Back to Journals » Drug Design, Development and Therapy » Volume 11
Phenotypic evaluation and in silico ADMET properties of novel arylimidamides in acute mouse models of Trypanosoma cruzi infection
Authors da Silva CF, Batista DDGJ, de Araújo JS, Cunha-Junior EF, Stephens CE, Banerjee M, Farahat AA, Akay S, Fisher MK, Boykin DW, Soeiro MDNC
Received 25 August 2016
Accepted for publication 2 November 2016
Published 3 April 2017 Volume 2017:11 Pages 1095—1105
DOI https://doi.org/10.2147/DDDT.S120618
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Cristiane França da Silva,1 Denise da Gama Jaén Batista,1 Julianna Siciliano de Araújo,1 Edézio Ferreira Cunha-Junior,2 Chad E Stephens,3 Moloy Banerjee,4 Abdelbasset A Farahat,4,5 Senol Akay,4 Mary K Fisher,3 David W Boykin,4 Maria de Nazaré Correia Soeiro1
1Laboratory of Cellular Biology, 2Laboratory of Biochemistry of Trypanosomatids, Fundação Oswaldo Cruz, Rio de Janeiro, Brazil; 3Department of Chemistry and Physics, Augusta University, Augusta, 4Department of Chemistry, Georgia State University, Atlanta, GA, USA; 5Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt
Abstract: Arylimidamides (AIAs), previously termed as reversed amidines, present a broad spectrum of activity against intracellular microorganisms. In the present study, three novel AIAs were evaluated in a mouse model of Trypanosoma cruzi infection, which is the causative agent of Chagas disease. The bis-AIAs DB1957, DB1959 and DB1890 were chosen based on a previous screening of their scaffolds that revealed a very promising trypanocidal effect at nanomolar range against both the bloodstream trypomastigotes (BTs) and the intracellular forms of the parasite. This study focused on both mesylate salts DB1957 and DB1959 besides the hydrochloride salt DB1890. Our current data validate the high activity of these bis-AIA scaffolds that exhibited EC50 (drug concentration that reduces 50% of the number of the treated parasites) values ranging from 14 to 78 nM and 190 to 1,090 nM against bloodstream and intracellular forms, respectively, also presenting reasonable selectivity indexes and no mutagenicity profile predicted by in silico absorption, distribution, metabolism, excretion, and toxicity (ADMET). Acute toxicity studies using murine models revealed that these AIAs presented only mild toxic effects such as reversible abdominal contractions and ruffled fur. Efficacy assays performed with Swiss mice infected with the Y strain revealed that the administration of DB1957 for 5 consecutive days, with the first dose given at parasitemia onset, reduced the number of BTs at the peak, ranging between 21 and 31% of decrease. DB1957 was able to provide 100% of animal survival, while untreated animals showed 70% of mortality rates. DB1959 and DB1890B did not reduce circulating parasitism but yielded >80% of survival rates.
Keywords: Chagas disease, arylimidamides, experimental chemotherapy, in vivo assays
Introduction
American trypanosomiasis or Chagas disease (CD) caused by Trypanosoma cruzi affects >6 million people worldwide, mostly in Latin America.1 Another public health concern is that the prevalence of T. cruzi infection in non-endemic countries such as Australia, Canada, Japan, Spain, and the US is increasing due to global migration of Latin American immigrants unknowingly carrying the infection.2,3
After more than a hundred years since its discovery, this pathology still presents several challenges. The silent chronic morbidities and high mortalities, the prolonged and expensive treatment courses without being effective for the late-stage disease and the current barriers to the access to essential medicines demand urgent attention for this neglected disease.4,5
Only two drugs are available for the treatment of CD: nifurtimox (Nf), a nitrofuran with a fast plasmatic clearance profile (half-life of 3 h),2 and benznidazole (Bz), a nitroimidazole, both of which show variable activity across different T. cruzi strains.6
The occurrence of T. cruzi strains exhibiting a naturally resistant profile to Bz and Nf may be related, at least in part, to ABCG-like transporters, which are overexpressed in several naturally resistant strains.7 Other significant drawbacks in the current medicines for CD are that both drugs require sustained periods of administration (30–60 days) and give low curative rates for the late-chronic stage.8–10
Pentamidine, an aromatic diamidine, has been extensively used as an antiparasitic drug for sleeping sickness and leishmaniasis.8 Several reports have demonstrated its effect toward T. cruzi in vitro and in vivo leading to a reduction in parasitemia levels and providing enhanced mice survival compared to untreated controls.11 Many of its derivatives, especially the arylimidamides (AIAs), show potent efficacy against intracellular parasites such as T. cruzi.12
AIAs have extraordinary biological action toward different trypanosomatids, both in vitro13–16 and in vivo.17–19 An important structural characteristic of AIAs is that the imino group is linked to the “anilino” nitrogen, while in classical amidines, it is directly attached to an aryl ring.15 Recent findings demonstrated that bis-AIAs are more effective than mono-AIAs and that some of these bis-AIAs are able to reduce the parasitemia in addition to protecting against mortality induced by acute infection with T. cruzi.18–21 In a previous report, our group demonstrated the in vitro efficacy of several bis-AIAs, including DB1867, DB1862 and DB1890B, which exhibited high trypanocidal activity against bloodstream and intracellular forms of T. cruzi superior to that of Bz.22
The excellent trypanosomicidal action of these molecules encouraged us to perform further studies to ascertain in vivo safety and potency of these three bis-AIAs by using mesylate salts for two of them (DB1957 and DB1959) in an effort to increase solubility. In this study, the bis-AIAs DB1957, DB1959 and DB1890B were evaluated using an experimental mouse model for acute T. cruzi infection with the goal of identifying novel antiparasitic drug candidates for possible future alternative therapies for CD.
Materials and methods
Compounds
The syntheses of DB1957, DB1959 and DB1890B (Figure 1) were performed according to methods (Scheme S1) previously described.23–25 Bz, purchased from Laboratório Farmacêutico do Estado de Pernambuco (Laboratório Farmacêutico do Estado de Pernambuco), Brazil, was used as the reference drug.18 Stock solutions were prepared in dimethyl sulfoxide (DMSO), with the final concentration for the in vitro experiments never exceeding 0.6%, which did not exert any toxicity toward the parasite or mammalian host cells. For in vivo studies, a stock solution of the AIA compounds was first prepared in DMSO and then diluted using distilled and sterile water. The final concentration of DMSO never exceeded 10%, which did not provide detectable mice toxicity.18,21 Bz was dissolved in sterilized distilled water supplemented with 3% Tween 80, which did not cause any detectable effect on the mice.
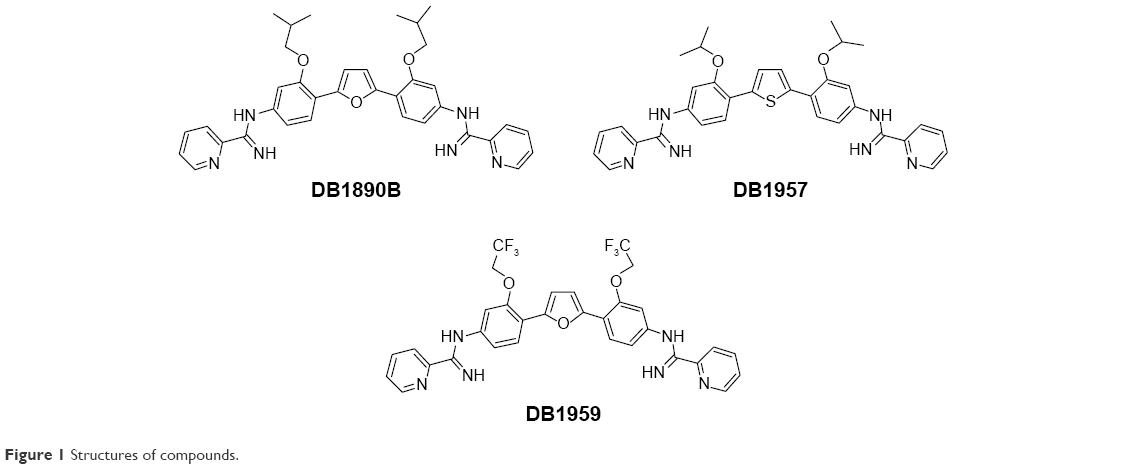 | Figure 1 Structures of compounds. |
Parasites
The Y strain of T. cruzi was used throughout the study. Bloodstream trypomastigote (BT) forms were obtained from T. cruzi-infected Swiss mice at the peak of parasitemia.26 During the analyses using intracellular forms, the parasites lodged within cardiac cell (CC) cultures were employed, as previously reported.20
In silico study of the druglikeness and absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties
In silico analysis of the pharmacokinetics properties (absorption, distribution, metabolism, and excretion [ADME]) and toxicity of the AIAs were evaluated using the pkCSM approach,27 which uses graph-based signatures to predict these parameters. The platform validation was performed using cross-validation scheme and sets of external validation data. pkCSM presents a statistically significant improvement in predictive power in comparison with the methods available.27
CC cultures and cytotoxicity assays
For the evaluation of cytotoxicity and compound activity against intracellular parasites, primary cultures of embryonic CCs were obtained from Swiss mice and purified following the method previously described.26 In order to rule out toxic effects upon mammalian host cells, uninfected CC cultures were exposed to each compound studied at 37°C for 24 and 48 h (1.18–32 μM). Untreated cultures were used as control samples. The cell death rates were measured by the MTT colorimetric assay allowing the determination of LC50 values (compound concentration that reduces 50% of cellular viability).22 All cell cultures were maintained in an atmosphere of 5% CO2 and air, and the assays were run at least three times in duplicates.
Trypanocidal assays
The effect of the compounds against BT was evaluated through assaying 5×106 parasites/mL for 24 h at 37°C in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% of fetal bovine serum, in the presence of serial dilutions of the AIAs (0.001–10.6 μM). Untreated parasites were used as control samples. Parasite death rates were determined by light microscopy using a Neubauer chamber that allowed the direct visualization and quantification of the number of motile and live parasites and allowed EC50 (drug concentration that reduce 50% of the number of the treated parasites) values to be calculated.22
For the analysis of the effect against intracellular parasites, after 24 h of parasite–host cell interaction (ratio 10:1), the infected CC cultures were washed to remove the free parasites and then maintained at 37°C in an atmosphere of 5% CO2 and air in the presence of nontoxic concentrations of each AIA (from 0.043 to 3.5 μM). Untreated infected cultures were used as control. After 48 h, all CC cultures were fixed using Bouin and stained with Giemsa solution. The infective index (II) was used to compare the compound activity calculated by multiplying the percentage of infected cells by the mean number of parasites per infected cell.17 Next, the EC50 values were calculated based on the drug concentration that reduced 50% of the II.
The selectivity index (SI) was calculated as follows: SI = LC50/EC50. The SI was calculated using the data for 24 and 48 h of incubation of the host cells with the three AIAs for BT and intracellular parasites, respectively.
Acute toxicity for mice
Acute toxicity levels were evaluated using female and male Swiss Webster mice (20–23 g) obtained from the animal facilities of the Oswaldo Cruz Foundation (CECAL, Rio de Janeiro, Brazil). Mice were housed per cage and were kept in a conventional animal room at 20°C–24°C under a 12/12-h light/dark cycle. The animals were provided with sterilized water and chow ad libitum. On day 1, one female mouse was treated with each compound by injection via intraperitoneal (ip) using escalating doses every 2 h, starting at 20 up to 200 mg/kg, and thus, the final accumulative dose reached a total of 400 mg/kg. The mice were inspected, 48 h post-final injection, for toxic and sub-toxic symptoms according to OECD guidelines as reported, and at least two assays were conducted with male and female animals.22
Mice infection and treatment schemes
Five male Swiss mice (18–21 g) were housed per cage and were maintained as described earlier. Infection was performed by ip injection of 104 (Y strain) BTs.18 The animals were divided into the following groups: uninfected (noninfected and untreated), untreated (infected with T. cruzi but treated only with vehicle), and treated (infected and treated – ip – with 25 mg/kg/day AIA or orally (po) 100 mg/kg/day Bz). Treated mice received daily dose (0.1 mL via ip) starting at 5 days postinfection (dpi) that corresponded to the parasitemia onset up to 9 dpi (1 day after the parasitemia peak) for all compounds (AIAs and Bz). For Bz treatment, infected mice received 0.2 mL oral dose (po) following the same therapeutic scheme as described earlier.
Parasitemia and mortality rates
Parasitemia was individually checked by direct microscopic counting of parasites in 5 μL of blood, as described earlier.18 Mortality levels were checked until 56 dpi (corresponding to 47 days after the last dose) and expressed as survival rates.20
Biochemical analysis
At the end point (56 dpi), the biochemical analysis of the plasmatic levels of creatine kinase (CK), blood urea nitrogen (BUN), and alanine aminotransferase (ALT) was determined in the CECAL/Fiocruz platform, using Vitros 250 (Ortho Clinical, Johnson & Johnson, Raritan, NJ, EUA) as reported.20
Statistical analysis
Statistical analysis was carried out using an analysis of variance (ANOVA) program with the level of significance set at P≤0.05. The data were representative of a minimum of two experiments run in duplicate.
All procedures were carried out in accordance with the guidelines established and approved by the Fiocruz Committee of Ethics for the Use of Animals (CEUA LW16/14 and CEUA 0099/01).
Results
Previous in vitro data22 showed the excellent activity of three AIAs (DB1867, DB1862, and DB1890B), providing EC50 values of 0.02, 0.06, and 0.013 μM, respectively, against BTs of T. cruzi after 24 h of incubation at 37°C.22 These AIAs did not exhibit toxicity against primary culture CCs and reduced the infection of T. cruzi-infected CCs, yielding EC50 values of 0.058, 0.016, and 0.9 μM, respectively, after 48 h of treatment.22 Thus, due to the low aqueous solubility profile for HCl salts of AIAs previously noted, we decided to explore mesylate salts for two of the compounds studied (DB1957 and DB1959). The pkCSM-calculated solubility for DB1957 (mesylate salt) was greater than that of the corresponding HCL salt: 69.4 vs 8.89 μM, respectively (data not shown). Similar results were observed with DB1959 (data not shown) and its corresponding HCl salt: 188.36 vs 24.43 μM, respectively (data not shown).
Prior moving to in vivo models, the in vitro activity of these three compounds was validated against bloodstream and intracellular forms of T. cruzi (Table 1). The findings confirmed that these AIAs presented good antiparasitic effects on intracellular forms with EC50 values ranging from 0.19 to 1.09 μM, all superior to that of Bz 3.7 μM (Table 1). The AIAs were more effective and selective against the trypomastigotes (BTs) compared to the amastigotes (intracellular forms), exhibiting EC50 values of 0.068, 0.078, and 0.014 μM and SI of 17, 45, and 760 after 24 h of incubation at 37°C for bloodstream forms using DB1957, DB1959, and DB1890B, respectively (Table 1). Statistical analysis demonstrated that all AIAs were more potent than Bz on BT forms (P=0.003, P=0.003, and P=0.0003, respectively).
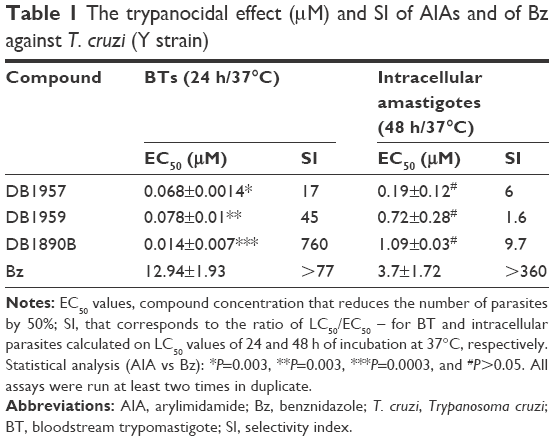 | Table 1 The trypanocidal effect (μM) and SI of AIAs and of Bz against T. cruzi (Y strain) |
Next, in silico analysis of physicochemical parameters and Lipinski’s rule of five (Table S1) besides the ADMET properties of these AIAs were predicted using the pkCSM methodology. The findings (Tables S1 and S2) showed that the three AIAs have quite similar physicochemical profile, with low probability of permeability of Caco-2 cells for DB1890B and DB1959 and a medium probability profile achieved for DB1957. However, all three compounds showed similar predicted permeability profiles (>95%) for human intestinal absorption, which were greater than those for Bz (69%). The high volume of distribution of DB1957 compared to DB1959 and DB1890B may reflect the importance of the thiophene group as the central ring instead of furan. These compounds are predicted to be metabolized by CYP3A4 and are predicted to be inhibitors of the same enzyme. Toxicity predictions (Table S3) suggest that these compounds are expected neither to be mutagenic nor to inhibit hERG1, but all were predicted to inhibit hERG2. DB1890B and DB1959 were not predicted to be hepatotoxic; however, both DB1957 and Bz were predicted to be hepatotoxic. These predictions indicate the need for evaluation of hepatic markers in biochemical analysis of treated animals that was next conducted in the efficacy mouse models.
Prior to this, to avoid toxic doses of the AIAs, preliminary acute toxicity studies were conducted in female and male (data not shown) mice, which displayed similar results. After 48 h of administration of the last dose, only mild toxic events were detected, such as reversible abdominal contractions and ruffled fur (data not shown). Subsequently, the effectiveness of the AIAs was investigated in male Swiss mice inoculated with 104 trypomastigotes (Y strain), using nontoxic doses of the three AIAs (25 mg/kg), via ip. Only animals that presented positive parasitemia were used in these studies. In this experimental acute mouse model that peaks at 8 dpi in the untreated and infected groups, the administration of DB1959 and DB1890B gave no reduction in the parasitemia levels (Figure 2A). However, treatment of the infected mice with DB1957 led to 21%–31% reduction in parasitemia peak (Figure 2A). DB1890B and DB1959 gave 80% survival rates, whereas DB1957 yielded 100% animal survival. The infected and untreated mice groups experienced only 30% survival (Figure 2B). The reference drug used in the same therapeutic scheme resulted in parasitemia suppression and gave mice survival rates of 80% (Figure 2A and B). Statistical analysis demonstrated that all groups demonstrated no different significance (P≤0.05).
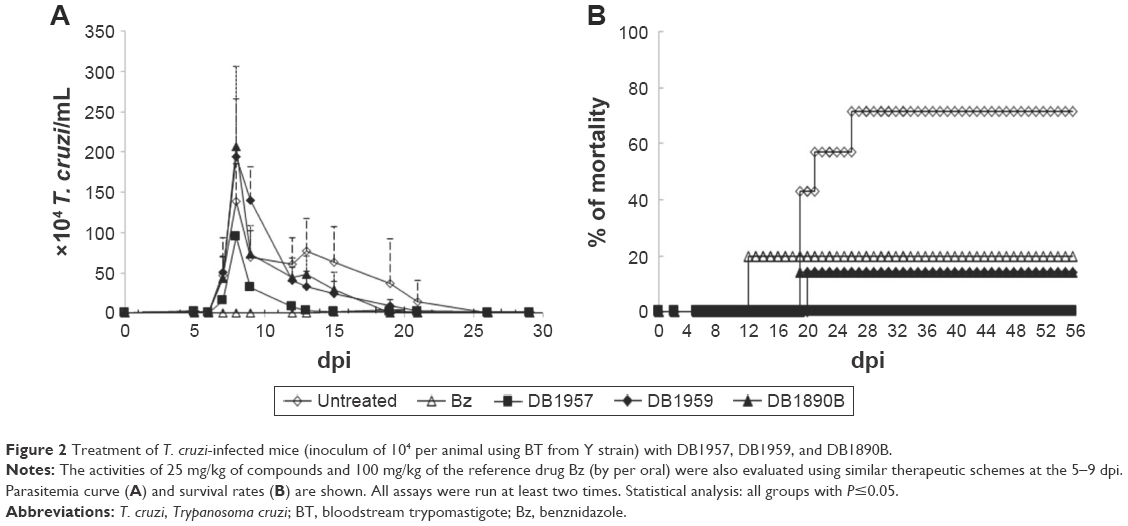 | Figure 2 Treatment of T. cruzi-infected mice (inoculum of 104 per animal using BT from Y strain) with DB1957, DB1959, and DB1890B. |
Biochemical analysis performed at the end point (56 dpi) showed only mild alterations in BUN levels when the mice were exposed to 25 mg/kg of the three AIAs (Table 2). Also, at 56 dpi (46 days after the last drug dose), only DB1890B resulted in a slight increase in the ALT plasma biochemical values, while the other two AIAs did not lead to altered levels (Table 2). Regarding CK measurements, DB1890B showed the higher levels although still under the limits of the reference values and only demonstrated statistical significance compared with DB1959 (P=0.03; Table 2).
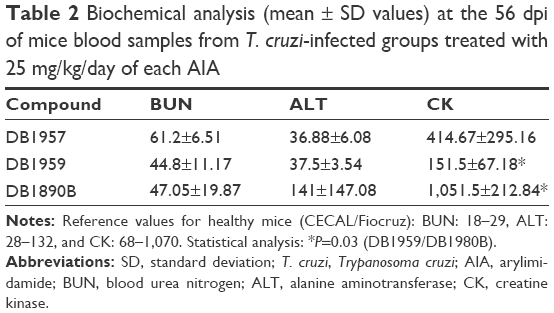 | Table 2 Biochemical analysis (mean ± SD values) at the 56 dpi of mice blood samples from T. cruzi-infected groups treated with 25 mg/kg/day of each AIA |
Discussion
CD is one of the leading causes of infection-induced heart failure in Latin America28 and remains a serious health problem.29 The drugs currently used lead to adverse events that frequently prevent the completion of the therapy regimen of the infected patients.10 Nf is currently produced and used predominantly in Central America,30 and Bz is now being produced only by Fundación Mundo Sano and ELEA Laboratory in Argentina. A new pediatric formulation of Bz is available improving the dose accuracy given to infected children.3 Unfortunately, a recent report from the BENEFIT clinical trial (a prospective, multicenter, randomized study involving 2,854 patients with Chagas’ cardiomyopathy who received Bz or placebo [up to 80 days] and followed for a mean of 5.4 years) demonstrated that although Bz reduced the parasitism, it did not alter the cardiopathology clinical progression.31 Another important challenge is that, despite current recommendations, <1% of infected individuals are treated with the available drugs.32 Thus, there is an urgent need for new safer and more selective drugs and new strategies for treatment of this illness, especially for the chronic late phase.5,31
Many AIAs such as DB76618 and DB1831,20 among others,12 have superior in vitro trypanocidal activity to that of the related classical aromatic amidines as well as the reference drug for CD. Our previous study of the in vitro biological efficacy of seven novel AIAs on BTs and intracellular amastigotes of T. cruzi gave very promising results.22 In view of these positive data, three AIAs were advanced to in vivo studies. In addition, theoretical analysis of the druglikeness was predicted by in silico analysis. The development of computational approaches to estimate pharmacokinetic and toxicity properties facilitates the progression of novel drug candidates.33 The prediction of metabolism or inhibition by the CYP3A4 indicates the need to perform further studies of interaction with drugs that inhibit the enzymes such as ritonavir and lopinavir.34 These compounds are predicted neither to be mutagenic nor to inhibit hERG I; however, interaction with hERG2 is predicted. It is interesting to point out that these predictions are in line with the experimental data on cardiomyocytes, since hERG2 is not expressed in the heart.35
In vivo studies demonstrated a low acute toxicity profile for these three compounds using both healthy female and male mice, which only displayed mild and reversible effects up to 400 mg/kg. Despite showing in vitro potency, DB1890B, DB1957, and DB1959 did not suppress the parasitemia peak when nontoxic doses were used by the ip route (25 mg/kg), while the reference drug (dose of 100 mg/kg) suppressed it completely. It is worthy to note that DB1890B and DB1959 provided ≥80% mice survival rates, similar to that of Bz, while untreated animals presented with 30% survival. More importantly, DB1957, which had the highest predicted VDss, showed a 21%–31% reduction in peak parasitemia and provided a 100% survival rate, which is consistent with the control of the replication of the intracellular parasitism into the tissues.
As previously discussed, a novel anti-T. cruzi entity must be highly active against intracellular parasites (the proliferative forms in the vertebrate hosts) and bloodstream forms.12,36,37 The three AIAs studied here range from 2.7 to 77 times less active against intracellular amastigotes than the bloodstream forms. It is intriguing that DB1957 is the most active compound in vivo (regarding survival rates of 100%) and more potent (0.19 μM) against the intracellular forms. Perhaps, in the future, more attention should be focused on optimization of in vitro activity of target compounds against intracellular amastigotes, which are the multiplying forms present in the host cell cytoplasm that may be distributed to different organs and tissues forming residual parasite nests.
Conclusion
Since the current treatment for CD is far from being ideal, presenting serious drawbacks related to efficacy, especially at the later chronic stage, and low safety profile, a compelling need for new candidates for alternative therapy is urgent. In this sense, aromatic amidines represent a promising class of antiparasitic compounds that deserve further optimization and deeper pre-clinical analysis.
Acknowledgments
This work was supported by Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ; E-26/102.839/2011; 203636 and 200381), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; 555010/2008-2, 301372/2015-2), Fundação Oswaldo Cruz, Program for Technological Development in Tools for Health, PAEF/CNPq/Fiocruz, and CAPES (23038.009424/2012-50, 23038008979201/10-1). MDNCS is a research fellow of CNPq and CNE research. The authors thank the Program for Technological Development in Tools for Health-PDTIS-Fiocruz for use of its facilities. The present study was supported by grants from, in part by, the Bill & Melinda Gates Foundation through a subcontract with the Consortium for Parasitic Drug Development (to DWB – R01AI64200). The authors also thank Marcos Meuser Batista for his assistance in their experiments and Eduardo Caio Torres-Santos for his assistance in ADMET in silico analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization [webpage on the Internet]. WHO Fact Sheet No 340; 2015. Available from: http://www.who.int/mediacentre/factsheets/fs340/en/. Accessed November 3, 2016. | ||
Barrett MP, Croft SL. Management of trypanosomiasis and leishmaniasis. Br Med Bull. 2012;104:175–196. | ||
Drugs for Neglected Disease initiative (DNDi, 2016) [webpage on the Internet]. Paediatric dosage form of benznidazole (Chagas). Available from: http://www.dndi.org/diseases-projects/portfolio/benznidazole-paedriatric-dosage-form.html. Accessed November 3, 2016. | ||
Hotez PJ, Dumonteil E, Woc-Colburn L, et al. Chagas disease: “the new HIV/AIDS of the Americas”. PLoS Negl Trop Dis. 2012;6:e1498. | ||
Chatelain E. Chagas disease drug discovery: toward a new era. J Biomol Screen. 2015;20(1):22–35. | ||
Teston AP, Monteiro WM, Reis D, et al. In vivo susceptibility to benznidazole of Trypanosoma cruzi strains from the western Brazilian Amazon. Trop Med Int Health. 2013;18(1):85–95. | ||
Zingales B, Araujo RG, Moreno M, et al. A novel ABCG-like transporter of Trypanosoma cruzi is involved in natural resistance to benznidazole. Mem Inst Oswaldo Cruz. 2015;110(3):433–444. | ||
Soeiro MN, Dantas AP, Daliry A, et al. Experimental chemotherapy for Chagas disease: 15 years of research contributions from in vivo and in vitro studies. Mem Inst Oswaldo Cruz. 2009;104(suppl 1):301–310. | ||
Apt W. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Des Devel Ther. 2010;24:243–253. | ||
Maya JD, Orellana M, Ferreira J, Kemmerling U, López-Muñoz R, Morello A. Chagas disease: present status of pathogenic mechanisms and chemotherapy. Biol Res. 2010;43(3):323–331. | ||
Díaz MV, Miranda MR, Campos-Estrada C, et al. Pentamidine exerts in vitro and in vivo anti Trypanosoma cruzi activity and inhibits the polyamine transport in Trypanosoma cruzi. Acta Trop. 2014;134:1–9. | ||
Soeiro MN, Werbovetz K, Boykin DW, Wilson WD, Wang MZ, Hemphill A. Novel amidines and analogues as promising agents against intracellular parasites: a systematic review. Parasitology. 2013;140(8):929–951. | ||
Silva CF, Batista MM, Mota RA, et al. Activity of “reversed” diamidines against Trypanosoma cruzi in vitro. Biochem Pharmacol. 2007;73:1939–1946. | ||
Silva CF, Meuser MB, De Souza EM, et al. Cellular effects of reversed amidines on Trypanosoma cruzi. Antimicrob Agents Chemother. 2007;51(11):3803–3809. | ||
Rosypal AC, Werbovetz KA, Salem M, et al. Inhibition by Dications of in vitro growth of Leishmania major and Leishmania tropica: causative agents of old world cutaneous leishmaniasis. J Parasitol. 2008;94(3):743–749. | ||
Timm BL, da Silva PB, Batista MM, et al. In vitro and in vivo biological effects of novel arylimidamide derivatives against Trypanosoma cruzi. Antimicrob Agents Chemother. 2014;58(7):3720–3726. | ||
Da Silva CF, Batista MM, Batista DG, et al. In vitro and in vivo studies of the trypanocidal activity of a diarylthiophene diamidine against Trypanosoma cruzi. Antimicrob Agents Chemother. 2008;52(9):3307–3314. | ||
Batista DG, Batista MM, de Oliveira GM, et al. Arylimidamide DB766, a potential chemotherapeutic candidate for Chagas’ disease treatment. Antimicrob Agents Chemother. 2010;54(7):2940–2952. | ||
De Araújo JS, Da Silva CF, Batista DG, et al. In vitro and in vivo studies of the biological activity of novel arylimidamides against Trypanosoma cruzi. Antimicrob Agents Chemother. 2014;58(7):4191–4195. | ||
Da Silva CF, Batista DG, Oliveira GM, et al. In vitro and in vivo investigation of the efficacy of arylimidamide DB1831 and its mesylated salt form – DB1965 – against Trypanosoma cruzi infection. PLoS One. 2012;7(1):e30356. | ||
Guedes-da-Silva FH, Batista DG, Meuser MB, et al. In vitro and in vivo trypanosomicidal action of novel arylimidamides against Trypanosoma cruzi. Antimicrob Agents Chemother. 2016;60(suppl 4):2425–2434. | ||
Da Silva CF, Daliry A, Silva PB, et al. The efficacy of arylimidamides against Trypanosoma cruzi in vitro. Parasitology. 2011;138:1863–1869. | ||
Stephens CE, Brun R, Salem MM, et al. The activity of diguanidino and ‘reversed’ diamidino 2,5-diarylfurans versus Trypanosoma cruzi and Leishmania donovani. Bioorg Med Chem Lett. 2003;13(12):2065–2069. | ||
Wang MZ, Zhu X, Srivastava A, et al. Novel arylimidamides for treatment of visceral Leishmaniasis. Antimicrob Agents Chemother. 2010;54:2507–2516. | ||
Chai Y, Munde M, Kumar A, et al. Structure-dependent binding of arylimidamides to the DNA minor groove. Chembiochem. 2014;15(1):68–79. | ||
Meirelles MN, de Araujo-Jorge TC, Miranda CF, de Souza W, Barbosa HS. Interaction of Trypanosoma cruzi with heart muscle cells: ultrastructural and cytochemical analysis of endocytic vacuole formation and effect upon myogenesis in vitro. Eur J Cell Biol. 1986;41(2):198–206. | ||
Pires DE, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066–4072. | ||
Roman-Campos D, Sales-Júnior P, Duarte HL, et al. Cardiomyocyte dysfunction during the chronic phase of Chagas disease. Mem Inst Oswaldo Cruz. 2013;108(2):243–245. | ||
Soeiro MN, de Castro SL. Screening of potential anti-Trypanosoma cruzi candidates: in vitro and in vivo studies. Open Med Chem J. 2011;5:21–30. | ||
Marin-Neto JA, Rassi A Jr, Avezum A Jr, et al; BENEFIT Investigators. The BENEFIT trial: testing the hypothesis that trypanocidal therapy is beneficial for patients with chronic Chagas heart disease. Mem Inst Oswaldo Cruz. 2009;104(suppl 1):319–324. | ||
Morillo CA, Marin-Neto JA, Avezum A, et al. Randomized trial of benznidazole for chronic Chagas’ cardiomyopathy. N Engl J Med. 2015;373:1295–1306. | ||
Maguire JH. Treatment of Chagas’ disease – time is running out. N Engl J Med. 2015;373:1369–1370. | ||
Cumming JG, Davis AM, Muresan S, Haeberlein M, Chen H. Chemical predictive modelling to improve compound quality. Nat Rev Drug Discov. 2013;12(12):948–962. | ||
Li F, Lu J, Ma X. CPY3A4-mediated lopinavir bioactivation and its inhibition by ritonavir. Drug Metab Dispos. 2012;40:18–24. | ||
Gutman GA, Chandy KG, Grissmer S, et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57(suppl 4):473–508. | ||
Molina I, Gómez I, Prat J, et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N Engl J Med. 2014;370:1899–1908. | ||
Patterson S, Wyllie S. Nitro drugs for the treatment of trypanosomatid diseases: past, present, and future prospects. Trends Parasitol. 2014;30(6):289–298. |
Supplementary materials
2,5-Bis(2-isobutoxy-4-nitrophenyl)furan
2,5-Bis(trimethylstannyl)furan (606 mg, 1.5 mmol) was injected into a solution of 1-bromo-2-isobutoxy-4-nitrobenzene (880 mg, 3.1 mmol) and tetrakis-(triphenylphosphine)palladium(0) (54 mg, 0.04 mmol) in anhydrous 1,4-dioxane (10 mL) at room temperature under nitrogen. The mixture was allowed to reflux under nitrogen overnight. The orange-colored suspension was diluted with hexanes (8 mL) and cooled to room temperature. The residue was filtered and washed with hexanes. The orange fluffy residue was recrystallized from toluene (25 mL) to give yellow fluffy solid (72%), mp: 230°C–232°C. 1H NMR (400 MHz, CDCl3): δ 1.16 (d, J =6.8 Hz, 12H), 2.35–2.28 (m, 2H), 4.01 (d, J =6 Hz, 4H), 7.36 (s, 2H), 7.82 (s, 2H), 7.94 (d, J =8.4 Hz, 2H), and 8.10 (d, J =8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 19.5, 28.3, 75.8, 106.9, 116.1, 116.2, 124.9, 125.9, 147.1, 148.9, and 155.1; and ESI-MS: m/z calculated for C24H26N2O7: 454.1 and found: 455.2 (M +1). Analysis was calculated for C24H26N2O7–0.7H2O: C, 61.71; H, 5.91; and N, 5.99 and found: C, 61.61; H, 5.65; and N, 5.59.
2,5-Bis[2-isobutoxy-4-(2-pyridylimino)aminophenyl]furan hydrochloride (DB 1890B)
To a suspension of 2,5-bis(2-isobutoxy-4-nitrophenyl)furan (2.5 g, 5.21 mmol) in a mixture of ethyl acetate (50 mL) and anhydrous ethanol (20 mL), Pd/C (500 mg, 10%) was added. The suspension was bubbled with dry nitrogen for 15 min and hydrogenated overnight using a Parr apparatus with a starting pressure of 50 psi. The consumption of hydrogen gave a clear solution. The solution was filtrated over well-packed Celite, and the filtrate was removed under reduced pressure to give a light brown fluffy solid. This compound was used in the next step without further characterization. S-(2-Naphthylmethyl)-2-pyridylthioimidate hydrobromide (2.2 mmol) was added to a cooled solution of 2,5-bis-(2-isobutoxy-4-aminophenyl)furan (1 mmol) in a mixture of dry ethanol (30 mL) and dry acetonitrile (10 mL) in an ice bath. The reaction mixture was stirred at room temperature overnight. After the disappearance of the starting material, the organic solvent was evaporated under reduced pressure to yield a crude oil product. Dry ether (50 mL) was added to the crude material, and the mixture was stirred at room temperature for 4 h. The red precipitate was filtered and washed with dry ether. The solid was dissolved in ethanol (5 mL); the solution was cooled to 0°C in an ice bath, and 10% NaOH was added until pH reached ~10. The free base was extracted with ethyl acetate (3×200 mL). The organic layer was washed with distilled water, dried over dry K2CO3, filtered, and concentrated under reduced pressure. The resulting suspension was crystallized by adding dry ether and then filtered. The free base was suspended in dry ethanol (10 mL) and cooled to 0°C in an ice bath. Freshly prepared ethanolic HCl (2 mL) was added to the suspension, and the mixture was stirred at room temperature overnight. The resulting red solution was concentrated under reduced pressure. The red crude solid was crystallized from dry ethanol and dry ether and filtered (orange solid, yield [68%], mp 205°C–206°C). 1H NMR (400 MHz, DMSO-d6): 1.10 (d, J =5.6 Hz, 12H), 2.25 (m, 2H), 3.99 (d, J =5.6 Hz, 4H), 7.35 (brs, 2H), 7.87 (m, 2H), 8.16 (d, J =7.6 Hz, 2H), 8.24 (t, J =7.6 Hz, 2H), 8.55 (d, J =7.6 Hz, 2H), 8.90 (d, J =5 Hz, 2H), 9.41 (s, 2H), 10.19 (s, 2H), and δ 11.99 (s, 2H); 13C NMR (100 MHz, CD3OD): δ 18.4, 28.1, 75.2, 109.3, 113.1, 117.2, 120.1, 122.9, 126.7, 128.4, 133.4, 138.2, 144.1, 148.4, 150.0, 156.1, and 160.3; and ESI-MS: m/z calculated for C36H38N6O3 (base): 602.3 and found: 603.4 (M + H)+. Analysis calculated for C36H38N6O3-2HCl–2.85H2O: C, 59.47; H, 6.33; and N, 11.56 and found: C, 59.45; H, 6.36; and N, 11.19.
2,5-Bis(2-isopropoxy-4-nitrophenyl)thiophene
1-Bromo-2-isopropoxy-4-nitrobenzene (5.23 g, 20 mmol), 2,5-bis(trimethylstannyl)thiophene (4.10 g, 10 mmol), and Pd(PPh3)4 (0.3 g, 0.26 mmol) were heated in 1,4 dioxane (25 mL) overnight at 100°C (oil bath) under nitrogen. The dark red solution was then diluted with excess hexanes to give an orange precipitate, and the filtered product was recrystallized from DMF/MeOH to yield a bright orange fluffy solid (3.21 g, 74%), mp 162°C–164°C. 1H NMR (500 MHz, DMSO-d6): δ (ppm): 1.45 (d, J =5.7 Hz, 12H), 5.03 (m, 2H), 7.85–7.90 (m, 6H), and 8.08 (d, J =8.5 Hz, 2H) and 13C NMR (125 MHz, DMSO-d6): δ (ppm): 21.7, 72.0, 108.7, 115.9, 127.8, 128.3, 129.7, 139.2, 146.9, and 153.0. Analysis calculated for C22H22N2O6S: C, 59.72; H, 5.01; and N, 6.33 and found: C, 59.57; H, 5.02; N, 6.23.
2,5-Bis[2-isopropoxy-4-(2-pyridylimino)aminophenyl]thiophene and dimesylate (DB1957)
A mixture of 2,5-bis(2-isopropoxy-4-nitrophenyl)thiophene (1.42 g, 3.2 mmol) and SnCl2 dihydrate (5.87 g, 26 mmol) in EtOH (80 mL) and DMSO (20 mL) was heated overnight at 80°C (oil bath). The cooled reaction was then basified with aqueous NaOH and extracted with ethyl acetate (with addition of brine). The extract was dried (Na2SO4), evaporated in vacuo, and the residue purified by column chromatography (SiO2) using hexanes:EtOAc (1:1) as eluent. The pure fractions were combined and concentrated in vacuo to give, after standing, the diamine [2,5-bis(4-amino-2-isopropoxyphenyl)thiophene] as a green sold (0.52 g, 43%). 1H NMR (300 MHz, DMSO-d6): δ (ppm): 1.35 (d, 12H), 4.57 (m, 2H), 5.21 (s, 4H), 6.17 (dd, J =8.2 and 1.6 Hz, 2H), 6.30 (d, J =1.5 Hz, 2H), 7.12 (s, 2H), and 7.29 (d, J =8.2 Hz, 2H). Without further characterization, the diamine (0.75 g, 1.95 mmol) was dissolved in acetonitrile (15 mL) and EtOH (8 mL) and treated with S-(2-naphthylmethyl)-2-pyridylthioimidate hydrobromide (1.41 g, 3.9 mmol). After stirring overnight, the red solution was concentrated in vacuo and the resulting oil was triturated with ether to give a solid, which was collected by suction. The solid was then dissolved in a minimal amount of EtOH and basified with NaOH (1 N) to give a yellow orange solid after stirring. Recrystallization of this solid from EtOH gave the title compound as the free base as yellowish brown crystals (0.61 g, 79% from the diamine), mp 112°C–115°C. 1H NMR (300 MHz, DMSO-d6): δ (ppm): 1.37 (d, 12H), 4.76 (m, 2H), 6.56 (dd, J =8.2 and 1.7 Hz, 2H), 6.64 (d, J =1.5 Hz, 2H), 7.45 (s, 2H), 7.55 (m, 2H), 7.66 (d, J =8.3 Hz, 2H), 7.95 (m, 2H), 8.32 (d, J =7.9 Hz, 2H) and 8.63 (m, 2H) and 13C NMR (75 MHz, DMSO-d6): δ (ppm): 22.1, 70.0, 107.3, 114.2, 118.1, 121.4, 123.4, 125.4, 128.3, 137.1, 138.5, 148.1, 150.6, 151.4, 151.9, and 153.8. Finally, to prepare the title dimesylate salt (DB1957), a solution of the free base (0.296 g, 0.5 mmol) in EtOH (35 mL) was treated with methanesulfonic acid (0.10 g, 1.05 mmol) and then diluted with excess diethyl ether to give a yellow brown solid, which was collected and dried under high vacuum, 90–95 mp dec. 1H NMR (500 MHz, DMSO-d6): δ (ppm): 1.44 (d, 12H), 2.32 (s, 6H), 4.86 (m, 2H), 7.11 (d, J =9.4 Hz, 2H), 7.32 (s, 2H), 7.69 (s, 2H), 7.86 (m, 2H), 8.24 (m, 2H), 8.36 (d, J =7.5 Hz, 2H), 8.91 (s, 2H), 9.34 (s, 2H), 10.03 (s, 2H), and 11.68 (s, 2H); 13C NMR (125 MHz, DMSO-d6): δ (ppm): 22.0, 71.0, 111.6, 117.9, 123.5, 123.9, 125.4, 128.7, 128.8, 134.1, 138.5, 138.6, 144.5, 149.9, 153.8, and 159.5. Analysis calculated for C34H34N6O2S-2CH3SO3H–1.5H2O: C, 53.38; H, 5.60; and N, 10.38 and found C, 53.47; H, 5.48; and N, 10.20.
2,5-Bis(2-(2,2,2-trifluoroethoxy)-4-nitrophenyl)furan
A mixture of 1-bromo-4-nitro-2-(2,2,2-trifluoroethoxy)benzene (1.54 g, 5.13 mmol) and 2,5-bis(trimethylstannyl)-furan (0.96 g, 2.43 mmol) was added to 50 mL of degassed dioxane. Pd(PPh3)4 (0.3 g, 0.26 mmol) was added to it and refluxed for 24 h under the exclusion of light after which the yellow solution was cooled and diluted with hexane to give a red precipitate. The precipitate was filtered and washed with hexane and dried to give 1.02 g (83%) of the title compound, mp 282°C–286°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm): 5.21 (q, J =8.8 Hz, 4H), 7.29 (s, 2H), 8.06–8.10 (m, 4H), and 8.33 (d, J =8.4 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ (ppm): 65.4 (q, JC-F = 35 Hz), 108.4, 115.6, 117.2, 123.4 (q, JC-F =276 Hz), 124.1, 126.4, 146.8, 147.9, and 152.4. Analysis calculated for C20H12F6N2O7: C, 47.44; H, 2.39; and N, 5.53 and found: C, 47.61; H, 2.34; and N, 5.44.
2,5-Bis[2-(2,2,2-trifluoroethoxy)-4-(2-pyridylimino)aminophenyl]furan and dimesylate (DB1959)
2,5-Bis(2-(2,2,2-trifluoroethoxy)-4-nitrophenyl)furan (0.94 g, 1.86 mmol) and 150 mg of 10% Pd/C as a catalyst was suspended in 150 mL of a degassed mixture of EtOAc–EtOH (9:1) and kept under a hydrogen pressure of 50 psi with shaking for 5 h. The solution was then filtered to remove the catalyst by passing through a bed of Celite, and the bed was washed with dichloromethane. The filtrate was concentrated under reduced pressure to viscous oil, and hexane was added to give a yellowish-white precipitate of the diamine (yield: 0.8 g [96%], mp 196°C–198°C). The diamine was used directly in the next step. 2,5-Bis(2-(2,2,2-trifluoroethoxy)-4-aminophenyl)furan (0.7 g, 1.58 mmol) was dissolved in a mixture of 40 mL of MeCN–EtOH (1:3) and cooled to 0°C, and S-(2-naphthylmethyl)-2-pyridylthioimidate hydrobromide (1.44 g, 4 mmol) was added to it and after 30 min allowed to warm to room temperature. The yellow solution was stirred for 2 days after which it was concentrated in vacuo and diethyl ether was added to provide a yellow precipitate of the hydrobromide salt, which was filtered and washed with a mixture of 1:1 ethanol–diethyl ether. The solid was then dissolved in a minimum volume of ethanol, and a dilute aqueous solution of sodium hydroxide was added to generate the free base. The organic layer was extracted with dichloromethane, dried with Na2SO4, filtered, and concentrated in vacuo to yield the free base 0.82 g (79%) as a yellow solid, mp 207°C–209°C. The imidamide free base was dissolved in 20 mL dichloromethane–ethanol (1:1) mixture, and (154 mg, 1.60 mmol) of methanesulfonic acid diluted with 5 mL diethyl ether was added to it with immediate darkening of the yellow solution to red. The mixture was stirred for 1 h, the solvents were evacuated to a small volume, and diethyl ether was added to the mixture to precipitate the bis(methanesulfonate) salt as an orange-red solid (yield: 0.62 g [96%], mp 273°C–275°C dec). 1H NMR (400 MHz, DMSO-d6): δ (ppm): 2.32 (s, 6H), 5.01 (q, J =8.4 Hz, 4H), 7.11 (s, 2H), 7.30 (d, J =8.4 Hz, 2H), 7.51 (s, 2H), 7.87–7.90 (m, 2H), 8.24–8.28 (m, 4H), 8.39 (d, J =8 Hz, 2H), 8.93 (d, J =4.4 Hz, 2H), 9.42 (brs, 2H), 10.15 (brs, 2H), and 11.85 (brs, 2H); 13C NMR (100 MHz, DMSO-d6): δ (ppm): 65.2 (q, JC-F =34 Hz), 111.1, 113.0, 118.7, 119.8, 123.9, 124.0 (q, JC-F =276 Hz), 127.0, 128.7, 134.3, 138.5, 144.5, 147.8, 150.0, 153.1, and 159.6. LRMS ESI (+): calculated for C32H24F6N6O3 (base): 654.2 and found (M + H+): 655.3. Analysis calculated for C32H24F6N6O3-2CH3SO3H-2H2O: C, 46.26; H, 4.11, and N, 9.52 and found C, 45.88; H, 3.98; and N, 9.26.
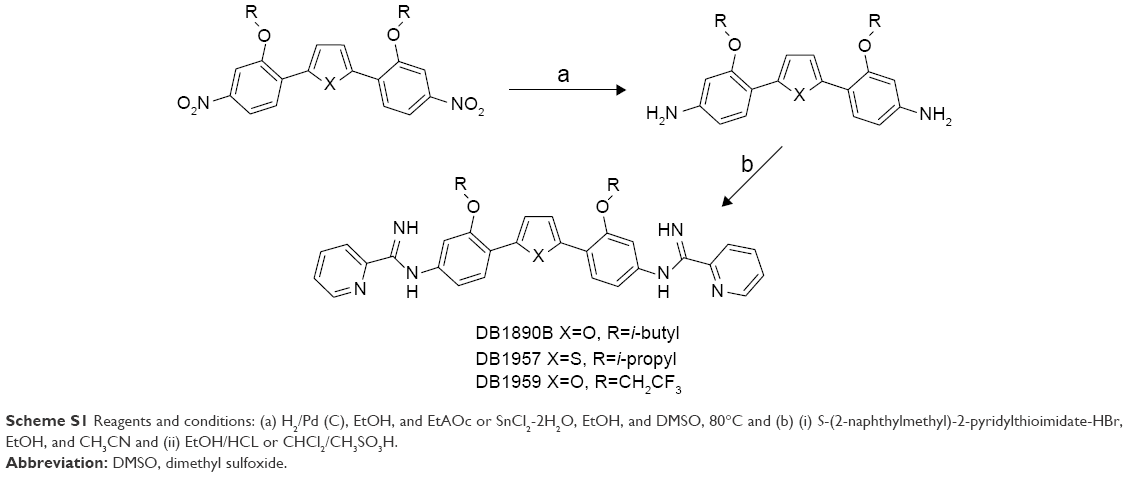 | Scheme S1 Reagents and conditions: (a) H2/Pd (C), EtOH, and EtAOc or SnCl2-2H2O, EtOH, and DMSO, 80°C and (b) (i) S-(2-naphthylmethyl)-2-pyridylthioimidate-HBr, EtOH, and CH3CN and (ii) EtOH/HCL or CHCl2/CH3SO3H. |
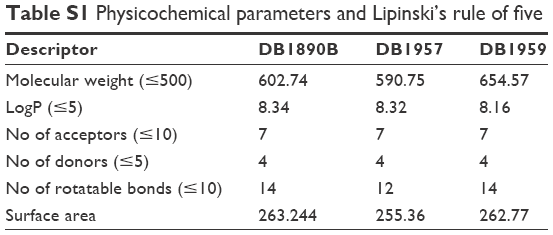 | Table S1 Physicochemical parameters and Lipinski’s rule of five |
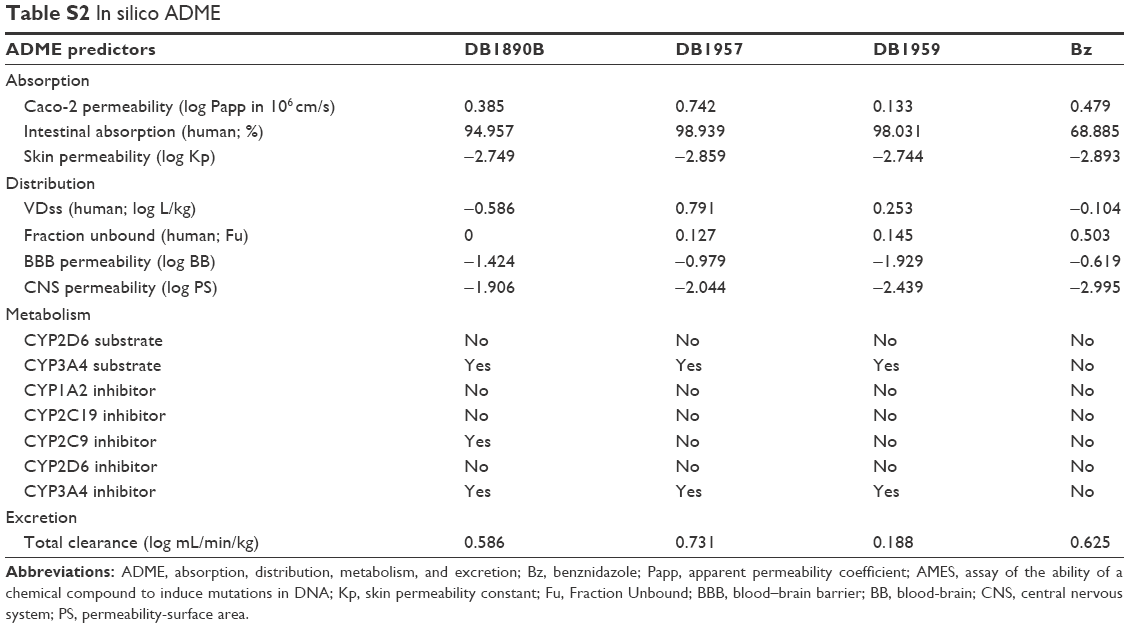 | Table S2 In silico ADME |
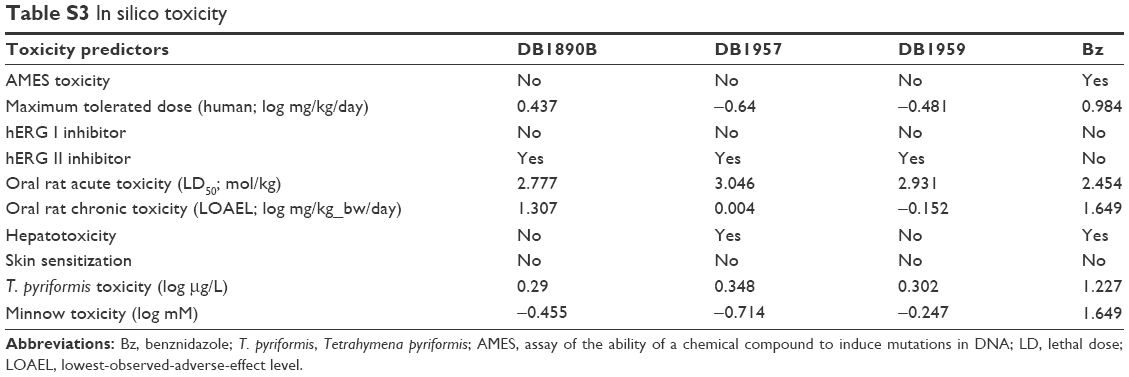 | Table S3 In silico toxicity |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.