Back to Journals » Drug Design, Development and Therapy » Volume 9
Phase III study of pasireotide long-acting release in patients with metastatic neuroendocrine tumors and carcinoid symptoms refractory to available somatostatin analogues
Authors Wolin E, Jarzab B, Eriksson B, Walter T, Toumpanakis C, Morse MA, Tomassetti P, Weber MM, Fogelman D, Ramage J, Poon D, Gadbaw B, Li J, Pasieka JL, Mahamat A, Swahn F, Newell-Price J, Mansoor W, Öberg K
Received 7 March 2015
Accepted for publication 29 May 2015
Published 3 September 2015 Volume 2015:9 Pages 5075—5086
DOI https://doi.org/10.2147/DDDT.S84177
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Edward M Wolin,1 Barbara Jarzab,2 Barbro Eriksson,3 Thomas Walter,4 Christos Toumpanakis,5 Michael A Morse,6 Paola Tomassetti,7 Matthias M Weber,8 David R Fogelman,9 John Ramage,10 Donald Poon,11 Brian Gadbaw,12 Jiang Li,12 Janice L Pasieka,13 Abakar Mahamat,14 Fredrik Swahn,15 John Newell-Price,16 Wasat Mansoor,17 Kjell Öberg3
1Markey Cancer Center, University of Kentucky, Lexington, KY, USA; 2Department of Nuclear Medicine and Endocrine Oncology, Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Gliwice Branch, Gliwice, Poland; 3Department of Medical Sciences, Endocrine Oncology Unit, University Hospital, Uppsala, Sweden; 4Department of Medical Oncology, Edouard Herriot Hospital, Lyon, France; 5Gastroenterology and Neuroendocrine Tumours, Royal Free Hospital, London, UK; 6Department of Medical Oncology, Duke University Medical Center, Durham, NC, USA; 7Department of Medical and Surgical Sciences, University Hospital St Orsola, Bologna, Italy; 8Medizinische Klinik und Poliklinik, Johannes Gutenberg-Universität Mainz, Mainz, Germany; 9Department of Gastrointestinal Medical Oncology, MD Anderson Cancer Center, Houston, TX, USA; 10Gastroenterology Unit, North Hampshire Hospital, Basingstoke, UK; 11Department of Medical Oncology, Raffles Hospital and Duke–NUS Graduate Medical School, Singapore; 12Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA; 13Surgery and Oncology Faculty of Medicine, Foothills Hospital, Calgary, AB, Canada; 14Department of Gastrointestinal Oncology, CHU de Nice Hôpital de l’Archet 1, Nice, France; 15Department of Clinical Science, Intervention and Technology, Karolinska Universitatssjukhuset, Huddinge, Stockholm, Sweden; 16Department of Human Metabolism, School of Medicine and Biomedical Science, The University of Sheffield, and Sheffield Teaching Hospitals NHS Foundation Trust, Sheffield, UK; 17Department of Medical Oncology, The Christie NHS Foundation Trust, Manchester, UK
Abstract: In a randomized, double-blind, Phase III study, we compared pasireotide long-acting release (pasireotide LAR) with octreotide long-acting repeatable (octreotide LAR) in managing carcinoid symptoms refractory to first-generation somatostatin analogues. Adults with carcinoid tumors of the digestive tract were randomly assigned (1:1) to receive pasireotide LAR (60 mg) or octreotide LAR (40 mg) every 28 days. Primary outcome was symptom control based on frequency of bowel movements and flushing episodes. Objective tumor response was a secondary outcome. Progression-free survival (PFS) was calculated in a post hoc analysis. Adverse events were recorded. At the time of a planned interim analysis, the data monitoring committee recommended halting the study because of a low predictive probability of showing superiority of pasireotide over octreotide for symptom control (n=43 pasireotide LAR, 20.9%; n=45 octreotide LAR, 26.7%; odds ratio, 0.73; 95% confidence interval [CI], 0.27–1.97; P=0.53). Tumor control rate at month 6 was 62.7% with pasireotide and 46.2% with octreotide (odds ratio, 1.96; 95% CI, 0.89–4.32; P=0.09). Median (95% CI) PFS was 11.8 months (11.0 – not reached) with pasireotide versus 6.8 months (5.6 – not reached) with octreotide (hazard ratio, 0.46; 95% CI, 0.20–0.98; P=0.045). The most frequent drug-related adverse events (pasireotide vs octreotide) included hyperglycemia (28.3% vs 5.3%), fatigue (11.3% vs 3.5%), and nausea (9.4% vs 0%). We conclude that, among patients with carcinoid symptoms refractory to available somatostatin analogues, similar proportions of patients receiving pasireotide LAR or octreotide LAR achieved symptom control at month 6. Pasireotide LAR showed a trend toward higher tumor control rate at month 6, although it was statistically not significant, and was associated with a longer PFS than octreotide LAR.
Keywords: neuroendocrine tumors, carcinoid syndrome, somatostatin analogues, pasireotide, symptom control, progression-free survival
Introduction
Neuroendocrine tumors (NET) are rare and heterogeneous neoplasms arising from the diffuse neuroendocrine cell system.1 NET produce and secrete bioactive amines, peptides, and polypeptides.2 Excessive release of bioactive substances, primarily serotonin and substance P, is associated with classic carcinoid syndrome in approximately 20% of patients with metastatic NET arising from jejunum or ileum (functional NET).3,4 Clinical manifestations of carcinoid syndrome include episodic flushing, diarrhea, and bronchoconstriction; 50%–66% of patients later develop valvular cardiac complications resulting from higher levels of 5-hydroxyindoleacetic acid and duration of the elevated 5-hydroxyindoleacetic acid.3,5,6
Somatostatin analogues (SSA) remain the gold standard treatment for patients with functional NET.5,7 Octreotide and lanreotide effectively reduce the frequency of diarrhea and flushing in 70%–90% of patients with carcinoid symptoms.5,7–9 Loss of symptom response due to tachyphylaxis has been reported approximately 6–18 months after initiation of treatment.10
Pasireotide (SOM230) is expected to have a distinct pharmacodynamic profile given its broader specificity and high affinity for somatostatin receptors 1–3 (sst1–3) and sst5 compared with octreotide and lanreotide, which have greater specificity for sst2.11,12 In a recent Phase II study, subcutaneous (SC) pasireotide effectively treated symptoms of patients with metastatic functional carcinoid tumors resistant to octreotide long-acting repeatable (octreotide LAR) therapy.13
Our Phase III study compared the efficacy and safety of pasireotide long-acting release (pasireotide LAR) with those of octreotide LAR in controlling diarrhea and flushing in patients with metastatic NET whose disease-related symptoms were inadequately controlled by the highest recommended doses of first-generation SSA. A post hoc progression-free survival (PFS) analysis was performed following the results of objective tumor response.
Methods
Study design
This was a multicenter, randomized, blinded, efficacy and safety, Phase III study of pasireotide LAR versus octreotide LAR in patients with metastatic NET of the digestive system who had inadequately controlled carcinoid symptoms. Planned enrollment was 216 patients (108 in each arm; see the “Sample size and interim analysis” section in supplementary materials, Table S1) from 47 centers in 15 countries (Argentina, Austria, Belgium, Brazil, Canada, France, Germany, Italy, Norway, Poland, Singapore, Spain, Sweden, UK, and USA) (Figure 1; ClinicalTrials.gov identifier, NCT00690430).
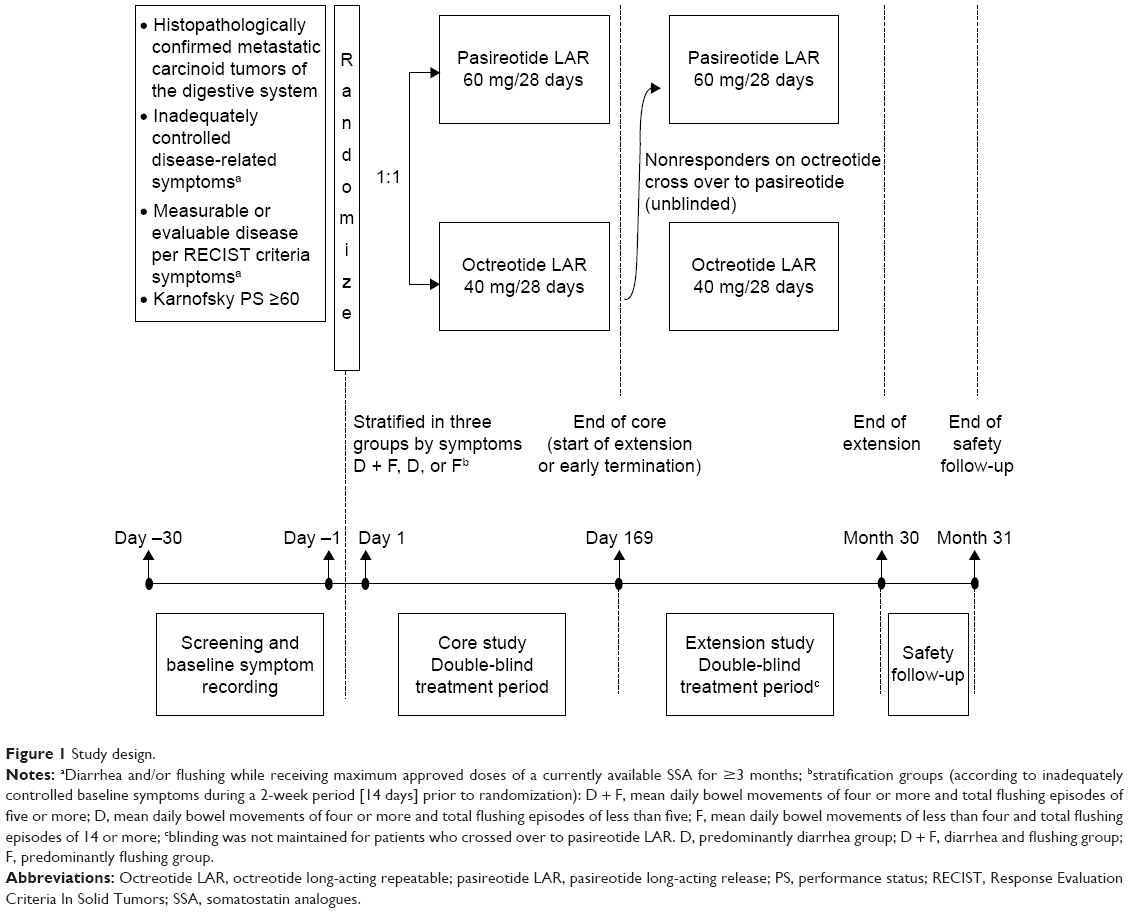 | Figure 1 Study design. |
The study protocol was reviewed by an independent ethics committee or institutional review board at each site and met the principles of the Declaration of Helsinki. The study was monitored by an independent data monitoring committee and was overseen by a protocol steering committee. All patients provided written informed consent before participation in the study.
Patients
Adult patients (age ≥18 years) with carcinoid tumors of the digestive tract were eligible for study enrollment if they had a histopathologically confirmed metastatic tumor and one evaluable lesion as assessed according to Response Evaluation Criteria In Solid Tumors (RECIST; version 1.0).14 For study participation, disease progression prior to study entry was not mandatory. Other key inclusion criteria included inadequately controlled diarrhea and/or flushing (as defined under patient screening and treatment) while receiving maximum approved doses of the currently available SSA for 3 months prior to study entry; Karnofsky performance status ≥60; and adequate bone marrow, renal, and hepatic function. Previous SSA doses to establish inadequate control during screening period included octreotide LAR 30 mg every 28 days, octreotide SC 600 μg (total daily dose), lanreotide autogel 120 mg every 28 days, or lanreotide SR 30 mg every 14 days. Patients with impaired fasting glucose or diabetes mellitus were eligible. In such patients, HbA1c and fasting glucose were routinely measured throughout the study period, additional monitoring was performed, and antidiabetic therapy was started as clinically indicated per investigator’s discretion.
Patients were ineligible if they received an SSA at a higher than approved dose (except a short-acting formulation) within 3 months before screening; were receiving radiolabeled SSA therapy (within 3 months before recording baseline symptoms); received any cytotoxic chemotherapy or interferon therapy (within 4 weeks); underwent major surgery (within 1 month before recording baseline symptoms) or surgical therapy of locoregional metastases (within 3 months); underwent hepatic artery embolization, chemoembolization, or radioembolization (yttrium 90 microspheres) within 6 months (or 1 month if there were other disease sites) or cryoablation or radiofrequency ablation of hepatic metastases within 2 months before recording baseline symptoms; received prior therapy with pasireotide; or had diabetes and poorly controlled blood glucose levels (glycosylated hemoglobin >8%).
Patient screening and treatment
Inadequate control of carcinoid symptoms was defined as a daily mean of four or more bowel movements over a 2-week period and five or more flushing episodes during the same period within the screening phase. Patients recorded symptoms during screening on a touch-tone telephone system.
After screening, eligibility confirmation, and a washout period (see the “Washout period” section in supplementary materials), patients were randomly assigned 1:1 to receive blinded treatment with pasireotide LAR 60 mg or octreotide LAR 40 mg. An interactive voice response system was used for randomization. True double blinding was not feasible due to the different appearances of the LAR formulations. Blinding was achieved by an independent nurse who knew the treatment assignment (see the “Treatment blinding” section in supplementary materials).
Treatments were administered via intragluteal depot injections once every 28 days (±3 days) by an independent study nurse. For patients who experienced breakthrough symptoms after the first injection, SC rescue medication was permitted (ie, pasireotide 600 μg bid SC for patients randomized to pasireotide LAR and octreotide 100 μg tid SC for patients randomized to octreotide LAR). Dose reductions to pasireotide LAR 40 mg and octreotide LAR 30 mg for safety and tolerability were allowed (see the “Permitted study drug adjustments” section in supplementary materials).
The treatment and evaluation periods for the core study were 6 months. A blinded treatment extension of up to 2 years was available in all participating countries except the UK. Patients not benefiting from octreotide by the end of the 6-month core phase were allowed to switch to pasireotide LAR treatment on entry into the extension phase and the blind was broken (see the “Extension study” section in supplementary materials).
Assessments
Efficacy
The primary efficacy outcome was symptom control (diarrhea and/or flushing) based on patient reports of daily frequency of bowel movements and flushing episodes. A patient was considered to be achieving symptom improvement according to the following subgroup-specific criteria (over a 28-day period comprising month 6): stratum D, less than four daily mean bowel movements and ≥20% reduction in the daily mean number of bowel movements from baseline; stratum F, ≥30% reduction in the total number of flushing episodes from baseline; and stratum D + F, less than four mean bowel movements, and ≥20% reduction in the daily mean number of bowel movements from baseline plus any reduction in the total number of flushing episodes compared with baseline.
Secondary analyses included evaluating the frequency of bowel movements alone and the number of flushing episodes alone during month 6 (the last 28 days of treatment) relative to the baseline assessment and the objective tumor response rate (complete response or partial response) and tumor control rate (complete response, partial response, or stable disease) at month 6 using RECIST criteria (based on the tumor responses assessed by the investigators). Tumors were measured using computed tomography or magnetic resonance imaging at baseline and every 3 months thereafter.
Safety
Safety assessments included monitoring and recording all adverse events (AEs) based on the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0.15
Statistical analysis
The full analysis set consisted of all randomized patients. Following the intent-to-treat principle, these patients were analyzed according to the treatment assigned at randomization. Demographic and baseline characteristics as well as tumor response were evaluated based on the full analysis set.
The efficacy analyzable set consisted of the subset of full analysis set patients randomized at least 6 months before the futility data monitoring committee data cutoff. It was used to assess primary and secondary efficacy outcomes, with the exception of tumor response. A two-sided Cochran–Mantel–Haenszel test (adjusted for the inadequately controlled symptom subgroups; significance level, 0.05) compared proportions of patients experiencing symptom improvement. Change from baseline in mean daily bowel movements and flushing episodes at month 6 was compared using an analysis of covariance model. A two-sided Fisher’s exact test compared objective tumor response rate. Odds ratios (ORs) and corresponding 95% confidence intervals (CIs) were calculated using logistic regression.
The safety analysis set consisted of patients who received at least one dose of study drug and had at least one post-baseline safety assessment, regardless of whether they entered the extension phase.
The extension analysis set consisted of patients receiving at least one dose of study medication in the extension phase and was used for safety analyses in the extension phase. Patients in the extension analysis set were stratified by the treatment received in the extension phase, as follows: pasireotide LAR, patients continuing on pasireotide LAR in the extension phase; octreotide LAR, patients continuing on octreotide LAR in the extension phase; crossover to pasireotide LAR, patients receiving octreotide LAR in the core phase and pasireotide LAR in the extension phase. Patients were excluded if no treatment information was available on entry into the extension phase.
An exploratory analysis of PFS was performed to evaluate the durability of the tumor response data. Distributions were assessed by the Kaplan–Meier method and were compared between treatment arms using a two-sided unstratified log-rank test. Hazard ratio (HR) was estimated with unstratified Cox regression.
Results
One hundred ten patients were enrolled between April 2008 and April 2012, before the study was halted following a data monitoring committee recommendation due to a low predictive probability of showing superiority of pasireotide over octreotide for symptom control (Figure 2). Baseline demographic and clinical characteristics of the patients in the two treatment arms were similar (Table 1).
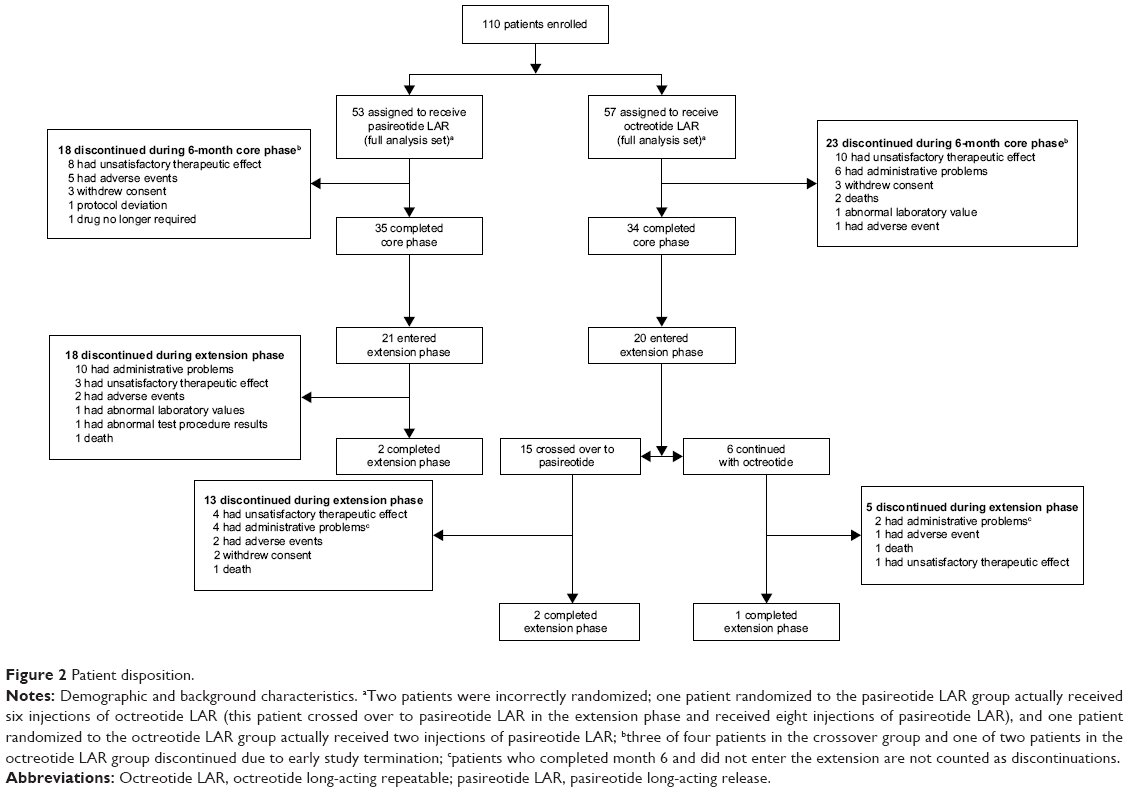 | Figure 2 Patient disposition. |
 | Table 1 Demographic and baseline characteristics of the patients |
Efficacy
Similar proportions of patients receiving pasireotide LAR (20.9%) or octreotide LAR (26.7%) achieved symptom control at month 6 (OR, 0.73; 95% CI, 0.27–1.97; P=0.53) (Table 2). At month 6, the magnitude of reduction in frequency of bowel movements and flushing episodes was comparable between treatments (Table S2).
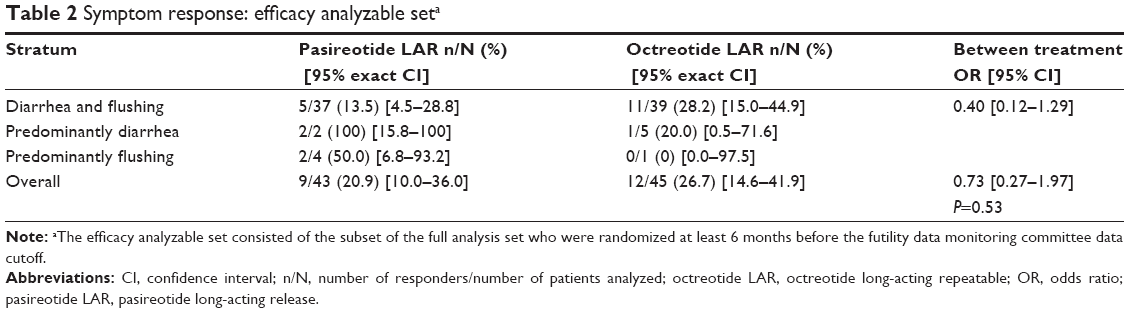 | Table 2 Symptom response: efficacy analyzable seta |
Objective tumor response
At month 6, a higher proportion of patients in the pasireotide LAR arm (60.8%) than in the octreotide LAR arm (42.3%) achieved stable disease, with a comparable objective response rate between arms (2.0% in the pasireotide LAR vs 3.8% in the octreotide LAR arm; OR, 0.50; 95% CI, 0.04–5.69; P=0.57). The tumor control rate at month 6 was 62.7% with pasireotide LAR and 46.2% with octreotide LAR (OR, 1.96; 95% CI, 0.89–4.32; P=0.09). At 48 months, the objective tumor response and disease control rates were similar between treatment arms (Table S3).
Progression-free survival
In a post hoc analysis, the median PFS based on investigator-assessed tumor response was 11.8 months (95% CI, 11.0 – not reached) in patients receiving pasireotide LAR versus 6.8 months (95% CI, 5.6 – not reached) in patients receiving octreotide LAR (unstratified Cox regression model HR, 0.46; 95% CI, 0.20–0.98; P=0.045) (Figure 3).
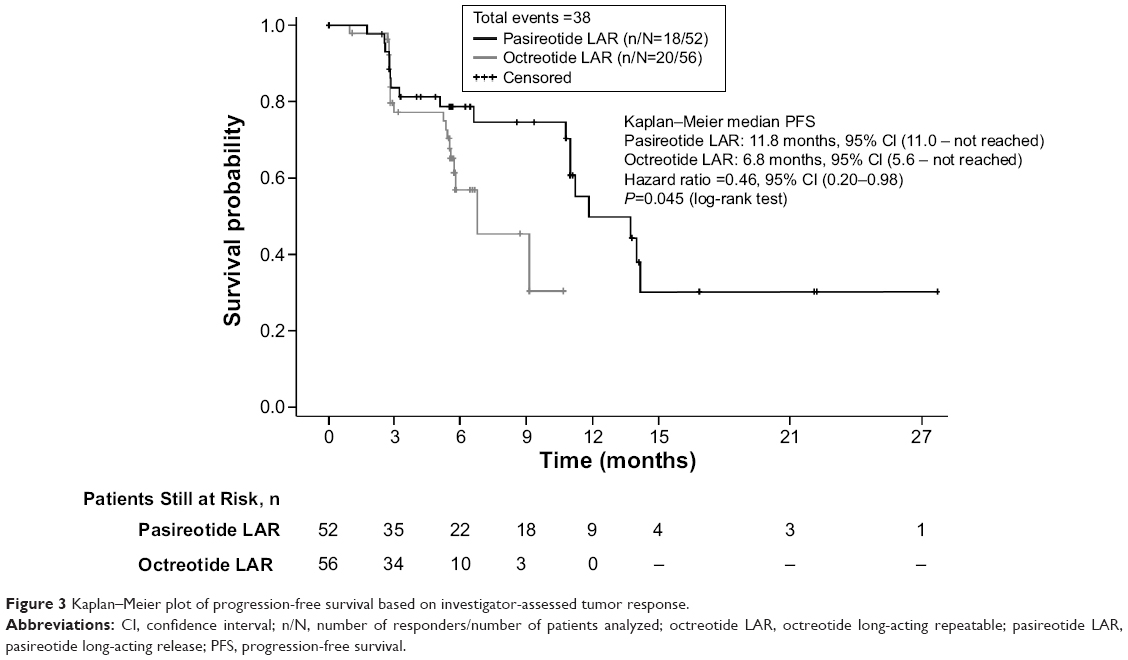 | Figure 3 Kaplan–Meier plot of progression-free survival based on investigator-assessed tumor response. |
Safety and tolerability
The most common drug-related AEs of any grade among patients who received pasireotide LAR were hyperglycemia, fatigue, and nausea (Table 3). The rate of grade 3 or 4 hyperglycemia was higher with pasireotide LAR (9.4%) than with octreotide LAR (1.8%).
 | Table 3 Drug-related adverse events reported in at least 5% of patients |
AEs leading to treatment discontinuation were more frequent with pasireotide LAR (17.0%) than with octreotide LAR (7.0%). Two patients receiving pasireotide LAR discontinued treatment because of grade 4 hyperglycemia, which was manageable with antidiabetic medication. During the core phase, AEs leading to study drug discontinuation occurred in seven patients (13.2%) in the pasireotide LAR arm and were considered study drug related in four patients (7.5%): one patient each with diarrhea and flushing, diarrhea and bradycardia, hyperglycemia, or liver enzyme elevation. In the octreotide LAR arm, no events leading to study discontinuation were considered to be drug related. During the extension phase, six patients discontinued the study drug due to an AE: two patients in the pasireotide LAR arm, one patient in the octreotide LAR arm, and three patients in the crossover to pasireotide LAR arm. For two patients from the crossover arm, the drug-related AEs leading to study discontinuation included orthostatic hypotension and dysesthesia and liver enzyme elevation.
Serious AEs occurred with comparable frequency in the pasireotide LAR (41.5%) and octreotide LAR (36.8%) arms, although more serious AEs in the pasireotide LAR arm were drug related (17% vs 3.5% with octreotide LAR). Five deaths were reported: two in the octreotide LAR arm during the core phase due to health deterioration or disease progression and three during the extension phase due to disease progression (pasireotide LAR), hepatic failure (octreotide LAR), and carcinoid tumor of the gastrointestinal tract (crossover to pasireotide LAR). All deaths were attributed to disease progression or underlying malignancy.
Discussion
SSA therapy remains the standard of care for treatment of hormonal symptoms in functional NET. Many patients with improved carcinoid symptoms experience recurrence and/or worsening of symptoms despite continued treatment with octreotide or lanreotide, and effective management of refractory carcinoid syndrome remains an unmet medical need.7,16 Although options such as combining interferon-alpha with SSA or locoregional treatment (such as hepatic transcatheter arterial embolization) could be considered in refractory patients, the lack of data from well-controlled studies and poor tolerance due to AEs with such therapies substantiate the need for new agents. Pasireotide LAR was as effective as high-dose octreotide LAR in controlling symptoms refractory to the highest recommended doses of first-generation SSA.
It was anticipated that pasireotide LAR would have greater efficacy than octreotide LAR due to its broader receptor-binding profile. During the planned interim futility analysis, however, the study showed similar efficacy for symptom control in an SSA-refractory population despite ~40% of patients in each treatment arm discontinuing the study. The higher discontinuation rates due to unsatisfactory therapeutic effect, 15% in the pasireotide LAR arm and 17% in the octreotide LAR arm, is not unexpected considering that the study population were refractory to SSA therapy for symptom control at study entry. In addition, imbalances between treatment arms in baseline demographic characteristics (particularly proportions of patients with a shorter period since initial diagnosis, Karnofsky score, and heterogeneity of primary tumor sites) might have confounded results.
Earlier, in an open-label, multicenter, Phase II study, Kvols et al13 evaluated pasireotide SC (600–900 μg bid) in 45 patients with metastatic NET resistant to octreotide LAR. They showed that pasireotide effectively controlled carcinoid syndrome symptoms in 27% of patients (symptom control for diarrhea and flushing over any consecutive 15-day period at a fixed dose of pasireotide, efficacy population).13 Consistent with these findings, we found that after 6 month’s treatment, pasireotide LAR controlled symptoms in 21% of patients refractory to octreotide or lanreotide. Collectively, the results strongly support pasireotide LAR as an effective medical therapy for symptom control in patients with carcinoid syndrome inadequately controlled by currently available SSA. The applicability of the findings to SSA treatment-naive patients merits investigation.
SSA have demonstrated antitumor activity in addition to their effects on symptom control in patients with functional NET. Octreotide is the first SSA shown to have antitumor efficacy in a Phase III randomized controlled trial. In the PROMID (Placebo-controlled, double-blind, prospective, Randomized study on the effect of Octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine MIDgut tumors) study, octreotide LAR significantly prolonged median time to tumor progression in patients with newly diagnosed advanced metastatic midgut NET with or without secretory symptoms (14.3 months vs 6.0 months with placebo; HR, 0.34; 95% CI, 0.20–0.59; P=0.000072).17 In the same study, 66.7% of patients receiving octreotide LAR experienced stable disease after 6 months (vs 37.2% of those from the placebo arm). Recently, the CLARINET study showed that lanreotide improved PFS in patients with stable disease gastroenteropancreatic NET versus placebo (lanreotide, PFS not reached; placebo, PFS 18 months; HR, 0.47; 95% CI, 0.30–0.73; P=0.0002).18
This study was designed to evaluate efficacy of pasireotide for symptom control, and therefore documentation of disease progression prior to study entry was not required. An improved, though not statistically significant, tumor control rate at 6 months for patients receiving pasireotide LAR in our study (62.7% vs 46.2% in patients receiving octreotide LAR; OR, 1.96; 95% CI, 0.89–4.32; P=0.09) drove an exploratory PFS analysis. In the post hoc analysis, pasireotide LAR prolonged median PFS by 5 months (11.8 months vs 6.8 months with octreotide LAR), corresponding to a 54% reduction in estimated risk for disease progression or death (HR, 0.46; 95% CI, 0.20–0.98; P=0.045). This effect of pasireotide LAR on PFS is very encouraging given the refractory population. Data from a recent open-label, Phase II study of pasireotide LAR have also shown promising antitumor efficacy with PFS of 12.2 months (95% CI, 7.7–17.5) in patients with metastatic NET without prior systemic therapy, including octreotide or lanreotide.19 The antitumor activity of pasireotide LAR warrants further exploration in a larger study with PFS and additional tumor control metrics as predefined end points.
The tumor control rate at 6 months observed in the octreotide LAR arm of our study was 46.2%, whereas that observed in the PROMID trial was 66.7%.17 Our study is not easily comparable with the PROMID study, in which most patients were treatment naive, whereas all patients in our study were symptomatic while being treated with the highest recommended doses of available SSA.
The safety profiles of pasireotide and octreotide were consistent with the known safety profiles of both agents. Although the incidence of hyperglycemia was higher in the pasireotide LAR arm, it was manageable. One patient discontinued due to hyperglycemia suspected to be related to the study drug. The increased incidence of hyperglycemia among patients receiving pasireotide has been reported.13,19 No cases of ketosis or ketoacidosis were observed in this study.
Studies of pasireotide LAR in patients with NET are ongoing. The Phase I COOPERATE-1 study has been extended to evaluate the long-term safety and tolerability of pasireotide LAR combined with everolimus (NCT01590199). The randomized, open-label, Phase II COOPERATE-2 study will investigate the efficacy of pasireotide LAR plus everolimus in patients with advanced progressive pancreatic NET (NCT01374451).
Conclusion
In conclusion, our findings show that pasireotide LAR and high-dose octreotide LAR have similar efficacy for symptom control in patients with functional NET and symptoms inadequately controlled with the highest recommended doses of available SSA. At month 6, a trend toward higher tumor control rate was observed with pasireotide LAR, although it was statistically not significant. Pasireotide LAR was associated with a longer PFS compared with octreotide LAR. Safety findings were consistent with known AE profiles for both drugs. These results support further investigation of the antitumor activity of pasireotide LAR in adequately powered prospective Phase III studies and establish its role in tumor control for patients with NET.
Acknowledgments
The authors thank Bhavik Shah, PhD, of Novartis Healthcare Pvt. Ltd., Hyderabad, India, for medical writing and editorial assistance and Jeffrey M Palmer, PhD, of ApotheCom, Yardley, Pennsylvania, USA, for editorial assistance. The paper was presented at the 2013 American Society of Clinical Oncology (ASCO) Annual Meeting; May 31–June 4, 2013; Chicago, IL, USA.
Author contributions
EMW was involved in every aspect of the study, including study design, recruitment of patients, interpretation of data, and drafting, critically revising, and final approval of the manuscript. BJ, BE, TW, CT, MM, PT, MMW, DF, JR, DP, BG, JL, JLP, AM, FS, JNP, WM, and KÖ also critically reviewed and approved the final manuscript. BJ, WM, and KÖ also contributed to the study design or protocol revisions. EMW, BJ, BE, TW, CT, MM, PT, MMW, DF, JR, JLP, AM, FS, JNP, WM, and KÖ recruited study patients. BJ, BE, TW, MM, MMW, DF, DP, BG, JL, FS, JNP, WM, and KÖ participated in data acquisition and analysis. All authors contributed to the interpretation of data and subsequent writing, reviewing, and amending of the manuscript. All authors also approved the final version of the manuscript and made the decision to submit for publication.
Funding
This study was sponsored by Novartis Pharmaceuticals Corporation. The study was designed by the academic investigators and by representatives of the sponsor. Data were collected with the use of the sponsor’s data management systems and were analyzed by the sponsor’s statistical team complying with the study protocol and statistical analysis plan. Writing assistance funded by the sponsor was provided.
Disclosure
BG is an employee and stockholder of Novartis AG. BJ has received honoraria from AstraZeneca, Bayer HealthCare Pharmaceuticals, SOBI, sanofi, Novartis Pharmaceuticals Corporation, Ipsen, Pfizer Inc., Roche, Eisai, and OxiGene. BG has also received research support from Bayer HealthCare Pharmaceuticals and consultancy fees from AstraZeneca and SOBI. JL is an employee of Novartis Pharmaceuticals Corporation. MM has received personal fees from Novartis Pharmaceuticals Corporation. JNP has received grants and consultancy fees from Novartis Pharmaceuticals Corporation. KÖ has received personal fees from Novartis Pharmaceuticals Corporation and Ipsen. JR has received speaker and advisory board fees and research grants from Novartis Pharmaceuticals Corporation. CT has received travel grants and honoraria from Novartis Pharmaceuticals Corporation. MMW has received a research grant and honoraria from Novartis Pharmaceuticals Corporation. EMW has served on advisory boards for Novartis Pharmaceuticals Corporation, Ipsen, and Celgene. All other authors declare that they have no conflicts of interest.
References
Vinik AI, Woltering EA, Warner RR, et al; North American Neuroendocrine Tumor Society (NANETS). NANETS consensus guidelines for the diagnosis of neuroendocrine tumor. Pancreas. 2010;39(6):713–734. | ||
Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–3072. | ||
Klöppel G. Tumour biology and histopathology of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21(1):15–31. | ||
National Cancer Institute. General Information about Gastrointestinal Carcinoid Tumors. NCI Web site. Available from: http://www.cancer.gov/cancertopics/pdq/treatment/gastrointestinalcarcinoid/HealthProfessional/page1#Section_231. Accessed August 25, 2014. | ||
National Comprehensive Cancer Network. Clinical practice Guideline in Oncology. Neuroendocrine Tumors Version 2. 2014. NCCN Web site. Available from: http://www.nccn.org/professionals/physician_gls/pdf/neuroendocrine.pdf. Accessed August 25, 2014. | ||
Modlin IM, Kidd M, Latich I, Zikusoka MN, Shapiro MD. Current status of gastrointestinal carcinoids. Gastroenterology. 2005;128(6):1717–1751. | ||
Pavel M, Baudin E, Couvelard A, et al; Barcelona Consensus Conference participants. ENETS consensus guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology. 2012;95(2):157–176. | ||
Kvols LK, Moertel CG, O’Connell MJ, Schutt AJ, Rubin J, Hahn RG. Treatment of the malignant carcinoid syndrome: evaluation of a long-acting somatostatin analogue. N Engl J Med. 1986;315(11):663–666. | ||
Vinik A, Wolin EM, Audry H, Gomez-Panzani EL; ELECT Study Group. ELECT: a phase 3 study of efficacy and safety of lanreotide autogel/depot (LAN) treatment for carcinoid syndrome in patients with neuroendocrine tumors (NETs) [abstract]. J Clin Oncol. 2014;32(3 suppl):268. | ||
Öberg KE. The management of neuroendocrine tumours: current and future medical therapy options. Clin Oncol (R Coll Radiol). 2012;24(4):282–293. | ||
Schmid HA. Pasireotide (SOM230): development, mechanism of action and potential applications. Mol Cell Endocrinol. 2008;286(1–2):69–74. | ||
Plöckinger U, Wiedenmann B. Treatment of gastroenteropancreatic neuroendocrine tumors. Virchows Arch. 2007;451(suppl 1):S71–S80. | ||
Kvols LK, Oberg KE, O’Dorisio TM, et al. Pasireotide (SOM230) shows efficacy and tolerability in the treatment of patients with advanced neuroendocrine tumors refractory or resistant to octreotide LAR: results from a phase II study. Endocr-Relat Cancer. 2012;19(5):657–666. | ||
Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors: European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–216. | ||
National Cancer Institute. Common Terminology Criteria for Adverse Events version 3.0 (CTCAE v3.0). Cancer Therapy Evaluation Program Web site. Available from: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed August 25, 2014. | ||
Toumpanakis C, Caplin ME. Update on the role of somatostatin analogs for the treatment of patients with gastroenteropancreatic neuroendocrine tumors. Semin Oncol. 2013;40(1):56–68. | ||
Rinke A, Müller HH, Schade-Brittinger C, et al; PROMID Study Group. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656–4663. | ||
Caplin ME, Pavel M, Ćwikła JB, et al; CLARINET Investigators. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224–233. | ||
Strosberg JR, Jump H, Valone T, Weber J, Khandelwal V, Kunz P. Phase II clinical trial of pasireotide LAR in patients with metastatic neuroendocrine tumors [abstract]. J Clin Oncol. 2014;32(3 suppl):241. |
Supplementary materials
Sample size and interim analysis
Among patients with inadequately controlled symptoms in both diarrhea and flushing (D + F), the response rate for octreotide long-acting repeatable (LAR; 40 mg) treatment is estimated by medical experts to be ~20%. Given that the primary analysis is based on the full analysis set according to intent-to-treat principle, the treatment effect may be diluted due to early dropouts since early dropouts are considered treatment failures. With an estimated 20% dropout rate, the response rate based on full analysis set was expected to be 32% for pasireotide long-acting release (LAR) versus 16% for octreotide LAR. The primary efficacy end point was to be analyzed using a two-sided Cochran–Mantel–Haenszel test adjusting for the inadequately controlled symptom subgroups. This allows for detection of an odds ratio of 2.5. The estimated patient population from each subgroup is listed in Table S1.
 | Table S1 Estimated patient population by inadequately controlled symptom subgroup |
A 20% difference was considered clinically meaningful to demonstrate the superiority of pasireotide over octreotide among this group of patients. With one interim analysis using 15% predictive power boundary, when 40% of patients complete the study, a sample size of 216 patients (108 patients per treatment arm) will be able to detect a common odds ratio of 2.5 between pasireotide LAR and octreotide LAR with 77% power, adjusting for the three inadequately controlled symptom groups.
Interim analysis
A futility analysis was planned to be performed when 40% of patients (approximately 86) completed the month 6 assessment. This included all patients who were randomized at least 6 months before the data cutoff date for the futility analysis. The primary intent of this futility analysis was to assess the risk–benefit ratio and to assess whether the study should be stopped in case futility was demonstrated based on the primary end point: proportion of patients achieving response at month 6. Analysis results were to be reviewed by the data monitoring committee, which was then to recommend whether the study should continue. A futility boundary defined in terms of predictive power (PP) was used for the futility analysis. PP was the probability of rejecting the null hypothesis at the final analysis given the data observed at the interim. The response rate affects the PP only through its prior distribution. For calculating PP, it was assumed that the response rate came from a noninformative prior (ie, beta (1, 1) distribution), which is equivalent to a prior sample size of 1. The futility analysis had the following characteristics:
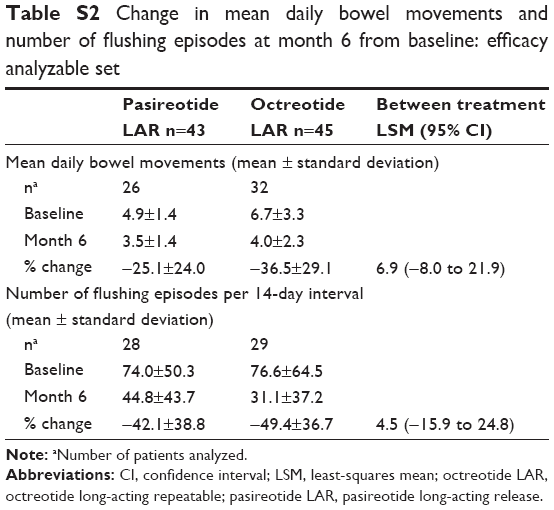 | Table S2 Change in mean daily bowel movements and number of flushing episodes at month 6 from baseline: efficacy analyzable set |
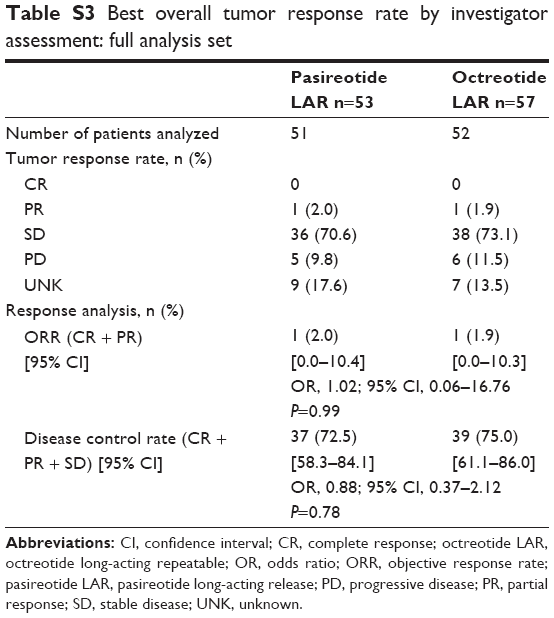 | Table S3 Best overall tumor response rate by investigator assessment: full analysis set |
- It was planned to be performed when 40% of patients (~86) had had the chance to complete the month 6 assessment.
- It was nonbinding (ie, the P-value of rejecting the null hypothesis was not impacted by the futility analysis and remained at two-sided 0.05).
- Predictive power boundary of 15% was the futility boundary.
At interim, if PP was ≤15%, then the trial could be discontinued due to lack of efficacy.
Washout period
For patients on a long-acting somatostatin analogues (SSA), symptom recordings started immediately after patients received the last dose of SSA and continued for 4 weeks (28 days). The last 2-week period (days 15–28) was used to assess the frequency of diarrhea and flushing events at baseline. Use of octreotide subcutaneous (SC) or a short-acting antidiarrheal agent was permitted during the first 2 weeks (ie, before baseline assessment) as needed for symptom control. However, no additional treatment was allowed during the last 2-week period (ie, during baseline assessment, days 15–28). In addition, patients were to observe an 8-hour interval between the last octreotide SC dose or a short-acting antidiarrheal agent before the start of the baseline assessment period on day 15.
For the patients who were receiving a short-acting SSA before study entry, baseline assessment started immediately after their enrollment and continued for 2 weeks (14 days). These patients continued their prior treatment while baseline assessments were being recorded. No other medication to control carcinoid symptoms was permitted during baseline assessment for these patients.
Treatment blinding
Due to the different appearances of the LAR formulations, a true double-blind treatment was not feasible. Blinding was achieved by having an independent study nurse/coordinator (to whom the treatment assignment was known) administer the LAR injections. The patient, investigator, and sponsor were blinded to treatment assignment. The independent study nurse/coordinator was required to keep the treatment information confidential and was instructed not to discuss or release information on treatment identities to the patient, the investigator, the sponsor’s clinical monitor, or other unauthorized personnel. Before study drug administration, the independent nurse/coordinator had to document the date and time of administration, detach the outer part of the label from the packaging, record the patient number on the label, and affix it to the source document (drug label form) containing that patient’s unique patient number. This document was kept strictly concealed from the patient, the investigator, and the sponsor’s clinical monitor. Immediately after study drug administration, the nurse/study coordinator was instructed to remove vials, ampules, reconstitution vials, and the outer packing and to lock them in a location inaccessible to others. In addition, the persons performing the assessments and the data analysts were blinded to the identity of the treatment from the time of randomization until the study had been completed, using the following methods: randomization data were kept strictly confidential until the time of unblinding and were not accessible by anyone else involved in the treatment; the identity of the LAR intramuscular depot treatments was concealed by the independent study nurse, who prepared and administered the injections. Investigators who wished to continue a patient in the extension-treatment period remained blinded if the decision was for the patient to continue the same treatment as in the core phase of the study. Unblinding was allowed after the patient completed 2 years in the extension after all end-of-study assessments had been completed and recorded; however, sponsor personnel and groups performing central assessments remained blinded to treatment until the database was locked. The identity of the SC treatments was concealed by the use of study drugs that were identical in packaging, labeling and schedule of administration, appearance, and odor.
Permitted study drug adjustments
After the first injection, patients who experienced breakthrough symptoms were permitted rescue medications as needed (ie, pasireotide 600 μg bid SC for patients randomized to pasireotide LAR and octreotide 100 μg tid SC for patients randomized to octreotide LAR). SC injections were interrupted if they led to tolerability issues but could be resumed if needed once the tolerability issue resolved. The pasireotide and octreotide rescue medications were supplied in double-blind packaging and were self-administered by patients. Dose reductions to pasireotide LAR 40 mg and octreotide LAR 30 mg were permitted to improve tolerability. Patients requiring dose reductions returned to the higher dose once the tolerability issue resolved.
Extension study
The treatment and evaluation period for core study was 6 months. Patients benefiting from either treatment per investigator assessment and not experiencing unacceptable toxicity were permitted to continue the same treatment and dose for up to 2 years in the extension phase in a blinded manner. Patients not benefiting from pasireotide during the core phase were discontinued, and those not benefiting from octreotide by the end of the core phase were switched to pasireotide LAR treatment on entry into the extension phase and the blind was broken. Patients in the extension phase recorded their symptoms daily in a paper diary. The treatment extension stage was stopped once the patient had achieved 2 years of the treatment extension or when pasireotide was commercially available or the development program was discontinued, whichever came first.
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.