Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Phenotypic assessment of pulmonary hypertension using high-resolution echocardiography is feasible in neonatal mice with experimental bronchopulmonary dysplasia and pulmonary hypertension: a step toward preventing chronic obstructive pulmonary disease
Authors Reynolds C, Zhang S, Shrestha A, Barrios R, Shivanna B
Received 4 April 2016
Accepted for publication 23 May 2016
Published 14 July 2016 Volume 2016:11(1) Pages 1597—1605
DOI https://doi.org/10.2147/COPD.S109510
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Corey L Reynolds,1 Shaojie Zhang,2 Amrit Kumar Shrestha,2 Roberto Barrios,3 Binoy Shivanna2
1Mouse Phenotyping Core, 2Section of Neonatology, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA; 3Department of Pathology and Genomic Medicine, Houston Methodist Hospital, Houston, TX, USA
Abstract: Bronchopulmonary dysplasia (BPD) and chronic obstructive pulmonary disease (COPD) are chronic lung diseases of human infants and adults, respectively, that are characterized by alveolar simplification. One-third of the infants with severe BPD develop pulmonary hypertension (PH). More importantly, PH increases morbidity and mortality in BPD patients. Additionally, COPD is a common respiratory morbidity in former BPD patients. The lack of an appropriate small animal model wherein echocardiography (Echo) can demonstrate PH is one of the major barriers to understand the molecular mechanisms of the disease and, thereby, develop rational therapies to prevent and/or treat PH in BPD patients. Thus, the goal of this study was to establish a model of experimental BPD and PH and investigate the feasibility of Echo to diagnose PH in neonatal mice. Since hyperoxia-induced oxidative stress and inflammation contributes to the development of BPD with PH, we tested the hypothesis that exposure of newborn C57BL/6J mice to 70% O2 (hyperoxia) for 14 days leads to lung oxidative stress, inflammation, alveolar and pulmonary vascular simplification, pulmonary vascular remodeling, and Echo evidence of PH. Hyperoxia exposure caused lung oxidative stress and inflammation as evident by increased malondialdehyde adducts and inducible nitric oxide synthase, respectively. Additionally, hyperoxia exposure caused growth restriction, alveolar and pulmonary vascular simplification, and pulmonary vascular remodeling. At 14 days of age, Echo of these mice demonstrated that hyperoxia exposure decreased pulmonary acceleration time (PAT) and PAT/ejection time ratio and increased right ventricular free wall thickness, which are indicators of significant PH. Thus, we have demonstrated the feasibility of Echo to phenotype PH in neonatal mice with experimental BPD with PH, which can aid in discovery of therapies to prevent and/or treat BPD with PH and its sequelae such as COPD in humans.
Keywords: hyperoxia, oxidative stress, malondialdehyde adducts, inflammation, inducible nitric oxide synthase
Introduction
Bronchopulmonary dysplasia (BPD) is a chronic lung disease of infancy that results from interrupted lung alveolar and vascular growth.1,2 The pathogenesis and pathophysiology of BPD is identical to chronic obstructive pulmonary disease (COPD), which is a chronic lung disease of human adults.3 Alveolar simplification is a unique histological pattern of BPD that is characterized by larger but fewer alveoli with decreased septation.2 Despite major advances in the respiratory care of premature infants, BPD remains among the most prevalent condition in these patients.4,5 Infants with BPD are more likely to have long-term pulmonary and neurodevelopmental morbidities.4,6 Importantly, COPD is a common long-term respiratory morbidity of former BPD patients.7–10
Evidence implicates hyperoxia-induced generation of reactive oxygen species (ROS) and lung inflammation as the major contributors to the development of BPD and its sequelae.11,12 Supplemental oxygen (O2) is commonly administered as a life-saving measure in patients with impaired lung function. Although it relieves the immediate life-threatening consequences transiently, O2 may also exacerbate lung injury.13 Excessive O2 exposure leads to increased ROS production, and the generated ROS react with nearby molecules (eg, protein, lipids, DNA, and RNA) and modify their structure and function, resulting in chronic pulmonary diseases such as BPD and COPD.14–18
Pulmonary hypertension (PH) is a severe form of pulmonary vascular disease that affects 25%–43% of infants with moderate-to-severe BPD.19,20 The pathogenesis of PH in BPD is complex and may result from interactions between antenatal risk factors such as pregnancy-induced hypertension, intrauterine growth restriction and infection, and postnatal risk factors such as oxidative stress, inflammation, infection, and mechanical ventilation in a preterm infant with underlying genetic susceptibility.21 Importantly, PH increases short- and long-term morbidity and mortality, including COPD, in BPD patients.21 Hence, there is an urgent need to improve therapies for BPD patients with PH.
Small animal models such as genetically modified mice offer a unique opportunity to understand the molecular mechanisms that contribute to the development of BPD and PH and, thereby, discover novel therapies. Although right heart catheterization is the gold standard to diagnose PH in adult mice,22,23 it is a terminal procedure and precludes long-term follow-up in these animals. Moreover, animal size is a major limitation to perform heart catheterization in newborn mice. Echocardiography (Echo) is a noninvasive technique that can reliably diagnose PH and can circumvent the catheterization-associated problems in mice. However, lack of Echo studies to diagnose PH in neonatal mice has precluded the development of reliable neonatal mouse models of PH-associated disorders such as BPD, which is pivotal to understand the pathogenesis and improving therapies for PH in human infants with BPD and preventing long-term respiratory morbidities such as COPD. Thus, the goal of this study was to provide a platform to discover early biomarkers and interventions to prevent BPD with PH and COPD in humans by investigating the feasibility of Echo to diagnose PH in newborn mice with experimental BPD and PH. Since hyperoxia-induced oxidative stress and inflammation contributes to the development of BPD with PH in human infants, we tested the hypothesis that exposure of newborn C57BL/6J mice to 70% O2 (hyperoxia) for 14 days leads to lung oxidative stress, inflammation, alveolar and pulmonary vascular simplification, pulmonary vascular remodeling, and Echo evidence of PH.
Materials and methods
Animals
This study was approved and conducted in strict accordance with the federal guidelines for the humane care and use of laboratory animals by the Institutional Animal Care and Use Committee of Baylor College of Medicine (Protocol number: AN-5631). The C57BL/6J wild-type mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Timed-pregnant mice raised in our animal facility were used for the experiments.
Exposure
Within 24 hours of birth, male and female pups from multiple litters were pooled before being randomly and equally redistributed to the dams, following which they were exposed to either 21% O2 (air, n=21) or 70% O2 (hyperoxia, n=21) for 14 days. During each experiment, four dams with 4–8 pups/dam were equally allocated to air and hyperoxic conditions. The dams were rotated between air- and hyperoxia-exposed litters every 48 hours to prevent oxygen toxicity in the dams and to eliminate maternal effects between the groups. Oxygen exposures were conducted in Plexiglas chambers, and the animals were monitored as described previously.24
Tissue preparation for lung morphometry
After completion of experiments, a subset of pups were euthanized, their lungs were inflated and fixed via the trachea with 10% formalin at 25 cm H2O pressure for at least 10 minutes, and sections of the paraffin-embedded lungs were obtained for the analysis of lung morphometry as described previously.24
Lung morphometry
As per American Thoracic Society/European Respiratory Society task force guidelines,25 a systematic, uniform, random sampling principle was used to evaluate the lung sections for morphometry. Alveolar development on selected mice (n=9/group) was evaluated by radial alveolar counts (RAC) and mean linear intercepts (MLI). The observers performing the measurements were masked to the slide identity. RAC was determined as described by Cooney and Thurlbeck.26 RAC measurements were made by dropping a perpendicular line from the center of a respiratory bronchiole to the edge of the septum or pleura and counting the number of alveoli traversed by this line. MLIs were assessed as described previously.27 Briefly, grids of horizontal and vertical lines were superimposed on an image and the number of times the lines intersected with the tissue was counted. The total length of the grid lines was then divided by the number of intersections to provide the mean linear intercept in micrometer. Photographs from at least ten random nonoverlapping lung fields (10× magnification) were taken from each animal for RAC and MLI measurements.
Analyses of pulmonary vascularization
Pulmonary vessel density was determined based on immunohistochemical staining for von Willebrand factor (vWF), which is an endothelial specific marker. At least ten counts from ten random nonoverlapping fields (20× magnification) were performed for each animal (n=9/group).
Analyses of pulmonary vascular remodeling
Pulmonary vascular remodeling, which reflects significant PH, was determined by the medial thickness index (n=9/group) of resistance pulmonary arteries (20–150 μm external diameter) calculated using the equation: Medial thickness index = [(areaext − areaint)/areaext] ×100, where areaext and areaint are the areas within the external and internal boundaries of the α-smooth muscle actin (α-SMA) layer, respectively.28 Additionally, α-SMA, which is a marker of vascular smooth muscle cells, was quantified by immunoblotting lung proteins using anti-α-SMA (Sigma-Aldrich Co., St Louis, MO, USA; A5228, dilution 1:1,000) and anti-β-actin (Santa Cruz Biotechnologies, Santa Cruz, CA, USA; sc-47778, dilution 1:2,000) antibodies.
Analysis of lung oxidative stress and inflammation
Malondialdehyde (MDA) is a stable end product of lipid peroxidation and is a generally accepted marker of oxidative stress.29 Likewise, inducible nitric oxide synthase (iNOS) is a well-known marker of lung inflammation.30 Hence, we performed immunoblotting on lung proteins using anti-MDA (Cell Biolabs, Inc., San Diego, CA, USA; STA-331, dilution 1:1,000), anti-iNOS (Santa Cruz Biotechnologies; sc-7271, dilution 1:1,000), and anti-β-actin (Santa Cruz Biotechnologies; sc-47778, dilution 1:2,000) antibodies. The primary antibodies were detected by incubation with the appropriate horseradish peroxidase-conjugated secondary antibodies. The immunoreactive bands were detected by chemiluminescence methods, and the band density was analyzed by Image J software (National Institutes of Health, Bethesda, MD, USA).
Echocardiography
PH was assessed by performing functional Echo in mice as described previously.31,32 Briefly, the mice (n=11/group) were anesthetized by using inhaled isoflurane via facemask and subjected to transthoracic two-dimensional, M-mode, and pulsed-wave Doppler (PWD) Echo using the VisualSonics Vevo 2100 machine (VisualSonics Inc., Toronto, ON, Canada) and a 40 MHz linear transducer. Right ventricular free wall (RVFW) thickness was measured during end diastole in the right parasternal long-axis view by two-dimensional and M-mode Echo. PWD recording of the pulmonary blood flow was obtained at the level of the aortic valve in the parasternal short axis view to measure pulmonary acceleration time (PAT, defined as the time from the onset of flow to peak velocity), and RV ejection time (ET, the time from the onset to the termination of pulmonary flow).
Statistical analyses
The results were analyzed by GraphPad Prism 5 software (La Jolla, CA, USA). At least three separate experiments were performed for each measurement (n= total animals from the three experiments), and the data are expressed as mean ± SD. The effects of exposure for the outcome variables were assessed using Student’s t-test. A P-value of <0.05 was considered significant.
Results and discussion
The hallmarks of a murine hyperoxia model that mimics BPD with PH in human preterm infants include growth restriction, lung oxidative stress and inflammation, interruption in alveolar and pulmonary vascular development (alveolar and pulmonary vascular simplification), pulmonary vascular remodeling, and decreased PAT/ET with increased RVFW thickness (PH). In this study, we investigated the effects of 70% O2 (hyperoxia) on these hallmarks and demonstrated that exposure of newborn wild-type mice to hyperoxia for 14 days increases lung oxidative stress and inflammation and causes alveolar and pulmonary vascular simplification, pulmonary vascular remodeling, and PH. Additionally, our study demonstrates the feasibility of Echo to elucidate PH in these mice at 14 days postnatal age. Our studies are important to identify novel interventions to prevent and/or treat BPD and PH and, thereby, prevent COPD in former human preterm infants with BPD and PH.
The concentration of oxygen used in this study was comparable to those used in previous studies.33–35 We chose 70% oxygen for our experiments because it was the lowest oxygen concentration that caused significant alveolar and pulmonary vascular simplification and PH in our mouse model. The survival rate of pups exposed to air and hyperoxia was identical. Hyperoxia exposure is known to restrict growth by various mechanisms in neonatal mouse models of experimental BPD.36–40 We observed a similar finding. Although the body weights were comparable at birth, 70% O2 (hyperoxia) exposure for 14 days decreased the body weight by 21% when compared to corresponding air-breathing animals (Figure 1).
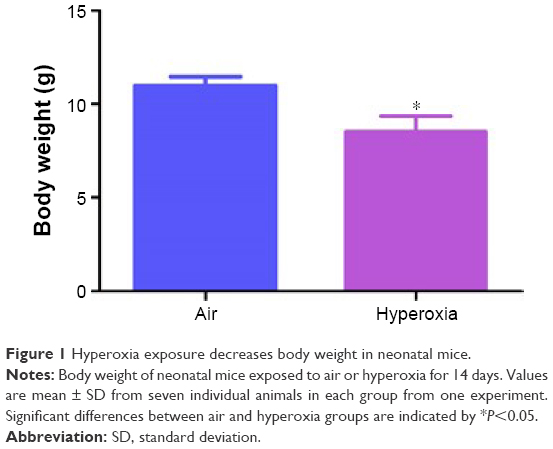 | Figure 1 Hyperoxia exposure decreases body weight in neonatal mice. |
Oxidative stress contributes to the development of BPD with PH. It is difficult to measure and characterize hyperoxia-induced ROS in real time as they are very unstable. Hence, we determined the expression of MDA-protein adducts, which are stable aldehyde end products of ROS-mediated lipid peroxidation.41 Our finding of hyperoxia induced MDA adducts in mice (Figure 2A and B) are consistent with previous studies42,43 and suggests that hyperoxia exposure causes oxidative stress in the lungs. Interestingly, hyperoxia induced MDA adducts in two specific regions between 40 and 80 kDa. This finding suggests that hyperoxia causes oxidative modifications of specific lung proteins in our model. The other possibility is that these proteins are abundant in the mouse lungs and are susceptible to hyperoxia-induced oxidative modification. Further studies are needed to identity these specific proteins because they could serve as potential biomarkers for oxidative stress in human infants with BPD. In addition to oxidative stress, inflammation plays a key role in the pathogenesis of BPD.44 NO is used as a marker of respiratory tract inflammation in patients with asthma.45 Three isoforms of NOS, neuronal NOS (nNOS, NOS-1), inducible NOS (iNOS, NOS-2), and endothelial NOS (eNOS, NOS-3), generate NO from the amino acid L-arginine. Although NO is critical for the homeostasis of lungs, excessive NO production increases nitrative stress and exerts proinflammatory effects. Inflammatory stimuli such as cytokines, chemokines, bacterial toxins, viral infections, allergens, hypoxia, and hyperoxia augment lung iNOS expression,30,46 which suggests that increased iNOS expression is biomarker of ongoing inflammation. Consistent with other murine hyperoxia models,46,47 hyperoxia exposure increased lung iNOS protein expression (Figure 3A and B), which indicates the presence of underlying lung inflammation.
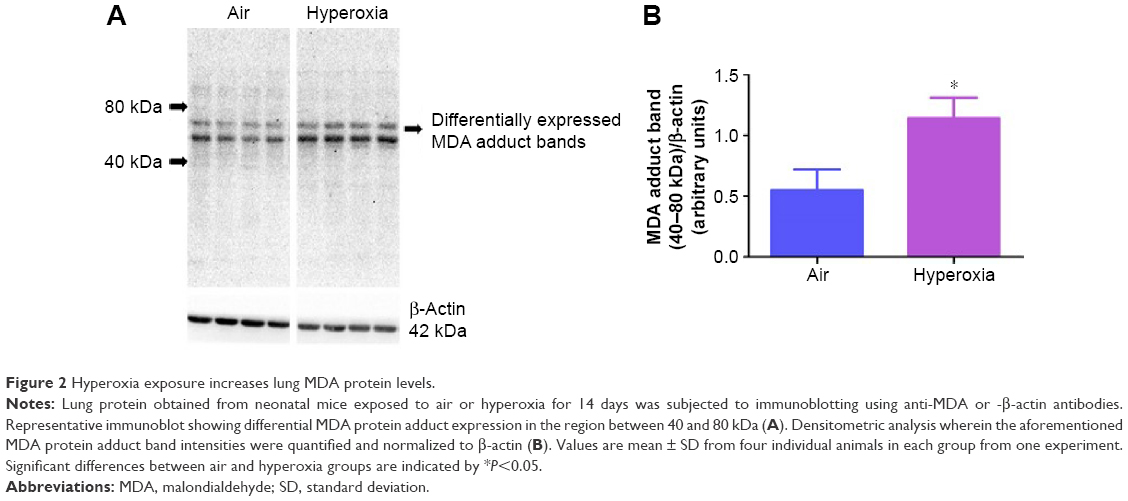 | Figure 2 Hyperoxia exposure increases lung MDA protein levels. |
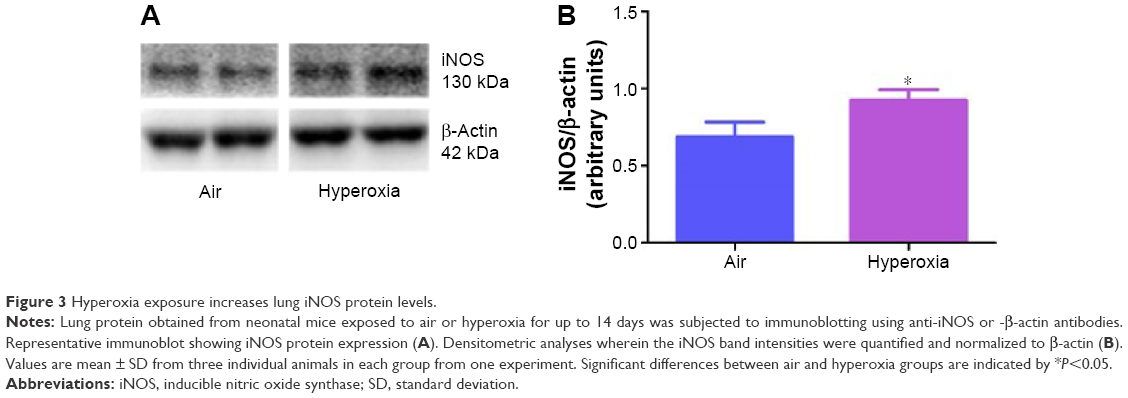 | Figure 3 Hyperoxia exposure increases lung iNOS protein levels. |
Hyperoxia is known to interrupt alveolar development by mechanisms entailing cell proliferation, cell death, and disruption of lung developmental signaling pathways,48 which collectively results in alveolar simplification in preterm infants1,2 and newborn mice.49 In line with these studies, exposure to hyperoxia for 14 days decreased RAC by 35% (Figure 4B and C) and increased MLI by 50% (Figure 4B and D), indicating that their alveoli were fewer in number and larger in diameter (alveolar simplification), respectively, when compared to corresponding air-breathing animals (Figure 4A, C, and D).
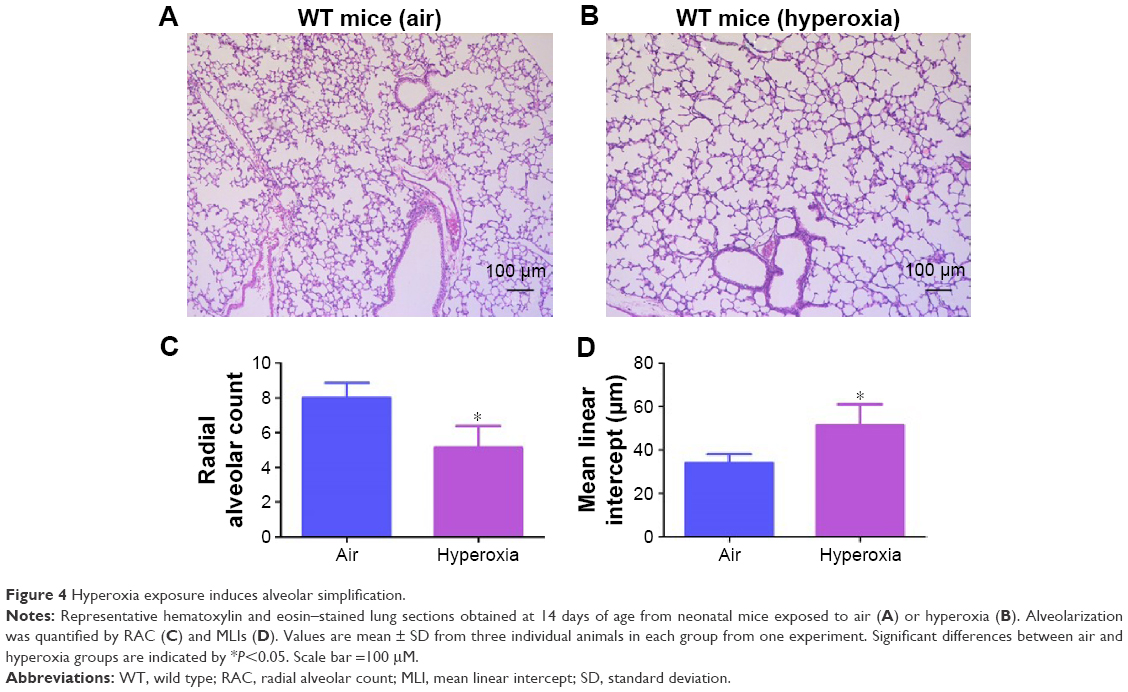 | Figure 4 Hyperoxia exposure induces alveolar simplification. |
Pulmonary vascular and alveolar development are highly orchestrated interdependent processes and studies clearly support this concept by demonstrating that an interruption of distal lung angiogenesis secondary to decreased expression of vascular endothelial growth factor (VEGF) and/or its signaling receptor, vascular endothelial growth factor receptor2, leads to alveolar simplification.50,51 Additionally, pulmonary vascular simplification decreases pulmonary vascular surface area leading to high pulmonary vascular resistance and PH.52 Our findings of hyperoxia-induced decrease in vWF-stained lung blood vessels (Figure 5) suggest that hyperoxia causes pulmonary vascular simplification, which is consistent with studies in human preterm infants1,2 and newborn mice.53
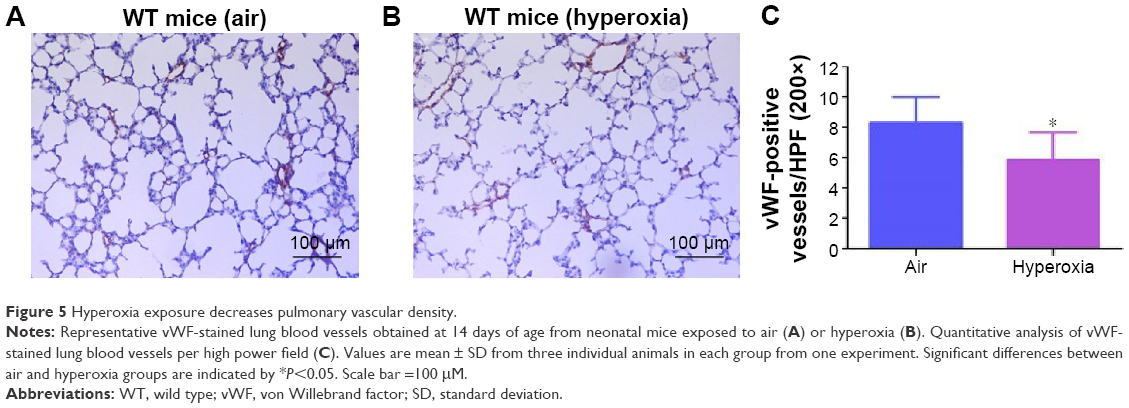 | Figure 5 Hyperoxia exposure decreases pulmonary vascular density. |
Pulmonary vascular remodeling secondary to increased smooth muscle cell proliferation of resistance pulmonary arteries is an additional risk factor that increases pulmonary vascular resistance and contributes to PH in BPD patients.21 Consistent with several rodent models of experimental BPD with PH,28,35,54 hyperoxia increased pulmonary vascular remodeling (Figure 6), which indicates the presence of significant PH in our experimental animals. Two-dimensional and Doppler Echo have been used in several murine models to effectively assess heart function, chamber dimensions and thickness, and vascular properties.55 It is a commonly used imaging modality in small animals because it is noninvasive, inexpensive, versatile, and is ideal for serial studies. PH is characterized by increased pulmonary artery (PA) and right ventricular systolic pressure, which is usually estimated indirectly from the tricuspid regurgitation (TR) peak flow velocity using Echo in human patients.56 However, because of technical limitations preventing proper flow alignment, the measurement of TR by Doppler is inaccurate in mice. Moreover, TR is rare in rodents and occurs only with severe PH.57 Right ventricular systolic time intervals such as PAT (the time from the onset of pulmonary flow to peak velocity) and ET (the time from onset to end of systolic flow) can be accurately obtained by high-resolution PWD Echo in rodents and are used as alternative indices of PA pressure in these animals.31,32 Although Echo has been the standard to diagnose PH in older murine models, there is lack of data on its feasibility to demonstrate PH in neonatal mice. In neonatal mouse models of BPD with PH, Echo has been used to diagnose PH several weeks later in the postneonatal period.34,58 This comes with a limitation of missing important earlier time points where the interventions can be targeted to successfully prevent and/or treat BPD with PH. Hence, we conducted Echo studies in neonatal mice with experimental BPD with PH.
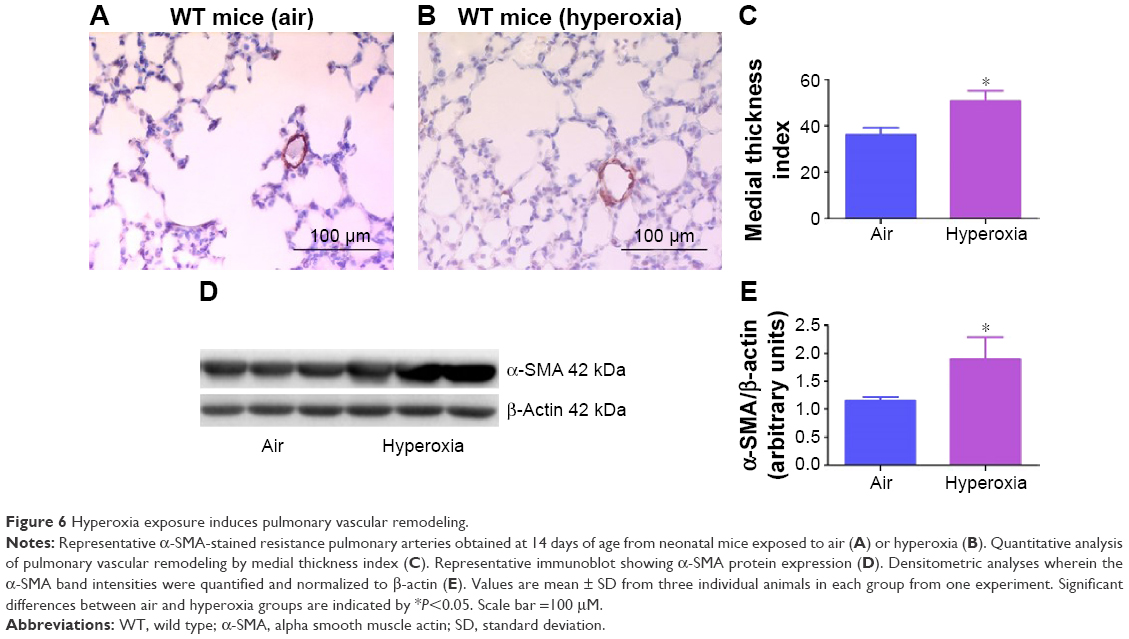 | Figure 6 Hyperoxia exposure induces pulmonary vascular remodeling. |
At a normal systolic pressure, the Doppler Echo pattern of pulmonary systolic flow is symmetric. With PH, the Doppler flow pattern becomes asymmetric, with the peak velocity occurring earlier because the PAT is decreased as the pulmonary valve closes prematurely due to high PA pressure. The reduced PAT also leads to a decrease in the ratio of PAT/ET. The normalization of PAT by ET offsets some of the confounders such as heart rate59 and cardiac output60 that might independently affect the PAT. PAT and PAT/ET ratio estimated using high-frequency Echo have been shown to correlate with PA pressure measured by cardiac catheterization. In agreement with studies in older rodents, Doppler Echo of PA showed that exposure of neonatal mice to hyperoxia decreased PAT by 27% (Figure 7B and C) and the ratio of PAT/ET by 28% (Figure 7B and D), resulting in an asymmetric flow pattern (Figure 7B) when compared to air-breathing animals (Figure 7A, C, and D). Additionally, right ventricular hypertrophy (RVH) reflects severe PH, and RVFW thickness measured in diastole is shown to strongly correlate with RVH.32 Therefore, we determined RVFW thickness in diastole using M-mode Echo in our experimental animals. Consistent with its effects on other indices of PH, hyperoxia exposure increased RVFW by 70% (Figure 8B and C) compared to air-breathing animals (Figure 8A and C). The Echo findings clearly demonstrate that our experimental animals have significant PH.
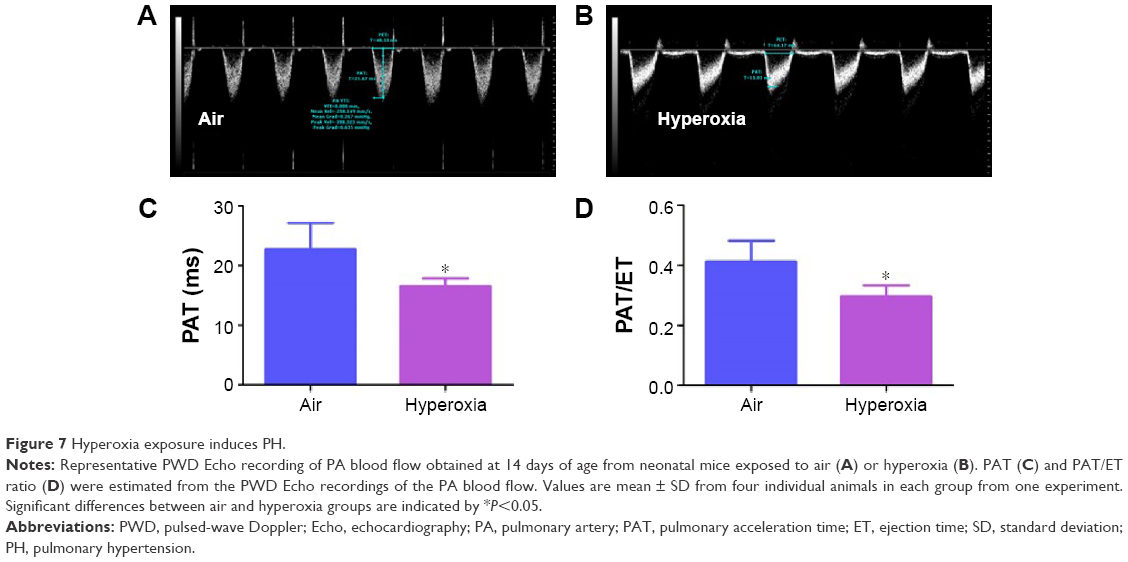 | Figure 7 Hyperoxia exposure induces PH. |
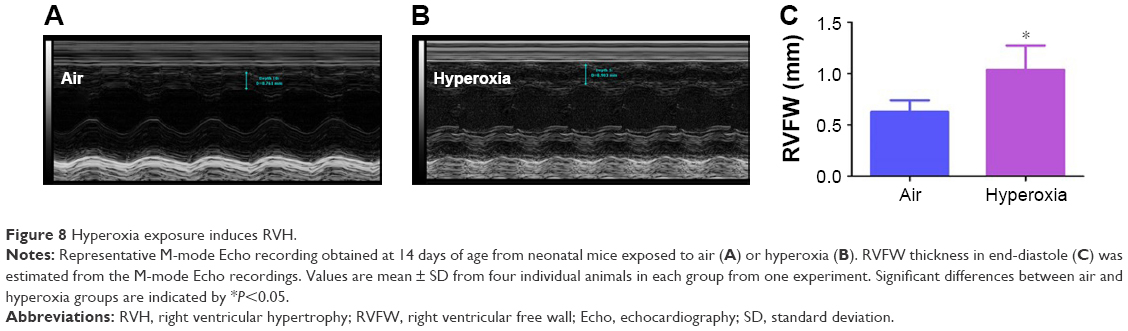 | Figure 8 Hyperoxia exposure induces RVH. |
In summary, we have established a model of experimental BPD with PH and demonstrated that noninvasive assessment of PH is feasible in neonatal C57BL/6J mice using high-resolution Echo. To the best of our knowledge, this is the first study to demonstrate PH using high-resolution Echo in neonatal mice at 14 days postnatal age. This animal model may offer the unique opportunity to identify pathophysiological mechanisms that contribute to PH and to develop therapeutic strategies to prevent and/or treat BPD with PH in human preterm infants and thereby prevent adult-onset COPD in former BPD patients. Additionally, our findings have important implications for research in the prevention and treatment of other congenital disorders such as pulmonary hypoplasia, congenital diaphragmatic hernia, and congenital heart diseases that are associated with PH in human infants.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH) HD-073323, American Heart Association BGIA-20190008, and American Lung Association RG-349917 to BS, and by the Mouse Phenotyping Core at Baylor College of Medicine with funding from the NIH (U54 HG006348). We thank Pamela Parsons for her timely processing of histopathology slides.
Disclosure
The authors report no conflicts of interest in this work.
References
Jobe AJ. The new BPD: an arrest of lung development. Pediatr Res. 1999;46(6):641–643. | ||
Husain AN, Siddiqui NH, Stocker JT. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum Pathol. 1998;29(7):710–717. | ||
Bourbon JR, Boucherat O, Boczkowski J, Crestani B, Delacourt C. Bronchopulmonary dysplasia and emphysema: in search of common therapeutic targets. Trends Mol Med. 2009;15(4):169–179. | ||
Fanaroff AA, Stoll BJ, Wright LL, et al. Trends in neonatal morbidity and mortality for very low birthweight infants. Am J Obstet Gynecol. 2007;196(2):147 e141–e148. | ||
Van Marter LJ. Epidemiology of bronchopulmonary dysplasia. Semin Fetal Neonatal Med. 2009;14(6):358–366. | ||
Short EJ, Klein NK, Lewis BA, et al. Cognitive and academic consequences of bronchopulmonary dysplasia and very low birth weight: 8-year-old outcomes. Pediatrics. 2003;112(5):e359. | ||
Bhandari A, McGrath-Morrow S. Long-term pulmonary outcomes of patients with bronchopulmonary dysplasia. Semin Perinatol. 2013;37(2):132–137. | ||
Boucherat O, Morissette MC, Provencher S, Bonnet S, Maltais F. Bridging lung development with chronic obstructive pulmonary disease. Relevance of developmental pathways in chronic obstructive pulmonary disease pathogenesis. Am J Respir Crit Care Med. 2016;193(4):362–375. | ||
Carraro S, Filippone M, Da Dalt L, et al. Bronchopulmonary dysplasia: the earliest and perhaps the longest lasting obstructive lung disease in humans. Early Hum Dev. 2013;89(Suppl 3):S3–S5. | ||
Gibson AM, Doyle LW. Respiratory outcomes for the tiniest or most immature infants. Semin Fetal Neonatal Med. 2014;19(2):105–111. | ||
Bhandari V. Hyperoxia-derived lung damage in preterm infants. Semin Fetal Neonatal Med. 2010;15(4):223–229. | ||
Wright CJ, Kirpalani H. Targeting inflammation to prevent bronchopulmonary dysplasia: can new insights be translated into therapies? Pediatrics. 2011;128(1):111–126. | ||
Thiel M, Chouker A, Ohta A, et al. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 2005;3(6):e174. | ||
Bhandari V, Choo-Wing R, Lee CG, et al. Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat Med. 2006;12(11):1286–1293. | ||
Bagdonas E, Raudoniute J, Bruzauskaite I, Aldonyte R. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:995–1013. | ||
Bernardo I, Bozinovski S, Vlahos R. Targeting oxidant-dependent mechanisms for the treatment of COPD and its comorbidities. Pharmacol Ther. 2015;155:60–79. | ||
Carpagnano GE, Kharitonov SA, Foschino-Barbaro MP, Resta O, Gramiccioni E, Barnes PJ. Supplementary oxygen in healthy subjects and those with COPD increases oxidative stress and airway inflammation. Thorax. 2004;59(12):1016–1019. | ||
Rahman I. The role of oxidative stress in the pathogenesis of COPD: implications for therapy. Treat Respir Med. 2005;4(3):175–200. | ||
Khemani E, McElhinney DB, Rhein L, et al. Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: clinical features and outcomes in the surfactant era. Pediatrics. 2007;120(6):1260–1269. | ||
Mourani PM, Ivy DD, Gao D, Abman SH. Pulmonary vascular effects of inhaled nitric oxide and oxygen tension in bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2004;170(9):1006–1013. | ||
Mourani PM, Abman SH. Pulmonary hypertension and vascular abnormalities in bronchopulmonary dysplasia. Clin Perinatol. 2015;42(4):839–855. | ||
Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104(2):236–244, 228p following 244. | ||
Medoff BD, Okamoto Y, Leyton P, et al. Adiponectin deficiency increases allergic airway inflammation and pulmonary vascular remodeling. Am J Respir Cell Mol Biol. 2009;41(4):397–406. | ||
Shivanna B, Zhang W, Jiang W, et al. Functional deficiency of aryl hydrocarbon receptor augments oxygen toxicity-induced alveolar simplification in newborn mice. Toxicol Appl Pharmacol. 2013;267(3):209–217. | ||
Hsia CC, Hyde DM, Ochs M, Weibel ER. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med. 2010;181(4):394–418. | ||
Cooney TP, Thurlbeck WM. The radial alveolar count method of Emery and Mithal: a reappraisal 1 – postnatal lung growth. Thorax. 1982;37(8):572–579. | ||
van Eijl S, Mortaz E, Versluis C, Nijkamp FP, Folkerts G, Bloksma N. A low vitamin A status increases the susceptibility to cigarette smoke-induced lung emphysema in C57BL/6J mice. J Physiol Pharmacol. 2011;62(2):175–182. | ||
Aslam M, Baveja R, Liang OD, et al. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am J Respir Crit Care Med. 2009;180(11):1122–1130. | ||
Del Rio D, Stewart AJ, Pellegrini N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis. 2005;15(4):316–328. | ||
Ricciardolo FL, Sterk PJ, Gaston B, Folkerts G. Nitric oxide in health and disease of the respiratory system. Physiol Rev. 2004;84(3):731–765. | ||
Thibault HB, Kurtz B, Raher MJ, et al. Noninvasive assessment of murine pulmonary arterial pressure: validation and application to models of pulmonary hypertension. Circ Cardiovasc Imaging. 2010;3(2):157–163. | ||
Urboniene D, Haber I, Fang YH, Thenappan T, Archer SL. Validation of high-resolution echocardiography and magnetic resonance imaging vs. high-fidelity catheterization in experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2010;299(3):L401–L412. | ||
Wang H, Jafri A, Martin RJ, et al. Severity of neonatal hyperoxia determines structural and functional changes in developing mouse airway. Am J Physiol Lung Cell Mol Physiol. 2014;307(4):L295–L301. | ||
Hansmann G, Fernandez-Gonzalez A, Aslam M, et al. Mesenchymal stem cell-mediated reversal of bronchopulmonary dysplasia and associated pulmonary hypertension. Pulm Circ. 2012;2(2):170–181. | ||
Heilman RP, Lagoski MB, Lee KJ, et al. Right ventricular cyclic nucleotide signaling is decreased in hyperoxia-induced pulmonary hypertension in neonatal mice. Am J Physiol Heart Circ Physiol. 2015;308(12):H1575–H1582. | ||
Chen HJ, Chiang BL. Effect of hyperoxia on retinoid metabolism and retinoid receptor expression in the lungs of newborn mice. PloS One. 2015;10(10):e0140343. | ||
Fritzell JA Jr, Mao Q, Gundavarapu S, et al. Fate and effects of adult bone marrow cells in lungs of normoxic and hyperoxic newborn mice. Am J Respir Cell Mol Biol. 2009;40(5):575–587. | ||
Gortner L, Monz D, Mildau C, et al. Bronchopulmonary dysplasia in a double-hit mouse model induced by intrauterine hypoxia and postnatal hyperoxia: closer to clinical features? Ann Anat. 2013;195(4):351–358. | ||
Jones CV, Alikhan MA, O’Reilly M, et al. The effect of CSF-1 administration on lung maturation in a mouse model of neonatal hyperoxia exposure. Respir Res. 2014;15:110. | ||
Rogers LK, Tipple TE, Nelin LD, Welty SE. Differential responses in the lungs of newborn mouse pups exposed to 85% or >95% oxygen. Pediatr Res. 2009;65(1):33–38. | ||
Moore K, Roberts LJ 2nd. Measurement of lipid peroxidation. Free Radic Res. 1998;28(6):659–671. | ||
Li HD, Zhang ZR, Zhang QX, Qin ZC, He DM, Chen JS. Treatment with exogenous hydrogen sulfide attenuates hyperoxia-induced acute lung injury in mice. Eur J Appl Physiol. 2013;113(6):1555–1563. | ||
Shivanna B, Zhang S, Patel A, et al. Omeprazole attenuates pulmonary aryl hydrocarbon receptor activation and potentiates hyperoxia-induced developmental lung injury in newborn mice. Toxicol Sci. 2015;148(1):276–287. | ||
Saugstad OD. Bronchopulmonary dysplasia-oxidative stress and antioxidants. Semin Neonatol. 2003;8(1):39–49. | ||
Kharitonov SA, Yates D, Robbins RA, Barnes PJ, Logan-Sinclair R, Shinebourne EA. Increased nitric oxide in exhaled air of asthmatic patients. Lancet. 1994;343(8890):133–135. | ||
Radomski A, Sawicki G, Olson DM, Radomski MW. The role of nitric oxide and metalloproteinases in the pathogenesis of hyperoxia-induced lung injury in newborn rats. Br J Pharmacol. 1998;125(7):1455–1462. | ||
Demchenko IT, Atochin DN, Gutsaeva DR, et al. Contributions of nitric oxide synthase isoforms to pulmonary oxygen toxicity, local vs mediated effects. Am J Physiol Lung Cell Mol Physiol. 2008;294(5):L984–L990. | ||
Silva DM, Nardiello C, Pozarska A, Morty RE. Recent advances in the mechanisms of lung alveolarization and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2015;309(11):L1239–L1272. | ||
Warner BB, Stuart LA, Papes RA, Wispe JR. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol. 1998;275(1 Pt 1):L110–L117. | ||
Thebaud B, Abman SH. Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med. 2007;175(10):978–985. | ||
McGrath-Morrow SA, Cho C, Cho C, Zhen L, Hicklin DJ, Tuder RM. Vascular endothelial growth factor receptor 2 blockade disrupts postnatal lung development. Am J Respir Cell Mol Biol. 2005;32(5):420–427. | ||
Nair J, Lakshminrusimha S. Update on PPHN: mechanisms and treatment. Semin Perinatol. 2014;38(2):78–91. | ||
Park MS, Rieger-Fackeldey E, Schanbacher BL, et al. Altered expressions of fibroblast growth factor receptors and alveolarization in neonatal mice exposed to 85% oxygen. Pediatr Res. 2007;62(6):652–657. | ||
Chen S, Rong M, Platteau A, et al. CTGF disrupts alveolarization and induces pulmonary hypertension in neonatal mice: implication in the pathogenesis of severe bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2011;300(3):L330–L340. | ||
Collins KA, Korcarz CE, Lang RM. Use of echocardiography for the phenotypic assessment of genetically altered mice. Physiol Genom. 2003;13(3):227–239. | ||
McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250–2294. | ||
Jones JE, Mendes L, Rudd MA, Russo G, Loscalzo J, Zhang YY. Serial noninvasive assessment of progressive pulmonary hypertension in a rat model. Am J Physiol Heart Circ Physiol. 2002;283(1):H364–H371. | ||
Ramani M, Bradley WE, Dell’Italia LJ, Ambalavanan N. Early exposure to hyperoxia or hypoxia adversely impacts cardiopulmonary development. Am J Respir Cell Mol Biol. 2015;52(5):594–602. | ||
Leighton RF, Weissler AM, Weinstein PB, Wooley CF. Right and left ventricular systolic time intervals. Effects of heart rate, respiration and atrial pacing. Am J Cardiol. 1971;27(1):66–72. | ||
Chan KL, Currie PJ, Seward JB, Hagler DJ, Mair DD, Tajik AJ. Comparison of three Doppler ultrasound methods in the prediction of pulmonary artery pressure. J Am Coll Cardiol. 1987;9(3):549–554. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.