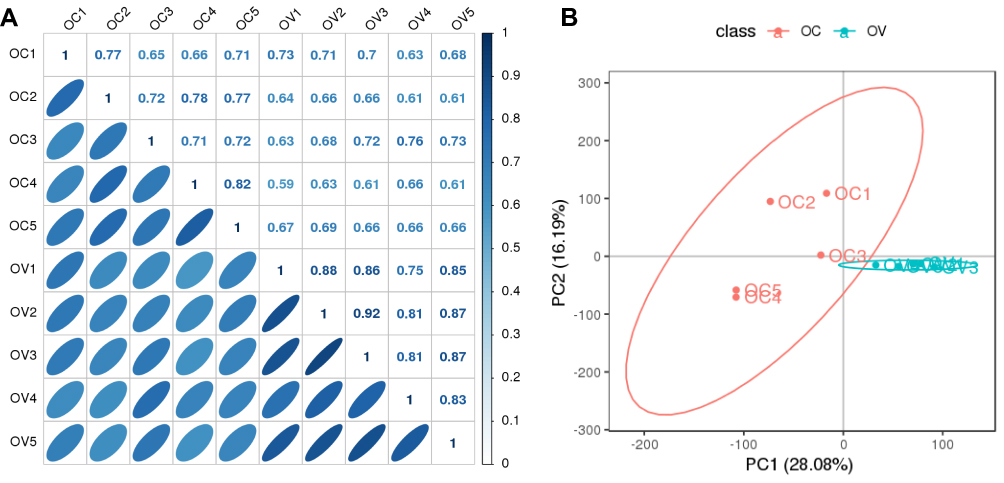 |
Figure 2 An overview of the relationship between patients with OC and the controls. (A) Pearson correlation analysis between those two groups. (B) PCA scores plot generated from the phosphorylation sites across all detected samples. |
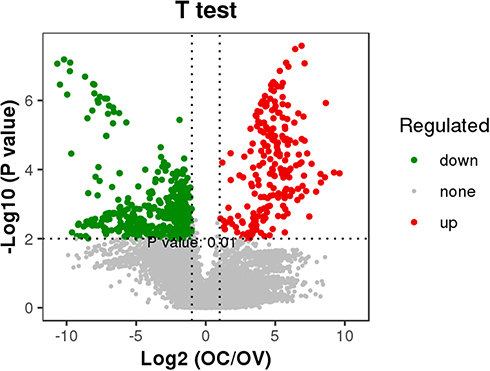 |
Figure 3 Volcano map of significantly different phosphorylated sites in OC patients. p values were adjusted using Bonferroni corrections. |
Functional Annotation of the Differentially Phosphorylated Proteins
Gene ontology-based functional enrichment analysis of 534 differentially phosphorylated proteins was firstly performed to identify their possible functional significance. The results showed that these phosphorylated proteins were significantly enriched in 99 terms from the Component Ontology, 63 terms from the Function Ontology, and 326 terms from the Process Ontology (Tables S6–S8). According to RichFactor values, the top 20 most significantly enriched GO terms were shown in Figure S1, including cellular component organization, cellular component organization or biogenesis, protein binding and binding. Moreover, the left 16 GO terms of the cellular component were also included, such as non-membrane-bounded organelle, intracellular non-membrane-bounded organelle, nuclear lumen and so on. To better understand metabolic pathways activated in the pathogenesis of OC, pathway enrichment analysis were further carried out. On one hand, all 534 differentially phosphorylated proteins were involved in 41 pathways, including signal transduction, cancers: overview, an infectious disease: viral (Figure S2). On the other hand, six pathways were finally determined to be significantly enriched: fructose & mannose metabolism, systemic lupus erythematosus, arrhythmogenic right ventricular cardiomyopathy (ARVC), spliceosome, RNA transport, and tight junction (Figure 4). Regarding the subcellular localization of 534 differentially phosphorylated proteins, 59.6% were localized in the nucleus, and 21.9% were localized in the cytoplasm (Figure S3). Moreover, among those differentially phosphorylated proteins, 83 transcription factors were included, most of which were transcription cofactors, zf-C2H2, and chromatin remodeling factors (Figure S4). Additionally, 29 kinases were also included in the differentially phosphorylated proteins, belonging to eight groups including AGC, CMGC, APK, and STE (Table S9). The top two groups were AGC, which mainly consists of protein kinases A, C and N, and CMGC, which mostly contains CDK kinases and DYRK families. Moreover, 12 kinases were with up-regulated sites, while the left 17 kinases were with down-regulated sites.
 |
Figure 4 Pathway enrichment analysis of significantly different phosphorylated sites in OC patients. |
Protein–Protein Interaction Network Analysis of Significantly Changed Kinases
Kinases play central roles in various cellular signaling processes, and dysregulated kinase signaling has been observed in cancer.15 Therefore, we accordingly employed protein–protein interaction network analysis of significantly changed kinases to provide insights into the regulatory pathways involved in the developmental and physiological processes underlying OC. Fifty-three significantly enriched pathways were determined (Table S10), the top 10 of which are shown in Figure 5, involving 16 associated kinases. Obviously, kinases including PRKCA, CDK2, and EGFR belonging to several important families such as PKC, CDK, and EGFR were connected to these cancer-related pathways such as prostate cancer, bladder cancer, and non-small cell lung cancer. Signaling pathways including MAPK, ErbB, and GnRH were also included in the interaction network. Additionally, different kinases belonging to the same family may change in the opposite direction. For instance, in the PKC family, PRKCA and PRKACB were significantly down-regulated in OC patients, while PRKCD and PRKCQ were significantly up-regulated. The two kinases EGFR and ERBB2, all belonging to the EGFR family, also displayed conversely regulatory roles in OC pathogenesis.
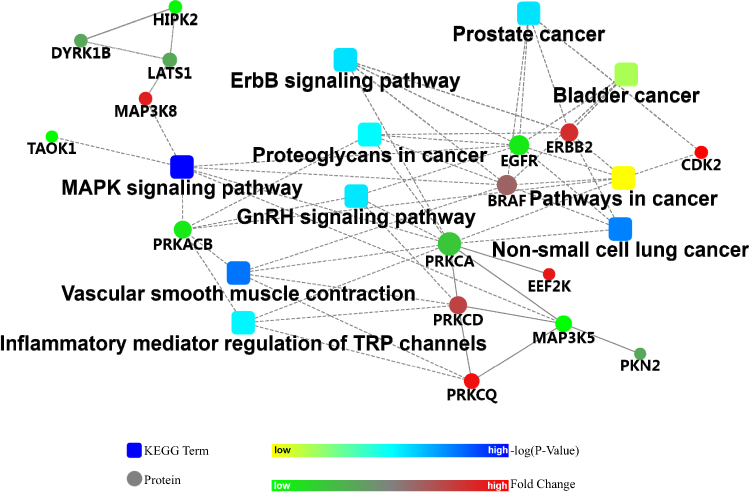 |
Figure 5 The top 10 significantly enriched pathways according to protein–protein interaction network analysis of 29 significantly different kinases. |
Validation of the Expression Levels of p-CDK and p-PRKCQ
To confirm the expression levels of those significantly changed proteins, two important kinases, p-CDK2 (T14) and p-PRKCQ (S695) were chosen for Western blot analysis in the other group of ovarian tissue samples (Table S1) using commercially available antibodies. As shown in Figure 6, p-CDK2 (T14) and p-PRKCQ (S695) had significantly higher levels in OC patients than those in the controls (p = 0.015 and p =0.016, respectively). The results here were obviously consistent with the findings in our phosphoproteomic study.
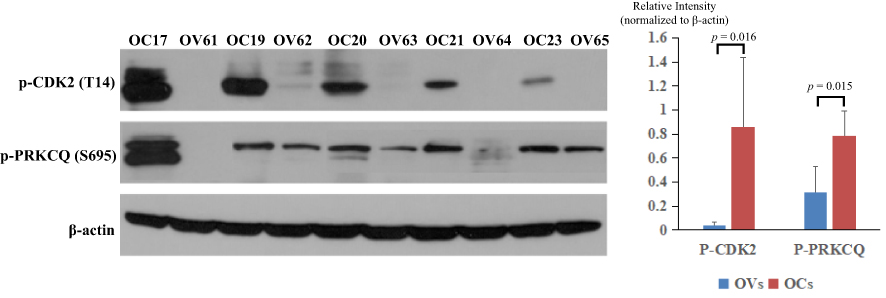 |
Figure 6 Validation of the expression levels of p-CDK2 (T14) and p-PRKCQ (S695). OCs represented patients with OC (OC17, OC19, OC20, OC21, and OC23), while OVs represented the controls (OV61, OV62, OV63, OV64, and OV65). |
Discussion
OC remains one of the top 10 cancers worldwide with a high mortality rate, highlighting the importance of understanding its pathogenesis given no sufficient diagnostic methods and few treatment options available. Therefore, to comprehensively reveal the molecular characterizations of OC, herein we employed Ti4+-IMAC coupled with nano-LC-MS/MS technology to perform phosphoproteomics analysis on tumor tissue samples and matched samples that served as the controls. The data revealed key phosphorylated sites, phosphorylated proteins and regulatory signal/metabolic pathways that are involved in OC pathogenesis, provided potential biomarkers for early diagnosis, and expanded the current knowledge of its physiopathology.
Phosphoproteomics has been demonstrated to be a very promising technology for the identification of diagnostic biomarkers and novel therapeutic targets for cancer. The success of phosphoproteomics especially relies on efficient phosphopeptide enrichment prior to mass spectrometric (MS) analysis.16 Among those selected methods, TiO2 and Ti4+-IMAC are typically applied to enrich phosphorylated peptides for phosphoproteomics analysis.17 Pinkse et al once revealed that TiO2 has a strong affinity for phosphopeptides and thus indicated its high potential in the field of phosphoproteomics.18 Moreover, several studies indicated that Ti4+-IMAC for enriching phosphopeptides could achieve a greater depth of coverage, with high specificity and very large dynamic ranges in large-scale phosphoproteome analysis.19,20 Particularly, Ti4+-IMAC was determined to be the most efficient when systematically compared with some developed isolation methods such as TiO2, ZrO2 Fe3+-IMAC, and Zr4+-IMAC for their specificity and efficiency.19 In the present study, we took advantage of the powerful tool, Ti4+-IMAC coupled with nano-LC-MS/MS, for an in-depth understanding of the biological functions of phosphoproteomes involved in OC development. To the best of our knowledge, there have been limited phosphoproteomics studies on OC by using the Ti4+-IMAC method. Francavilla et al recently employed TiO2-based chromatography for phosphoproteomics to report epithelial ovarian cancer signatures and 7658 distinct phosphorylation sites were determined from 13 independent patient samples.21 Here we identified 10,934 unique phosphorylation sites across the 10 tissue samples, much more than that in the previous study by Francavilla et al.21 Obviously, our phosphoproteomics study represented a larger number of phosphorylation sites in human OC tissues, corroborating the power of this method to reveal the molecular characteristics of the syndrome.
Altered metabolism in cancer cells, especially the Warburg effect, is well established as a hallmark of tumors.22 It is characterized by an increase in glucose uptake and lactate production, which is often accompanied by a decrease in oxidative phosphorylation.23 The aerobic glycolysis in tumor supports high energetic and biosynthetic demands to promote cell growth, proliferation, and metastasis. Although the pathway of glycolysis/gluconeogenesis was not significantly enriched in our study, another glycolysis-related pathway, fructose and mannose metabolism, was found to be significantly enriched based on 534 differentially phosphorylated proteins. Four glycolytic enzymes (PFKFB2, PFKL, ALDOA, and TPI1) mapped to fructose and mannose metabolism were significantly changed in OC, which may play important roles in glycolysis and further promote ovarian tumor growth. For example, PFKFB-2 is the cardiac isoform of the most critical glycolytic regulator, phosphofructokinase-2, while the PFKFB2 gene has determined to be overexpressed, ranking in the top 33% according to the TCGA database with more than 300 ovarian cancers.24 Meanwhile, knockdown of PFKFB2 could induce ovarian cancer cell growth inhibition and may enhance sensitivity to paclitaxel-based chemotherapy in OC patients. The results here revealed that the phosphorylation level of PFKFB2 on the Ser466 residue was much higher in OC patients than the controls, and its high activity probably further promotes glycolysis in ovarian cancer cells. Likewise, ALDOA plays an important role in glycolysis and keeping the maintenance of glucose homeostasis.25 We found a down-regulation of phosphorylated ALDOA protein in OC, though a large number of studies have reported high ALDOA expression in other various types of tumors including osteosarcoma, lung cancer, oral squamous cell carcinoma, and hepatocellular cell carcinoma. Additionally, other two significantly down-regulated enzymes, PFKL and TPI1, were also reported to be involved in targeting glucose metabolism of human OC cells by ABT737.26
Because of their important roles in cell growth and proliferation, these four glycolytic enzymes and the corresponding pathway may be promising targets for treating ovarian cancer. Besides, 29 differentially expressed kinases were also identified in OC patients, which are key phosphoproteins involved in various cellular processes including signal transduction.27 Importantly, dysregulated kinase signaling usually occurs in many diseases including cancer and thus targeting specific kinases is expected of particular interest in cancer therapy. Castillo et al performed human testis phosphoproteome and indicated kinases as potential targets in spermatogenesis and testicular cancer.28 As respect for OC, certain kinases including CDK7 have been identified to be potential players in EOC development and highly associated with poor survival.21,29 In the present study, 29 kinases belonged to 22 kinase families were found to be associated with OC development, some of which were previously reported including CDK2, EGFR, MAP3K5, BRAF, and PKCa.21,29 Four well-known kinase families such as PKC, DYRK, CDK and EGFR were also included and were expected to play crucial roles in ovarian tumor growth. For example, Zhang et al revealed the PKC family plays a key regulatory role in various cancer-associated signal transduction pathways and indicated PKCι as a potential oncogene in ovarian carcinoma.30 Protein–protein interaction network analysis of those 29 kinases also suggested that 16 key kinases could modulate key signaling pathways and regulate tumor proliferation. Notably, different kinases belonging to the same family changed in the opposite direction, indicating their different roles in the physiological processes underlying OC.
There were still some deficiencies in this study. Firstly, fallopian tube epithelium (FTE) and ovarian surface epithelium (OSE) were both considered candidates for the cell-of-origin of HGSOC.31 Here, no samples of FTE were used for the comparison to high-grade serous carcinoma. It would be more accurate to parallelly use both FTE and OSE as the controls. Secondary, Ti4+-IMAC beads do have high specificity and large dynamic ranges for the enrichment of various types of phosphopeptides without bias. However, because of its lower endogenous frequency of occurrence, the analysis of tyrosine phosphorylation still poses a larger challenge than threonine and serine phosphorylation.32 Therefore, other complementary methods such as immunoprecipitation (IP) should be employed to selectively probe tyrosine phosphorylation levels. Thirdly, the number and types of samples were limited. In the next steps, extensive validation of our findings on phosphoproteomics changes would be performed in a larger scale study that features adequate amounts of ovarian samples. Targeted strategies including parallel reaction monitoring assay are supposed to be used to demonstrate the changes of those important proteins and their phosphorylated forms. In addition, we would deeply reveal the biological roles of those important phosphorylated proteins involved in tumorigenesis of ovarian cancer according to gain and loss of function experiments in future work.
Conclusion
Altogether, the present study comprehensively revealed the molecular characteristics in relation to OC and identified significantly distinct phosphorylated sites and phosphorylated proteins underlying the syndrome. The results here dissected regulatory elements (especially kinases) and pathways involved in cancer-related pathways including fructose & mannose metabolism, MAPK, ErbB, and GnRH signaling pathway. These findings may not only provide potential biomarkers and novel therapeutic targets for OC but also bolstered our knowledge on the physiopathology of this syndrome.
Data Sharing Statement
All the raw mass spectrometry data in the present study have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD016183.
Acknowledgments
This work was supported by the Funds for Chinese Academy of Medical Sciences Initiative for Innovative Medicine (CAMS-2017-I2M-1-002), National Natural Science Foundation of China (81960865), Jiangxi provincial department of science (Grant No. 20171BBG70119 and 20181BAB205047), and Health & Family Planning Commission of Jiangxi Province (Grant No. 20203533).
Disclosure
The authors declared no conflicts of interest in this work.
References
1. Cabasag C, Arnold M, Bray F, Soerjomataram I. Incidence trends and age-period cohort effect on ovarian cancer in high-income countries. Rev Epidemiol Sante Publique. 2018;66:254. doi:10.1016/j.respe.2018.05.052
2. Torre LA, Trabert B, DeSantis CE, et al. Ovarian cancer statistics, 2018. Cancer J Clin. 2018;68(4):284–296. doi:10.3322/caac.21456
3. Jacobs IJ, Menon U, Ryan A, et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet. 2016;387(10022):945–956. doi:10.1016/S0140-6736(15)01224-6
4. Kopper O, de Witte CJ, Lõhmussaar K, et al. An organoid platform for ovarian cancer captures intra-and interpatient heterogeneity. Nat Med. 2019;25(5):838–849. doi:10.1038/s41591-019-0422-6
5. Zhang H, Liu T, Zhang Z, et al. Investigators. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 2016;166(3):755–765. doi:10.1016/j.cell.2016.05.069
6. Yang JY, Yoshihara K, Tanaka K, et al. Predicting time to ovarian carcinoma recurrence using protein markers. J Clin Invest. 2013;123(9):3740–3750. doi:10.1172/JCI68509
7. Creighton CJ, Hernandez-Herrera A, Jacobsen A, et al. The cancer genome atlas research network. Integrated analyses of microRNAs demonstrate their widespread influence on gene expression in high-grade serous ovarian carcinoma. PLoS One. 2012;7(3):34546. doi:10.1371/journal.pone.0034546
8. Coscia F, Lengyel E, Duraiswamy J, et al. Multi-level proteomics identifies CT45 as a chemosensitivity mediator and immunotherapy target in ovarian cancer. Cell. 2018;175(1):159–170. doi:10.1016/j.cell.2018.08.065
9. Tajmul M, Parween F, Singh L, et al. Identification and validation of salivary proteomic signatures for non-invasive detection of ovarian cancer. Int J Biol Macromol. 2018;108:503–514. doi:10.1016/j.ijbiomac.2017.12.014
10. Zeleznik OA, Eliassen AH, Kraft P, et al. A prospective analysis of circulating plasma metabolomics and ovarian cancer risk. BioRxiv. 2019;654962. doi:10.1101/654962
11. Selvan LDN, Danda R, Madugundu AK, et al. Phosphoproteomics of retinoblastoma: a pilot study identifies aberrant kinases. Molecules. 2018;23(6):1454. doi:10.3390/molecules23061454
12. Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6(5):359–362. doi:10.1038/nmeth.1322
13. Zagorac I, Fernandez-Gaitero S, Penning R, et al. In vivo phosphoproteomics reveals kinase activity profiles that predict treatment outcome in triple-negative breast cancer. Nat Commun. 2018;9(1):3501. doi:10.1038/s41467-018-05742-z
14. Zhao S, Feng J, Li C, et al. Phosphoproteome profiling revealed abnormally phosphorylated AMPK and ATF2 involved in glucose metabolism and tumorigenesis of GH-PAs. J Endocrinol Invest. 2019;42(2):137–148. doi:10.1007/s40618-018-0890-4
15. Cohen P. The role of protein phosphorylation in human health and disease. The Sir Hans Krebs Medal Lecture. Eur J Biochem. 2001;268(19):5001–5010. doi:10.1046/j.0014-2956.2001.02473.x
16. Piersma SR, Knol JC, de Reus I, et al. Feasibility of label-free phosphoproteomics and application to base-line signaling of colorectal cancer cell lines. J Proteomics. 2015;127:247–258. doi:10.1016/j.jprot.2015.03.019
17. Zou X, Jie J, Yang B. Single-step enrichment of N-Glycopeptides and phosphopeptides with novel multifunctional Ti4+-immobilized dendritic polyglycerol coated chitosan nanomaterials. Anal Chem. 2017;89(14):7520–7526. doi:10.1021/acs.analchem.7b01209
18. Pinkse MW, Uitto PM, Hilhorst MJ, Ooms B, Heck AJ. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal Chem. 2004;76(14):3935–3943. doi:10.1021/ac0498617
19. Zhou H, Ye M, Dong J, et al. Specific phosphopeptide enrichment with immobilized titanium ion affinity chromatography adsorbent for phosphoproteome analysis. J Proteome Res. 2008;7(9):3957–3967. doi:10.1021/pr800223m
20. Hou C, Ma J, Tao D, et al. Organic-inorganic hybrid silica monolith based immobilized titanium ion affinity chromatography column for analysis of mitochondrial phosphoproteome. J Proteome Res. 2010;9(8):4093–4101. doi:10.1021/pr100294z
21. Francavilla C, Lupia M
22. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi:10.1126/science.123.3191.309
23. Zheng Z, Xu L, Zhang S, et al. Peiminine inhibits colorectal cancer cell proliferation by inducing apoptosis and autophagy and modulating key metabolic pathways. Oncotarget. 2017;8(29):47619–47631. doi:10.18632/oncotarget.17411
24. Worku T, Bhattarai D, Ayers D, et al. Long non-coding RNAs: the new horizon of gene regulation in ovarian cancer. Cell Physiol Biochem. 2017;44(3):948–966. doi:10.1159/000485395
25. Chang YC, Yang YC, Tien CP, Yang CJ, Hsiao M. Roles of aldolase family genes in human cancers and diseases. Trends Endocrinol Metab. 2018;29(8):549–559. doi:10.1016/j.tem.2018.05.003
26. Xu Y, Gao W, Zhang Y, et al. ABT737 reverses cisplatin resistance by targeting glucose metabolism of human ovarian cancer cells. Int J Oncol. 2018;53(3):1055–1068. doi:10.3892/ijo.2018.4476
27. Yu LR, Issaq HJ, Veenstra TD. Phosphoproteomics for the discovery of kinases as cancer biomarkers and drug targets. Proteomics Clin Appl. 2007;1(9):1042–1057. doi:10.1002/prca.200700102
28. Castillo J, Knol JC, Korver CM, et al. Human testis phosphoproteome reveals kinases as potential targets in spermatogenesis and testicular cancer. Mol Cell Proteomics. 2019;18(Supplement 1):S132–S144. doi:10.1074/mcp.RA118.001278
29. Tong M, Yu C, Zhan D, et al. Molecular subtyping of cancer and nomination of kinase candidates for inhibition with phosphoproteomics: reanalysis of CPTAC ovarian cancer. EBioMedicine. 2019;40:305–317. doi:10.1016/j.ebiom.2018.12.039
30. Zhang L, Huang J, Yang N, et al. Integrative genomic analysis of protein kinase C (PKC) family identifies PKCι as a biomarker and potential oncogene in ovarian carcinoma. Cancer Res. 2006;66(9):4627–4635. doi:10.1158/0008-5472.CAN-05-4527
31. Zhang S, Dolgalev I, Zhang T, et al. Both fallopian tube and ovarian surface epithelium are cells-of-origin for high-grade serous ovarian carcinoma. Nat Commun. 2019;10(1):1–16. doi:10.1038/s41467-019-13116-2
32. Di Palma S, Zoumaro-Djayoon A, Peng M, et al. Finding the same needles in the haystack? A comparison of phosphotyrosine peptides enriched by immuno-affinity precipitation and metal-based affinity chromatography. J Proteomics. 2013;91:331–337. doi:10.1016/j.jprot.2013.07.024
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.