")
Back to Journals » Journal of Experimental Pharmacology » Volume 13
Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis
Authors Pinto MC , Silva IAL, Figueira MF, Amaral MD, Lopes-Pacheco M
Received 4 May 2021
Accepted for publication 30 June 2021
Published 23 July 2021 Volume 2021:13 Pages 693—723
DOI https://doi.org/10.2147/JEP.S255377
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Paola Rogliani
Madalena C Pinto,1,* Iris AL Silva,1,* Miriam F Figueira,2 Margarida D Amaral,1 Miquéias Lopes-Pacheco1,*
1Biosystems & Integrative Sciences Institute (BioISI), Faculty of Sciences, University of Lisboa, Lisboa, Portugal; 2Marsico Lung Institute/Cystic Fibrosis Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
*These authors contributed equally to this work.
Correspondence: Miquéias Lopes-Pacheco
Instituto de Biossistemas e Ciências Integrativas, Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edifício C8, Sala 8.2.50, Lisboa, 1749-016, Portugal
Tel +351 217 500 857
Email [email protected]
Abstract: Cystic fibrosis (CF) is a life-shortening monogenic disease caused by mutations in the gene encoding the CF transmembrane conductance regulator (CFTR) protein, an anion channel that transports chloride and bicarbonate across epithelia. Despite clinical progress in delaying disease progression with symptomatic therapies, these individuals still develop various chronic complications in lungs and other organs, which significantly restricts their life expectancy and quality of life. The development of high-throughput assays to screen drug-like compound libraries have enabled the discovery of highly effective CFTR modulator therapies. These novel therapies target the primary defect underlying CF and are now approved for clinical use for individuals with specific CF genotypes. However, the clinically approved modulators only partially reverse CFTR dysfunction and there is still a considerable number of individuals with CF carrying rare CFTR mutations who remain without any effective CFTR modulator therapy. Accordingly, additional efforts have been pursued to identify novel and more potent CFTR modulators that may benefit a larger CF population. The use of ex vivo individual-derived specimens has also become a powerful tool to evaluate novel drugs and predict their effectiveness in a personalized medicine approach. In addition to CFTR modulators, pro-drugs aiming at modulating alternative ion channels/transporters are under development to compensate for the lack of CFTR function. These therapies may restore normal mucociliary clearance through a mutation-agnostic approach (ie, independent of CFTR mutation) and include inhibitors of the epithelial sodium channel (ENaC), modulators of the calcium-activated channel transmembrane 16A (TMEM16, or anoctamin 1) or of the solute carrier family 26A member 9 (SLC26A9), and anionophores. The present review focuses on recent progress and challenges for the development of ion channel/transporter-modulating drugs for the treatment of CF.
Keywords: anionophores, CFTR modulators, drug development, ENaC, precision medicine, SLC26A9, TMEM16A
Introduction
Mutations in the gene encoding the cystic fibrosis (CF) transmembrane conductance regulator (CFTR) protein cause CF – one of the most common life-shortening autosomal recessive diseases.1–3 CFTR is a member of the ATP-binding cassette (ABC) transporter family and functions as a chloride (Cl–) and bicarbonate (HCO3–) channel expressed at the apical plasma membrane (PM) of epithelial cells in the airways, intestine, pancreas, sweat glands and other organs.4,5 This protein is composed of 1480 amino acid residues that are organized into five domains (Figure 1):6,7 two transmembrane domains (TMD1 and TMD2), two nucleotide binding-domains (NBD1 and NBD2) and an intrinsically disordered regulatory domain (RD). The latter connects the two homologous halves of the protein and is unique to CFTR among ABC transporters. The TMD segments cross the phospholipid bilayer and are connected by extracellular and intracellular loops, thus forming the channel pore through which anions are conducted.6,7 Conformational changes in the protein occur following ATP binding and/or hydrolysis in NBDs and phosphorylation of RD by protein kinase A (PKA) and protein kinase C (PKC), leading to channel opening.6–8 For this complex protein to attain its native functional state, domain folding and interdomain interactions have to occur by cooperative mechanisms.9,10
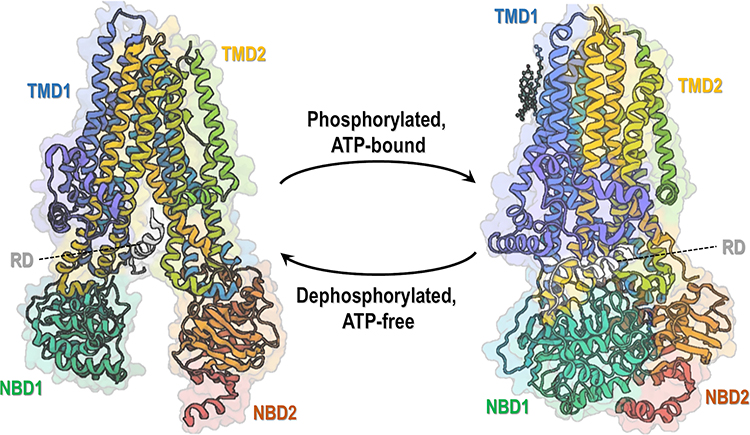 |
Figure 1 Overall structure of CFTR protein. CFTR structure is composed of five functional domains: two transmembrane domains (TMD1 and TMD2), two nucleotide-binding domains (NBD1 and NBD2) and an intrinsically disordered regulatory domain (RD). Ribbon diagram of two conformations of human CFTR: dephosphorylation, ATP-free conformation (left, PDB: 5UAK) (data from Liu et al)6 and phosphorylated, ATP-bound conformation (right, PDB: 6MSM) (data from Zhang et al).7 Notably, only a small portion of RD is depicted as most of its structure remains undetermined due to being intrinsically unstructured. |
CF affects over 90,000 individuals worldwide who are heterogeneously distributed, but with a higher incidence among Caucasians.11 Clinically, the disease has multi-organ involvement, being the respiratory disorder the major cause of morbidity and premature death.4,5,12,13 A cycle of airways dehydration and obstruction by a thick tenacious mucus, chronic inflammation and recurrent infections leads to epithelial injury, tissue remodeling and progressive loss of lung function, ultimately resulting in respiratory failure.4,5,12,13
Over the last decades, major clinical and therapeutic advances have been achieved to delay CF progression. These include mostly time-consuming symptomatic therapies that mitigate lung function deterioration and compensate intestinal malabsorption and pancreatic insufficiency (Table 1). Along with the implementation of newborn screening programs and specialized healthcare management, CF life expectancy has significantly increased with many individuals currently living in their 40s and beyond.14–16 However, these individuals are still overwhelmed by considerable clinical, economic and psychosocial issues, which have a negative impact on their quality of life.11 In order to further enhance life expectancy and significantly reduce therapeutic burdens, CF must be treated beyond its symptoms by addressing the primary defect associated to CFTR mutations, thus halting the detrimental effects downstream of CFTR dysfunction, as indeed has occurred over the last decade.
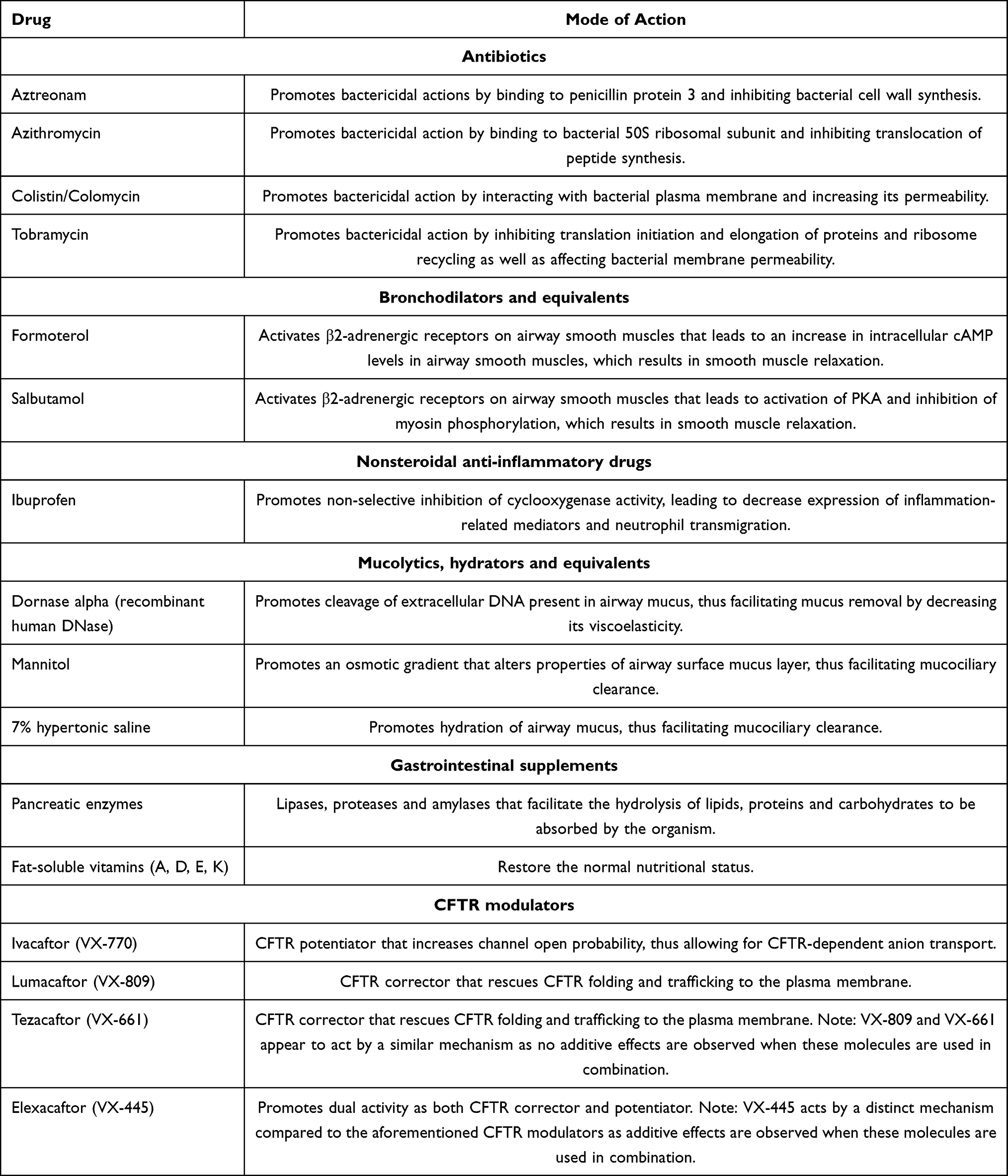 |
Table 1 Pharmacological Therapies Commonly Used in Therapeutic Regimens of Individuals with Cystic Fibrosis |
Numerous assays and high-throughput screens (HTS) have been developed and optimized to screen drug-like compound libraries and identify CFTR modulators.11,17 These specialized small molecules target the root cause of CF by rescuing the functional expression of several CFTR mutants. Significant success has been achieved in this field as a growing number of compounds are under experimental and early-stage clinical development, and four CFTR modulators are now approved for clinical use for individuals with specific CF genotypes (Table 1).18–23 However, the clinically available CFTR modulators, even the highly effective CFTR modulator therapies, only partially correct CFTR dysfunction,24–29 which suggests that there is scope for further enhancement. Moreover, a significant number of individuals with CF, who presumably carry very rare CFTR mutations (termed as “orphan mutations”), remain without any effective “on target” therapy.
More than 2,100 CFTR gene variants have been reported to date (Cystic Fibrosis Mutation Database, http://www.genet.sickkids.on.ca/), for which only 440 the associated disease liability is established and being approximately 360 confirmed as disease causing (Clinical and Functional Translation of CFTR, https://cftr2.org/). However, one single mutation – the deletion of a phenylalanine at position 508 (F508del) located in NBD1 – is found in at least one allele of 80–85% of individuals with CF worldwide.11,17 Nevertheless, such wide variety of CFTR mutations poses substantial challenges as specific drug development for every single mutation is unfeasible and most CFTR mutations are present in a very low number of individuals worldwide. Although all CF-causing mutations result in CFTR-dependent Cl–/HCO3– defective transport, these are due to distinct cellular/functional defects. Accordingly, CFTR mutations have been grouped into functional classes/theratypes,11,12,17,30 characterized by: (I) no production of full-length protein, (II) defective folding and trafficking, (III) defective gating, (IV) reduced anion conductance, (V) reduced protein production, (VI) reduced stability at the PM, and (VII) no mRNA production. Despite not all CFTR gene variants have been characterized according to their respective cellular/functional defect(s), this classification has been useful as mutations within the same group are expected to be treated by the same therapeutic strategy if not by the same drug.
In parallel to CFTR modulators, other pharmacological therapies have emerged aiming to modulate non-CFTR ion channels/transporters that may potentially compensate for CFTR dysfunction.16,31–33 These include strategies to inhibit the epithelial sodium (Na+) channel (ENaC), which is upregulated in CF epithelia, or modulate alternative Cl– channels/transporters, such as the calcium (Ca2+)-activated Cl– channels (CaCCs), namely transmembrane 16 (TMEM16A, or anoctamin 1 [ANO1]), or the solute carrier family 26A member 9 (SLC26A9). Modulating the activity of these ion channels/transporters offers the advantage of functioning agnostically (ie, regardless of the CFTR mutation class) and, therefore, may benefit the entire CF population. These drugs might also be used alone or in combination with CFTR modulators for improved clinical outcomes. Here, we review the recent advances and challenges in the development of pharmacological modulators of CFTR and of other ion channels for the treatment of CF. Furthermore, we summarize advances in the development of anionophores, which are small artificial transmembrane anion transporters, as potential therapeutic strategies for CF.
CFTR Modulator Drugs and Personalized Medicine
CFTR Modulator Drugs
The CF drug development pipeline has been expanding with the discovery of novel small molecules from a diversity of chemical series that are able to correct specific cellular/functional defect(s) generated by CF-causing mutations.17,30 Accordingly, CFTR modulator drugs may be grouped into five main types according to their actions on CFTR mutations: read-through agents, correctors, potentiators, amplifiers and stabilizers (Figure 2). Examples of promising CFTR modulators that are under both experimental and clinical investigation are described in the following sub-sections.
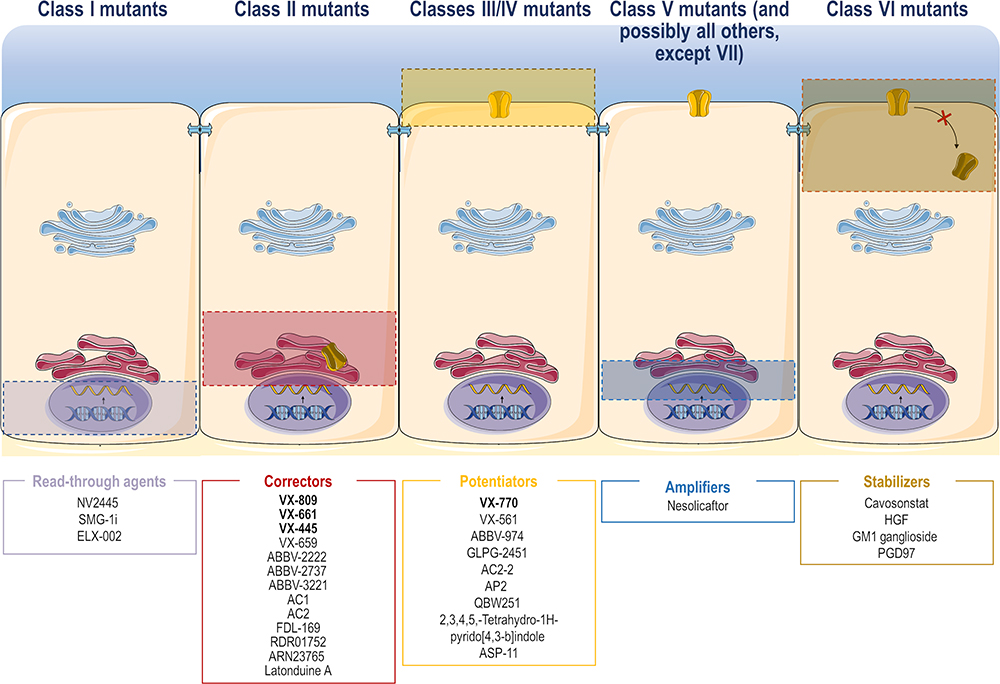 |
Figure 2 Site of action of the different CFTR modulator drugs. CFTR modulator drugs may be grouped into five main types according to their actions on CFTR mutations: read-through agents (for class I mutants), correctors (for class II mutants), potentiators (for classes III and IV mutants), amplifiers (for class V mutants, and possibly all others, except VII) and stabilizers (for class VI mutants). These molecules have a different putative site of action in order to correct specific defects in CFTR mutations. Some examples of promising CFTR modulators that are under experimental and clinical investigation have been provided (see text for further details).Notes: Adapted from Lopes-Pacheco M. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol. 2020;10:1662.11 Copyright © 2020 Lopes-Pacheco. Creative Commons Attribution License (CC BY). |
Class I Mutations: Read-Through Agents
Read-through agents are molecules that enable the incorporation of an amino acid in a site where a premature termination codon (PTC) was introduced into the CFTR mRNA (ie, class I CFTR mutation).34,35 These agents (also termed as PTC suppressors) may prevent protein translation from stopping at PTCs, ie, before the full-length CFTR is produced. PTC suppression is mediated by the base pairing of a near-cognate aminoacyl-tRNA to the PTC and subsequently, the respective amino acid becomes incorporated into the nascent polypeptide chain at the site of the PTC. Accordingly, the local mRNA sequence context plays a key role in near-cognate aminoacyl-tRNA selection during PTC suppression and different PTC mutations will incorporate distinct amino acids, despite treatment with the same read-through agent.36 Furthermore, PTC-carrying transcripts are susceptible to nonsense-mediated decay (NMD)-related degradation, which significantly reduces the abundance of these PTC-carrying transcripts.34,35 Therefore, both NMD and PTC should be suppressed in order to achieve therapeutically relevant levels of CFTR rescue for class I mutants.
Ataluren (PTC124, PTC Therapeutics) was identified in a HTS of ~800,000 molecules using firefly luciferase-based read-through reporters37 and was considered one of the leading compounds to rescue class I CFTR mutations. Despite its promising effects in the experimental setting, ataluren was unable to demonstrate efficacy in Phase III clinical trials involving individuals with CF and thus failed to reduce sweat Cl– concentration and improve the percentage of predicted forced expiratory volume in 1 sec (ppFEV1; a commonly used parameter to measure lung function).38,39 This lack of efficacy led to discontinuation of ataluren development for CF, albeit not for other conditions (eg, Duchenne muscular dystrophy), leaving an unmet need for novel drugs targeting CFTR PTC mutations.
In an effort to identify more effective read-through agents, three ataluren derivative compounds were found to have a higher read-through efficacy, while maintaining low toxicity in a human bronchial epithelial cell line (W1282X/F508del CF genotype).40 However, their efficacy in rescuing PTC CFTR function still needs further investigation. Alternatively, structural chemical replacements, such substitution of 1,2,4-oxadiazoles to 1,3,4-oxadiazoles, were demonstrated to be highly important in drug discovery and enhanced compound properties.41 A novel molecule with structural similarity to ataluren but with a 1,3,4-oxadiazole heterocycle core (termed as NV2445) revealed greater read-through activity when compared to the former molecule and rescued CFTR PM expression and function in cell models carrying different CFTR PTC mutations, including G542X and W1282X.41 NV2245 was also shown to have better pharmacological properties than ataluren,42 and its effects on in vivo experimental and early-stage clinical studies are under investigation.
A molecule able to inhibit the serine/threonine-protein kinase-1 (SMG-1i) was identified as a promising NMD inhibitor.43 SMG-1i was able to modestly increase CFTR mRNA abundance, protein expression and channel function in the 16HBEo– cell line CRISPR-edited to express W1282X-CFTR,44 although such effects were not observed in a following study by other group using this cell model.45 SMG-1i also demonstrated to rescue W1282X-CFTR in primary human nasal epithelial (HNE) cells,46 and synergistic effects were also observed when SMG-1i and other read-through agents (eg, gentamycin, G418/geneticin, paromomycin) were co-administered,47 reinforcing the idea that combination of read-through agents and NMD inhibitors may represent a potential therapeutic option for the treatment of individuals with CF carrying PTC mutations.
ELX-02 is a novel aminoglycoside analogue drug developed by Eloxx Pharmaceuticals that demonstrated read-through activity on CFTR PTC mutations.48 In Phase I clinical studies (NCT02807961, NCT03292302, NCT03309605), 62 healthy volunteers were treated with ELX-02 and no serious adverse events or deaths were reported.49 Two Phase II clinical studies have been initiated with 24 individuals with CF carrying the G542X mutation in at least one allele (NCT04126473, NCT04135495). Interestingly, ELX-02 demonstrated to rescue CFTR activity in the forskolin-induced swelling assay using intestinal organoids from individuals with CF carrying G542X-CFTR.50 An increase in CFTR mRNA abundance and appearance of the fully-glycosylated form of CFTR were also observed in this study.50 Such promising results have supported the ongoing clinical evaluation of ELX-02 for individuals with CF carrying the G542X mutation; however, its read-through activity for other CFTR PTC mutations needs to be further elucidated.
Several HTS have been performed over the last few years in an attempt to identify novel read-through agents for CFTR PTC mutations.11,30 A library containing ~85,000 molecules was screened by the CF Foundation Therapeutics laboratory and some promising hits were found to rescue the Y122X and W1282X mutations, including CFFT-0182812 and CFFT-0176974, which were able to increase both CFTR PM expression and transepithelial conductance.51 Notably, the five leading hits of W1282X increased CFTR PM expression to more than 20% of the WT-CFTR levels, which makes it encouraging to conclude that these hit compounds may yield full-length functional CFTR protein.51 Moreover, this study demonstrated that certain CF-causing mutations may benefit from development of mutation-specific modulators but efficacy ranking may differ significantly among different mutations in the same functional class. A more recent study screening over 660,000 molecules (Scripps Drug Discovery Library) for their ability to rescue G542X identified 188 compounds that rescue this PTC mutation when in combination with other modulators.52 These compounds are now being evaluated in primary cells from individuals with CF in order to validate the previous results and to analyze the translational read-through in a more physiologically relevant context. If successful, translational read-through would be an exciting approach to restore the expression of CFTR carrying PTC mutations to levels approaching those of WT-CFTR. However, as shown by previous studies,35,36,53 the combination of a read-through agent with other modulators that have complementary mechanisms may be required to efficiently rescue CFTR PTC mutations, as the incorporation of a random amino acid may produce a full-length protein which is still misfolded or dysfunctional. Accordingly, in the pursuit of therapeutic options for individuals with CF carrying PTC mutations, and the lack of any clinically approved therapy for this group of individuals, it will be of the outmost importance to exploit the possibility of developing an effective combination of modulators.
Class II Mutations: Correctors
Correctors are small molecules that rescue CFTR mutants with a traffic defect (ie, class II CFTR mutation) to the PM.4,12,54 Defective traffic occurs as a result of CFTR mutations that cause protein misfolding, thus being recognized by the endoplasmic reticulum quality control and targeted to be prematurely degraded in the proteasome.55,56 As the F508del mutation belongs to this class, a considerable number of CF drug programs has been focused on the development of small-molecule correctors that rescue its PM traffic.
Lumacaftor (VX-809) and tezacaftor (VX-661) are two correctors (first- and second-generation, respectively) developed by Vertex Pharmaceuticals that were already approved some years ago by both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA).19,20,57,58 However, despite their promising effects in rescuing F508del-CFTR in vitro,59,60 their effects on improving lung function in clinical studies was somewhat more modest than anticipated.19,20 Indeed, either VX-809 or VX-661 in combination with the potentiator VX-770 (see next sub-section) only led to a fairly small improvement in lung function of individuals with CF homozygous for F508del (~4 and ~7 ppFEV1, respectively).19,20 More recently, a next-generation corrector – elexacaftor (VX-445) – was approved in combination with VX-661/VX-770 and this triple combination led to greater therapeutic benefits in individuals with CF carrying one or two copies of F508del-CFTR,22,23 although this combination of two correctors still promotes only a partial rescue of F508del-CFTR traffic defect.61 A label extension of these drugs has been granted by the FDA to other CF-causing mutations based on in vitro data in cell lines,17 thus significantly increasing the number of individuals with CF who may benefit from these causative therapies. However, there are several class II CFTR mutations unresponsive or only modestly responsive (ie, below therapeutically relevant levels) to experimental and clinically approved correctors.62–68 The real-life and long-term effects of clinically available correctors are also being further investigated, namely regarding liver and kidney function and overall tolerability. Another major limitation of these drugs is their very high costs, which makes them inaccessible for many individuals with CF, particularly for those living in low- and middle-income countries.11,69 In parallel, the same company (Vertex Pharmaceuticals) is pursuing the development of novel correctors, such as VX-121, which is currently being evaluated in combination with VX-661/VX-561 in phase II clinical studies (NCT03912233).
Many other studies have been performed by several academic laboratories and pharmaceutical companies in order to identify more potent correctors that could rescue class II CFTR mutations alone or in combination with the clinically approved ones. Some promising drugs developed by Abbvie/Galapagos include ABBV-2222, ABBV-2737 and ABBV-3221. ABBV-2222 is a first-generation corrector that demonstrated greater efficacy in rescuing CFTR function in F508del homozygous primary human bronchial epithelial (HBE) cells, being 25% more potent than VX-809 or VX-661.70,71 However, ABBV-2222, VX-809 and VX-661 seem to share a similar mechanism in rescuing F508del-CFTR, as these molecules were not additive when tested together.68 Although ABBV-2222 was safe and reduced sweat Cl– concentration in individuals with CF homozygous for F508del, it showed no improvements in ppFEV1 in a phase IIa clinical trial,72 and therefore, another clinical study is currently ongoing (NCT03969888). ABBV-2737 is a second-generation corrector that was well-tolerated in phase I studies and demonstrated to rescue CFTR function in F508del/F508del HBE cells synergistically with VX-809, suggesting that these two molecules act by distinct mechanisms.73,74 In a phase IIa study, ABBV-2737 led to a reduction in sweat Cl– concentration and improved ppFEV1, albeit modestly, in individuals with CF homozygous for F508del.75 ABBV-3221 is another second-generation corrector that demonstrated rescue of F508del-CFTR function with greater effects when in combination with corrector ABBV-2222 and potentiator ABBV-974 (formerly GLPG1837).76 Abbvie also has two additional investigational correctors (AC1 and AC2) that were shown to rescue processing and trafficking of other class II CFTR mutations, including G85E, M1101K and N1303K.46,77
Other investigational correctors include FDL-169 (Flatley Discovery)68 and RDR01752,78 which also appear to share the rescue mechanism with VX-809 and VX-661. In addition, three small-molecule series were identified in a HTS of ~600,000 drug-like molecules: 6258, 3151 and 4172, which target defects at NBD1, NBD2 and TMD interfaces, respectively.79 Although their individual use demonstrated a modest rescue of F508del-CFTR, the combination of these three compounds demonstrated much greater effects and were also able to rescue other class II mutations located at different CFTR domains.79 ARN23765 is another novel corrector that demonstrated to be synergistic with other type of correctors in rescuing F508del-CFTR with the advantage of having picomolar potency.80 Synthetic analogues of the marine natural product latonduine A also demonstrated to rescue F508del-CFTR traffic by inhibiting function of Poly(ADP-ribose) polymerase 3 and 16 (PARP3 and PARP16).81 Novel pyrrolothiazole derivative compounds were recently synthesized and their ability to rescue F508del-CFTR was evaluated in a small-scale screen.82 Among these, compound 44 rescued F508del-CFTR processing and function being additive to VX-809 but not to VX-661.82 Proteostasis Therapeutics also developed a potent corrector – posenacaftor (PTI-801) – that was described as having a similar efficacy to VX-661 in rescuing F508del-CFTR traffic defect. Its safety, tolerability, and pharmacokinetics was evaluated in combination with other modulators from this company, namely a potentiator and an amplifier (see next sub-sections), in individuals with CF in a clinical trial (NCT03500263) with promising clinical outcomes. However, the clinical development of these compounds was discontinued after Proteostasis Therapeutics merged with another pharmaceutical company, Yumanity Therapeutics. Overall, all these compounds provide a good resource to further explore their mechanisms of action and pharmacophore structural space by medicinal chemistry to identify novel and more potent correctors.11,30
Classes III and IV Mutations: Potentiators
Potentiators are compounds that increase CFTR channel open probability, thus allowing for anion transport through CFTR.11,83 These compounds are useful for CFTR mutations in which channel gating or conductance is impaired (ie, classes III and IV, respectively), or for those in which protein translation or traffic are deficient and after rescue by another modulator type, the channel is still not functioning properly.
Ivacaftor (VX-770, Vertex Pharmaceuticals) was the first potentiator approved for clinical use.18,84 It was initially approved by both FDA and EMA for individuals with CF carrying G551D-CFTR in at least one allele after remarkable improvement in lung function (~10 ppFEV1) and significant reduction in sweat Cl– concentration.18,85 Thereafter, its approval was extended for other eight gating mutations.86 More recently, the FDA has extended the approval of VX-770 for 96 residual function mutations (among gating, conductance and splicing mutations) based on in vitro data in cell lines,17 with subsequent clinical studies also confirming therapeutic benefit in some of these mutations.87 As F508del-CFTR still exhibits a gating defect when the protein is rescued to the PM, VX-770 was also tested and approved in combination with VX-809,19 VX-66120 and VX-445/VX-66122,23 as described above (see previous sub-section). Nevertheless, VX-770 only partially reverses G551D-CFTR dysfunction24,84 and individuals with CF taking this modulator may benefit from a combination of potentiators with complementary mechanisms (termed as co-potentiators).28,88,89 Surprisingly, VX-445 was recently demonstrated to have dual activity as both corrector and potentiator,90 being its potentiator activity additive to VX-770 in rescuing F508del- and G551D-CFTR gating defect.29 Furthermore, a deuterated form of ivacaftor (VX-561, deuticaftor) is currently under clinical investigation (NCT03911713). Since the replacement of hydrogen by the heavier deuterium atoms, renders the molecule more stable, as a drug it could be taken once daily instead of twice as it is the case of VX-770.91
ABBV-974 and GLPG-2451 are two novel potentiators developed by Abbvie/Galapagos. These molecules were demonstrated to rescue mutant CFTR carrying G551D, G178R, R334W, S549N or F508del – the latter in combination with either VX-809 or ABBV-2222 – in both cell lines and CF HBE.73,92,93 Patch-clamp functional studies showed that ABBV-974 and GLPG-2451 reduce the closed time and increase the open time of CFTR channels by a similar mechanism to that of VX-770, as no additive effects were observed when these molecules were tested together.73,92,93 ABBV-3067 is another potentiator developed by Abbvie that was demonstrated to rescue F508del-CFTR in combination with the corrector ABBV-2222,74 and this combination is currently under clinical investigation (NCT03969888). Abbvie also has two other investigational potentiators that are being pre-clinically tested: AC2-2 and AP2. The former was found to have dual activity as both corrector and potentiator in rescuing G85E- and M1101K-CFTR, but it only functions as a potentiator for N1303K-CFTR.77 The combination of these two potentiators with the AC1 corrector efficiently rescued the functional expression of the ultra-rare I1234-R1239del-CFTR in HNE cells.94
Novartis has developed icenticaftor (QBW251), a novel potentiator that showed to improve lung function (6.5 ppFEV1) and reduce sweat Cl– concentration in individuals with CF carrying a class III or IV CFTR mutation in at least one allele.95 In parallel, QBW251 was evaluated in individuals with chronic obstructive pulmonary disease in a phase II trial and demonstrated to improve systemic inflammation and sputum bacterial colonization,96 indicating that CFTR potentiators may benefit individuals with other lung diseases. Proteostasis Therapeutics also developed a potentiator termed as Dirocaftor (PTI-808) that was shown to have a similar efficacy to VX-770, and its effect was also assessed in an aforementioned clinical trial (NCT03500263) in individuals with the F508del mutation in at least one allele. Despite promising outcomes, its clinical development was discontinued.
A considerable number of pre-drugs has been identified by HTS programs and are under further development by academic research groups. For instance, some thiazole derivatives were found to act as both correctors and potentiators of F508del-CFTR, although the potentiator effect was lower in comparison to VX-770.97,98 Compounds containing a 2,3,4,5-Tetrahydro-1H-pyrido[4,3-b]indole core were able to efficiently rescue the function of F508del- and G551D-CFTR.99 A spiro[piperidine-4,1-pyrido[3,4-b]indole compound demonstrated potentiation activity for N1303K and I1234-R1239del CFTR with additive effects when in combination with VX-770.100 ASP-11 is an arylsulfonamide-pyrrolopyridine co-potentiator that acts synergistically with VX-770 in the rescue of F508del-, G551D-, N1303K- and W1282X-CFTR mutations in cell lines.101 The effects of ASP-11 were confirmed for N1303K in HBE cells, but not for W1282X.28,101 This considerably high number of compounds with distinct chemical structures provide a great source for the development of novel potentiator drugs.
Class V Mutations: Amplifiers
In a HTS of a library containing ~54,000 drug-like compounds, a novel type of CFTR modulator was identified: the amplifiers.102 These molecules act on CFTR gene expression and, consequently, increase the total amount of protein produced.103 Such compounds may be beneficial not only for class V mutations that result in a reduction of protein production, but also as adjuvants for all other CFTR mutation classes (except for class VII mutations that do not produce CFTR mRNA), as amplifiers may increase the protein abundance to be rescued by other CFTR modulators.
Nesolicaftor (PTI-428) is an amplifier developed by Proteostasis Therapeutics and the first-in-class evaluated in clinical studies.102 In the experimental setting, this molecule was able to selectively increase CFTR expression with no alterations on cytosolic stress or endoplasmic reticulum-associated unfolded protein response pathways. PTI-428 also augmented the effects of VX-809 and VX-770 in rescuing F508del- and I1234-R1239del-CFTR.102,104 A more recent study demonstrated that PTI-428 enhances the stability of CFTR mRNA by directly enhancing the binding of poly(rC)-binding protein 1 to a consensus sequence present in the open reading frame of CFTR mRNA.103 This finding provides novel insights into the cellular regulation of CFTR biosynthesis along with mechanistic information on how amplifiers may be used to enhance the therapeutic benefits of other types of effective CFTR modulators.
Class VI Mutations: Stabilizers
Stabilizers are molecules that enhance the anchoring of CFTR channel to the PM, being thus able to significantly reduce its internalization rate. Even when CFTR mutants reach the PM, they may present intrinsic protein instability (ie, class VI CFTR mutations) that result in increased CFTR endocytosis105 and/or decreased recycling back to the PM.106 For instance, F508del-CFTR rescued either by low-temperature incubation107 or by VX-809,25,108 VX-661 and/or VX-44561 exhibits much lower PM stability in comparison to WT-CFTR, thus being rapidly removed from the PM by peripheral quality controls mechanisms.109
Cavosonstat (N91115) is a stabilizer developed by Nivalis Therapeutics and the first-in-class evaluated in clinical studies.110 It was demonstrated to enhance CFTR maturation and PM stability by inhibiting S-nitrosoglutathione reductase.111 Hepatocyte growth factor (HGF) was also demonstrated to facilitate the PM anchoring of F508del-CFTR by activating endogenous Rac1 signaling that promotes the interaction of CFTR with the Na+/H+ exchanger regulatory factor 1 (NHERF1).112 Furthermore, HGF treatment had a more significant effect in rescuing F508del-CFTR processing and PM stability when combined with VX-809.113,114
Chemical inhibition of calpain 1 was also found to rescue F508del-CFTR expression and function as this interactor prevents ezrin recruitment, a key player in facilitating PM anchoring of F508del-CFTR.115 GM1 ganglioside is a bioactive lipid that also demonstrated to increase PM stability of F508del-CFTR.116 In cells chronically treated with VX-809 and VX-770, GM1 increased total levels of NHERF1 and ezrin, as well as levels of the mature form of F508del-CFTR, which was accompanied by an augment in CFTR-dependent Cl– transport in cell lines and HBE cells.116
CFTR PM expression is negatively regulated by the CFTR-associated ligand (CAL)117,118 and PGD97 is a cyclic peptidyl inhibitor of the interaction between CAL and CFTR, leading to an increase in F508del-CFTR PM stability and function in cell lines and HBE cells with a greater effect when combined with VX-661.119 Such findings provide evidence for the interest in further developing PGD97 and other CAL inhibitors to rescue F508del-CFTR PM stability.
Personalized Medicine: Theratyping and Theranostics
The responsiveness of CFTR mutations to current available modulator drugs has been assessed in a process termed as theratyping, which consists in grouping mutations into classes to be treated by the same strategy (see above section and Figure 2).17,30,120 Although still incomplete, this process has been useful for the approval of label extension to less common CF mutations, thus clinically benefiting a larger population of individuals with CF.17 However, variability in clinical improvement was observed in clinical studies evaluating CFTR modulators even in individuals carrying the same CF genotype.18,57,58,85 While it is assumed that such differences may in part account for the structural tissue injury occurred over disease progression in each individual, it is currently known that responsiveness to a certain drug depends not only on the CF genotype but also on a number of modifier genes and epigenetic factors.11,30,121–123 Accordingly, it is generally accepted that every individual with CF is unique, not only in terms of prognosis for the same CFTR genotype but also regarding response (and benefit) from CFTR modulators. As it is also expected that more CFTR modulator drugs will become available for clinical use, there is a need to advance in the development of tools that enable predicting the clinical effectiveness of drugs by ex vivo assays performed in individual-derived specimens so as to select the best therapeutic option(s) for that individual – a process termed theranostics, the principle underlying precision medicine.30,124 Such fact becomes even more relevant when treating individuals who have CF genotypes combining two different rare CFTR mutations (one in each allele) or even a complex allele.
In order to assess individual responses to CFTR modulator drugs (approved and under development), several assays have been developed and optimized using specimens from individuals with CF.59,125–130 Primary human airway epithelial cells are considered the gold standard since they have been successfully used as pre-clinical data for the CFTR modulators currently approved for clinical use. Indeed, this ex vivo cell model provides responsiveness in a physiologically relevant tissue and allows for the measurement of CFTR-dependent Cl– transport.59,60,79 Primary HNE cells have emerged as a surrogate for bronchial epithelial cells as they share many phenotypic features59,126,130 and may be obtained by less invasive procedures (nasal scrapping instead of bronchoscopy or explanted lungs).
Intestinal organoids grown from stem cells obtained from biopsies have served as another robust and feasible model to evaluate the efficacy of CFTR modulators at an individual level, using the forskolin-induced swelling (FIS) assay.128,129 Using similar technology, airway (bronchial or nasal) organoids/spheroids have been developed based on the same FIS assay.127,131
Induced pluripotent stem cells (iPSCs) might also be a useful model since they can be differentiated in several cell types and recapitulate the variable responses of individuals with the same CF genotype to modulator drugs.132–134 Several studies have been performed to establish correlations between responses in ex vivo individual-derived specimens and clinical parameters to identify which are the most precise biomarkers.59,62,135–137 The possibility of evaluating drug effectiveness at an individual level using ex vivo individual-derived specimens enables precision medicine and therapeutic counseling – ie, theranostics. More importantly, it can reduce the prescription of drugs that would create high expectation to the individual and family members but which do not necessarily provide significant clinical benefits.
Modulation of Alternative (Non-CFTR) Channels/Transporters
Despite remarkable progress in developing highly effective CFTR modulator therapies that target the cellular/functional defects for different CFTR mutations, several issues still need to be resolved: 1) 10–15% of individuals with CF do not benefit from any clinically available CFTR modulator; 2) those who benefit, do so with different levels of responsiveness, which never reach levels of individuals without CF (or carriers); 3) some individuals cannot tolerate or respond poorly to available modulators; 4) there is no equitable access to these drugs due to their very high costs and lack of international regulatory issues; 5) real-world and long-term benefits and sequelae are yet to be demonstrated. Accordingly, an alternative therapeutic option that has gained increasing attention over the last few years consists in the modulation of other (non-CFTR) ion channels/transporters expressed at the PM of secretory epithelia.16,31–33 The main allure of this approach is that it may possibly be used for all individuals with CF, regardless of their CF genotypes, thus being a “mutation-agnostic” approach. Notably, CFTR modulators and drugs targeting alternative channels/transporters may also be combined for additive effects in improving fluid secretion in CF. Here, we have discussed three of these alternative targets: ENaC, TMEM16A and SLC26A9 (Figure 3).
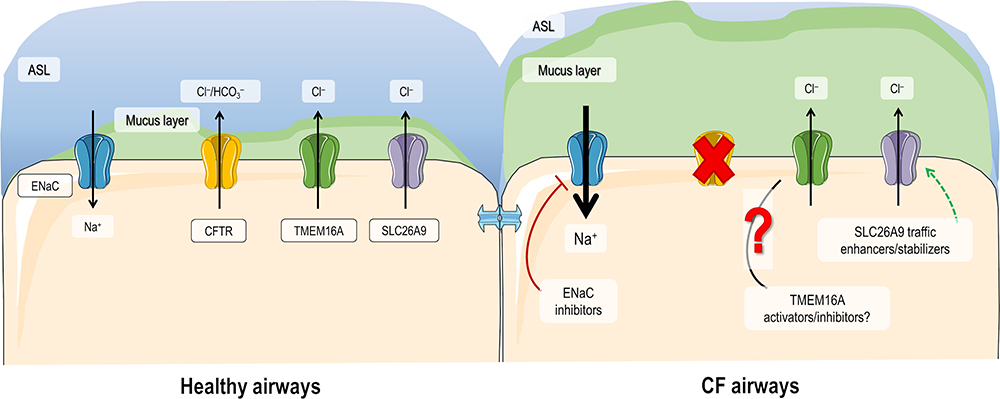 |
Figure 3 Modulation of ion channels/transporters as alternative therapies for CF. In healthy airways, CFTR, ENaC, TMEM16A and SLC26A9 are expressed at the plasma membrane (PM) of epithelial cells where they contribute to ion and water homeostasis. In CF airways, due to the absence of functional CFTR, Cl– secretion is compromised and Na+ absorption is upregulated, leading to a dehydrated air surface liquid (ASL) and impaired mucociliary clearance (MCC). Expression of TMEM16A and SLC26A9 at the PM are also diminished in CF ciliated cells, although the role of TMEM16A overexpression in secretory cells and its role in mucus secretion are still controversial. Alternative therapies for CF thus include blocking ENaC, enhancing SLC26A9 expression at the PM, and modulating TMEM16A. Although for the latter is still not clear whether activators or inhibitors are beneficial, a TMEM16A potentiator in currently under clinical investigation. |
Epithelial Na+ Channel (ENaC)
The idea of targeting ENaC as a therapy for CF came from studies of its role in both healthy and CF airways. However, there is still a debate that encompasses two different hypotheses (not mutually exclusive) regarding the underlying cause of CF pathogenesis. The first one proposes that there is altered pH of air surface liquid (ASL) in CF resulting from absence of CFTR-dependent HCO3– transport, which leads to inactivation of natural antimicrobial peptides in the ASL.138,139 The second model is based on the idea that loss of CFTR function leads to Na+ hyperabsorption by ENaC, leading to subsequent higher absorption of water and consequent ASL dehydration that causes impaired mucociliary clearance (MCC) in CF.140,141 As research data continue to support that a hyperactivation of ENaC occurs in CF cells, specific inhibition of ENaC function may represent a pathway to partially reverse the disruptive downstream effects of CF pathophysiology.
Numerous ENaC inhibitors have been developed with promising effects on the experimental setting but still failed to demonstrate clinical improvements in individuals with CF, including amiloride and SPX-101.32,141 There are several challenges that need to be overcome to successfully develop ENaC inhibitor compounds. These include the ability to penetrate the mucus barrier, the potency, the correct dose without off-target effects and a long half-life to achieve the best therapeutic effects. Accordingly, different strategies are under development to directly or indirectly inhibit ENaC function (Figure 4) and several novel molecules are advancing in the experimental pipeline, such as BI 1265162 and ARO-ENaC.142–144
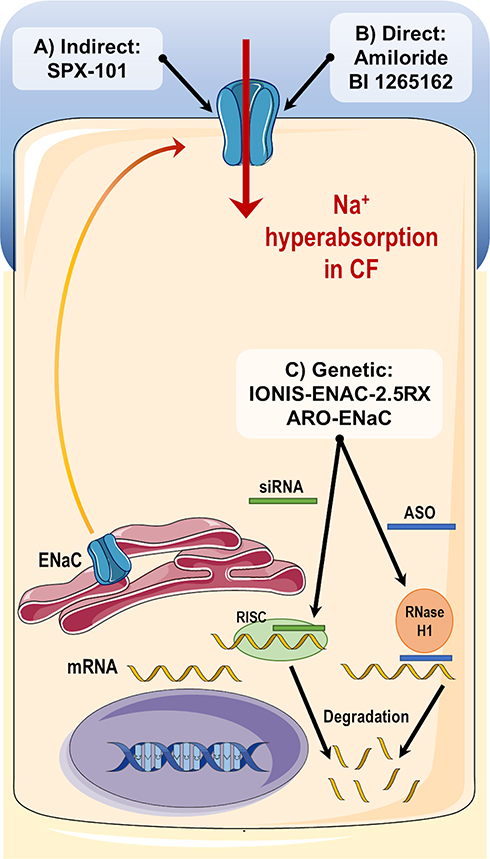 |
Figure 4 Summary of different strategies to inhibit ENaC. (A) Indirect inhibition: SPX-101 is a peptide analogue mimicking the inhibitory actions of SPLUNC1; (B) Direct inhibition: both amiloride and BI 1265162 promote direct inhibition of ENaC by binding to channel and decreasing its open probability; (C) Genetic inhibition: IONIS-ENAC-2.5Rx is an antisense oligonucleotide that recruits RNase H to degrade ENaC mRNA and consequently decrease ENaC protein synthesis and channel function; ARO-ENaC is a small interfering RNA (siRNA) that also promotes degradation of ENaC mRNA transcripts by the RNA-induced silencing complex (RISC) mechanism. |
Amiloride was the first ENaC inhibitor developed in the 1960s by Merck and it is still commonly used in the experimental setting. Furthermore, its structural motif is present in several novel ENaC inhibitors under development. Amiloride directly binds to ENaC and promotes a decrease in the channel open probability.32 This molecule was initially used as a diuretic due to its action on blocking Na+ absorption on the distal convoluted tubule of the kidney and in the fine tuning of Na+ and water levels in the human body. In order to avoid any kidney side-effects or in other ENaC-expressing organs, amiloride was evaluated as an inhaled therapy in CF clinical trials.141 However, results were modest and showed only short-term effects on MCC, which was mainly attributed to the short half-life of amiloride.
Due to clinical failure of amiloride and other first-generation ENaC inhibitor drugs, different strategies were pursued resulting in the discovery of SPX-101, a compound indirectly inhibiting ENaC by mimicking a protein named short palate lung and nasal epithelial clone 1 (SPLUNC1).145,146 Experimental data demonstrated that SPX-101 was able to significantly increase ASL height in primary HBE cultures and recover MCC in CF animal models145,146 and a phase I trial validated the safety of SPX-101 for clinical use.142 However, SPX-101 failed to promote any therapeutic benefit in individuals with CF in a phase II clinical trial (NCT03229252), resulting in the discontinuation of its clinical development. Other SPLUNC1-like compounds may become promising drugs to target ENaC for the treatment of CF.
BI 1265162 is a pre-drug developed by Boehringer Ingelheim that directly inhibits ENaC. It demonstrated a higher potency than amiloride with promising effects in experimental and phase I clinical studies.144 BI 1265162 dose was selected based on data from a CF rat model in which this compound was administered intratracheally and then the fluid absorption net was calculated. BI 1265162 was also studied in a CF sheep model and the deposition of this drug in human airways was estimated by using a “soft mist aerosol” (Respimat®) inhaler.143 The safety and efficacy of BI 1265162 are under investigation in a phase II clinical trial (NCT04059094).
IONIS-ENAC-2.5Rx is a novel genetic strategy developed by Ionis Pharmaceuticals to reduce ENaC function. It consists of an antisense oligonucleotide (ASO) sequence that targets and degrades the pre-mRNA α-subunit of ENaC, critical for channel formation. This degradation induced the RNase H1 activity that subsequently lead to a reduction in ENaC expression. In a phase I trial, IONIS-ENAC-2.5Rx demonstrated to be safe and reduced ENaC mRNA expression on airways of healthy volunteers, after product inhalation via a nebulizer. Subsequent phase II clinical studies were performed to evaluate the safety and efficacy of IONIS-ENAC-2.5Rx in individuals with CF. However, due to recent pre-clinical toxicological data, the company discontinued the development of this drug.
The small interfering RNAs (siRNAs) consist of important alternatives to ASOs. These are composed of 21–23 nucleotides containing a mRNA sequence capable of degrading the mRNA of a specific target gene. The target mRNA degradation occurs with the association between the developed siRNAs with the RNA-induced silencing complex (RISC).32,147 ARO-ENaC (from Arrowhead Pharmaceuticals) is the key siRNA under development to target ENaC.32,147 ARO-ENaC was recently shown to significantly reduce ENaC mRNA and protein levels in lung tissue of rats. Although further studies are necessary to evaluate and optimize the efficacy of this therapy, its safety is currently under investigation on a phase I clinical study (NCT04375514).
Despite the several challenges, ENaC inhibitors are still a promising strategy to treat prominent abnormalities in CF lungs: the CF dehydrated mucus and its downstream detrimental consequences in CF airways. In addition, as mucus hyperproduction has been implicated in the pathogenesis of a variety of lung diseases, ENaC inhibitors may be beneficial therapeutics far beyond CF. Notwithstanding, it has been argued that an intensive ENaC inhibition should not be pursued as this may lead to excessive liquid in the lungs, ie, pulmonary edema, an equally pathological condition. Therefore, modulators that normalize Na+ homeostasis and, consequently, water, are likely to constitute a more appropriate approach.148
Transmembrane Protein 16A (TMEM16A) or Anoctamin 1 (ANO1)
TMEM16A/ANO1 is a Ca2+-activated Cl– channel (CaCC) expressed in various epithelia, including the airways, large intestine, salivary glands, pancreas, kidney and liver.33,149 It is also expressed in the nervous system,150,151 smooth muscles152 and tumor cells.153 Such a broad tissue expression justifies its multiple physiological roles, which include airway and exocrine gland secretion, smooth muscle contraction, neuronal signaling control and peristalsis regulation of gastrointestinal system.154 Furthermore, TMEM16A upregulation has been described in various types of cancer, such as gastrointestinal squamous cancer,155 head and neck squamous cell carcinoma,156 breast cancer157 and lung cancer,158 and it is generally associated with a poor prognosis.159
Targeting TMEM16A for the treatment of CF has been a matter of debate. While some research groups support the need to activate TMEM16A in the context of CF in order to compensate for the absence of CFTR-mediated Cl–/HCO3– transport and thus increase airway hydration and MCC, others consider the opposite and suggest that inhibition of TMEM16A might decrease mucus secretion and bronchoconstriction. Efforts have been performed in both directions to discover TMEM16A modulators (activators and inhibitors) and to better understand the role of TMEM16A in CF.
TMEM16A Activators
The rationale for the activation of TMEM16A as a therapeutic strategy for CF was based on its role on fluid secretion in order to increase the hydration of the ASL, thus ameliorating MCC and recovering antimicrobial activities. In support of these arguments, reports demonstrated that TMEM16A knockout (KO) mice have low Cl– secretion and mucus accumulation in the airways, which are features in common with CF lung disease.160–162 TMEM16A also plays a role in intestinal fluid secretion and in protecting the intestinal epithelium from colitis.163 Furthermore, TMEM16A KO mice showed early signs of inflammation, and their airway cellular landscape is altered, lacking epithelial cell progenitors. Interestingly, in human cell models, inhibition of TMEM16A decreased ASL height in vitro, leading to airway dehydration, similarly to dysfunctional CFTR.164 A strong functional relationship between TMEM16A and CFTR has also been proposed. Namely, in the absence of TMEM16A, both CaCC- and CFTR-mediated currents are reduced in mouse intestine and airways.165 In human cells, genetic inactivation of TMEM16A led to dramatically reduction of WT-CFTR PM expression and function.165 Along these lines, inhibiting TMEM16A would also cause CFTR inhibition, an important fact if we consider that some people with CF have mutations still associated with residual function of CFTR.
Different approaches can be used to activate TMEM16A in CF.166 The most obvious would be to increase channel opening probability, enabling TMEM16A-dependent Cl– secretion, but in a Ca2+-independent way, as an increase in Ca2+ concentration may lead to activation of multiple signaling pathways. To date, several TMEM16A modulator drugs have been identified, although their specificity and mechanism of action remain poorly elucidated (Table 2). An alternative strategy would be to stabilize the channel in the open state or to prevent its desensitization. TMEM16A channels are characterized by a time-dependent current decay after prolonged Ca2+-dependent activation. This process is thought to be regulated by phosphatidylinositol 4,5-bisphosphate (PIP2).167 Indeed, PIP2 has been reported to bind to TMEM16A and promote its stabilization in the open pore conformation.167,168 Inositol phosphates, such as Ins(3,4,5,6)P4, have demonstrated to regulate Cl– channels in multiple epithelia. Furthermore, INO-4995 (a cell permeant InsP4 derivative compound) directly activates TMEM16A in overexpressing cells.169 Another approach to stimulate TMEM16A-dependent Cl– secretion consists in increasing its expression at the PM, either by increasing its anterograde traffic170 or by blocking its endocytosis. Examples of relevant TMEM16A activators are further described below.
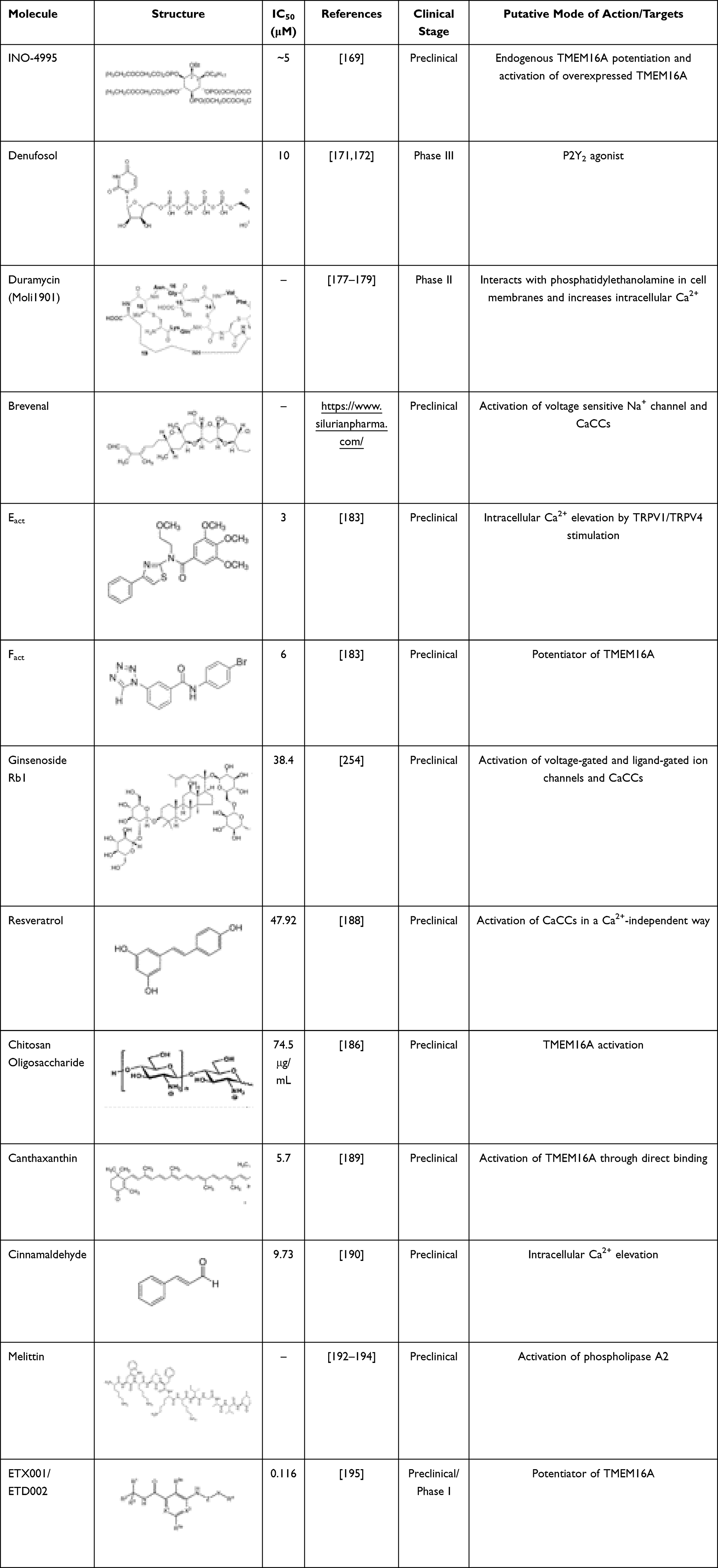 |
Table 2 TMEM16A Activators |
Some drugs able to indirectly modulate CaCCs in airway epithelia have already been investigated in clinical trials. Denufosol is a selective P2Y2 receptor agonist that was evaluated as an inhaled therapy for CF. Experimental and clinical data demonstrated promising effects by increasing both Cl– and water secretion, enhancing ciliary beat frequency and stimulating mucus release. Despite such effects, denufosol failed to demonstrate improvement in lung function in individuals with CF in phase III trials.171,172 The lack of clinical efficacy may be related to its limited time of action (shorter than its expected half-life in the airways) and receptor desensitization.173 Furthermore, purinergic stimulation induces a transient increase in Ca2+ concentration that leads to a short-term activation of CaCCs, which might be insufficient to compensate for the lack of CFTR-mediated anion secretion.174 An increase in intracellular Ca2+ concentration may also lead to undesired side effects, such as increased mucus release from airway secretory cells.173 Duramycin (Moli1901/lancovutide) is an antibiotic that indirectly promotes CaCC activation by interacting with phosphatidylethanolamine at the PM175 and raising intracellular Ca2+ concentration.176 Although it was demonstrated to be safe and to improve lung function in individuals with CF in a phase II clinical study,177–179 no further studies have evaluated the utility of duramycin for the treatment of CF.
Silurian Pharmaceuticals has developed brevenal, a brevetoxin antagonist and candidate drug for CF and other respiratory diseases. Brevenal demonstrated to bind to the voltage sensitive Na+ channel180 and mobilize ATP-dependent intracellular Ca2+ that results in the TMEM16A activation. In an animal model, brevenal was able to block brevetoxin-induced bronchoconstriction, increase mucus secretion181 and reduce lung inflammation.182 Silurian Pharmaceuticals has attempted to initiate a phase I clinical study with brevenal for the treatment of CF; however, there are no further reports so far.
Eact and Fact are two small molecules identified by HTS that were shown to activate TMEM16A.183 Eact promotes a strong increase in Cl– currents in the absence of Ca2+. On the other hand, Fact potentiates TMEM16A by reducing the EC50 for Ca2+-dependent activation of TMEM16A.183 Although Eact was initially described as a direct activator of TMEM16A, subsequent studies demonstrated that its targets are indeed transient receptor potential channels, namely TRPV1184 and TRPV4,185 which lead to increase in intracellular Ca2+ concentration and, consequently, indirect activation of TMEM16A and CFTR (via Ca2+-dependent adenyl cyclases).
Some natural and safe compounds have also been reported as TMEM16A activators with no apparent effects on intracellular Ca2+ concentration, which constitutes an advantage. These compounds include ginsenoside Rb1,186,187 resveratrol,188 chitooligosaccharide186 and canthaxanthin.189 Cinnamaldehyde is another natural compound that activates TMEM16A, but it does so by increasing Ca2+ concentration.190 These molecules may have therapeutic value for gastrointestinal disorders, such as hypomotility and constipation, as they demonstrated to increase ileum contractions in guinea pigs. Melittin, a major component of bee venom,191 is also a potent activator of TMEM16A and of other TMEM16 family members.192 It acts by phospholipase A2 stimulation and has anti-inflammatory properties, being widely used as an anti-cancer therapy.193,194 Notably, most of these compounds are not specific to TMEM16A and they actually have demonstrated a range of effects in different organs and modulate a number of other ion channels. Nevertheless, their chemical structures may serve as sources for medicinal chemistry in order to identify specific TMEM16A activators.
Enterprise Therapeutics identified a novel TMEM16A potentiator – ETX001 – that was able to enhance TMEM16A-dependent anion secretion in a Ca2+-independent way, ASL height, and MCC both in vitro (in CF primary HBE cells) and in vivo (in a CF sheep model).195 ETX001 demonstrated to potentiate TMEM16A-mediated Cl– secretion without negatively affecting CFTR or ENaC.195,196 This portfolio has been recently acquired by Roche, including ETD002, a novel molecule that is currently in phase I clinical trials. Results are expected to demonstrate the ability of this TMEM16A potentiator in reducing mucus congestion, improving lung function and reducing frequency of lung infections. Overall, activating TMEM16A can be of interest not only for the treatment of CF, but also for other obstructive respiratory diseases and motility disorders in the gastrointestinal tract. However, it is clear that most currently known TMEM16A activators act via non-specific pathways – some of them by increasing intracellular Ca2+ concentration – rather than by directly binding and activating TMEM16A. Accordingly, one cannot safely assume that cellular effects promoted by these compounds occur by TMEM16A-specific mechanisms.
TMEM16A Inhibitors
Some studies have suggested that, in the airways, TMEM16A is more abundant in mucus producing cells rather than in ciliated cells.33 TMEM16A expression and Ca2+-dependent Cl– secretion are also increased under inflammatory conditions, namely in asthmatic patients or in cells stimulated with Th2 interleukins (IL-4 or IL-13). Furthermore, as its expression is upregulated concomitantly with mucus hypersecretion, TMEM16A has been associated with goblet cell metaplasia.197,198 Following studies have demonstrated that inhibition of TMEM16A leads to a reduction in mucus secretion in airways152 and intestine,198 proposing a causal relationship between these two events. It is therefore tempting to conclude that inhibition of TMEM16A (rather than activation) might be beneficial in CF.
In addition to the possible effects of TMEM16A on mucus hypersecretion, activation of TMEM16A in airway smooth muscles was shown to result in membrane depolarization, causing muscular contraction and potentially bronchoconstriction.152 Accordingly, inhibition of TMEM16A may induce bronchodilation, which could be advantageous in airway inflammatory disease.199 Attenuation of intestinal contraction and diarrhea200,201 as well as nociception, itching and heat perception150 may also be achieved by inhibiting TMEM16A. Finally, it has been argued that inhibition of TMEM16A might induce anti-cancer effects by blocking proliferation, migration and invasion of cancer cells, while also increasing the sensitivity to chemotherapies.153 Nevertheless, the role of TMEM16A in cell proliferation is also controversial, with a recent report suggesting that TMEM16A is upregulated by cell proliferation and not the opposite.164 Indeed, the data showed that, during wound, expression levels of TMEM16A rise concomitantly with the proliferation marker Ki-67, indicating that it is proliferation that triggers TMEM16A upregulation.
A growing number of molecules able to inhibit TMEM16A has been identified (Table 3). These include niflumic acid (NFA),202 flufenamic acid (FFA),203 5-nitro-(3-phenylpropylamino)-benzoic acid (NPPB)204 and 4–4ʹ-diisothiocyanatostilbene-2-2ʹ-disulfonic acid (DIDS),205,206 which are broad spectrum Cl– channel blockers widely used for long to inhibit CaCCs and more recently demonstrated to act also on TMEM16A. However, they are non-selective and may elicit several other cellular responses. Talniflumate, a pro-drug of NFA, is a potent analgesic and anti-inflammatory that has been used for the treatment of rheumatoid disease. Experimental studies have demonstrated that talniflumate increases the survival rate of CF mice, which is a model of distal intestinal obstructive syndrome.207 Talniflumate has also been investigated as a mucoregulator for the treatment of various respiratory diseases,207 namely CF, chronic obstructive pulmonary disease and asthma; however, its clinical development has been discontinued.
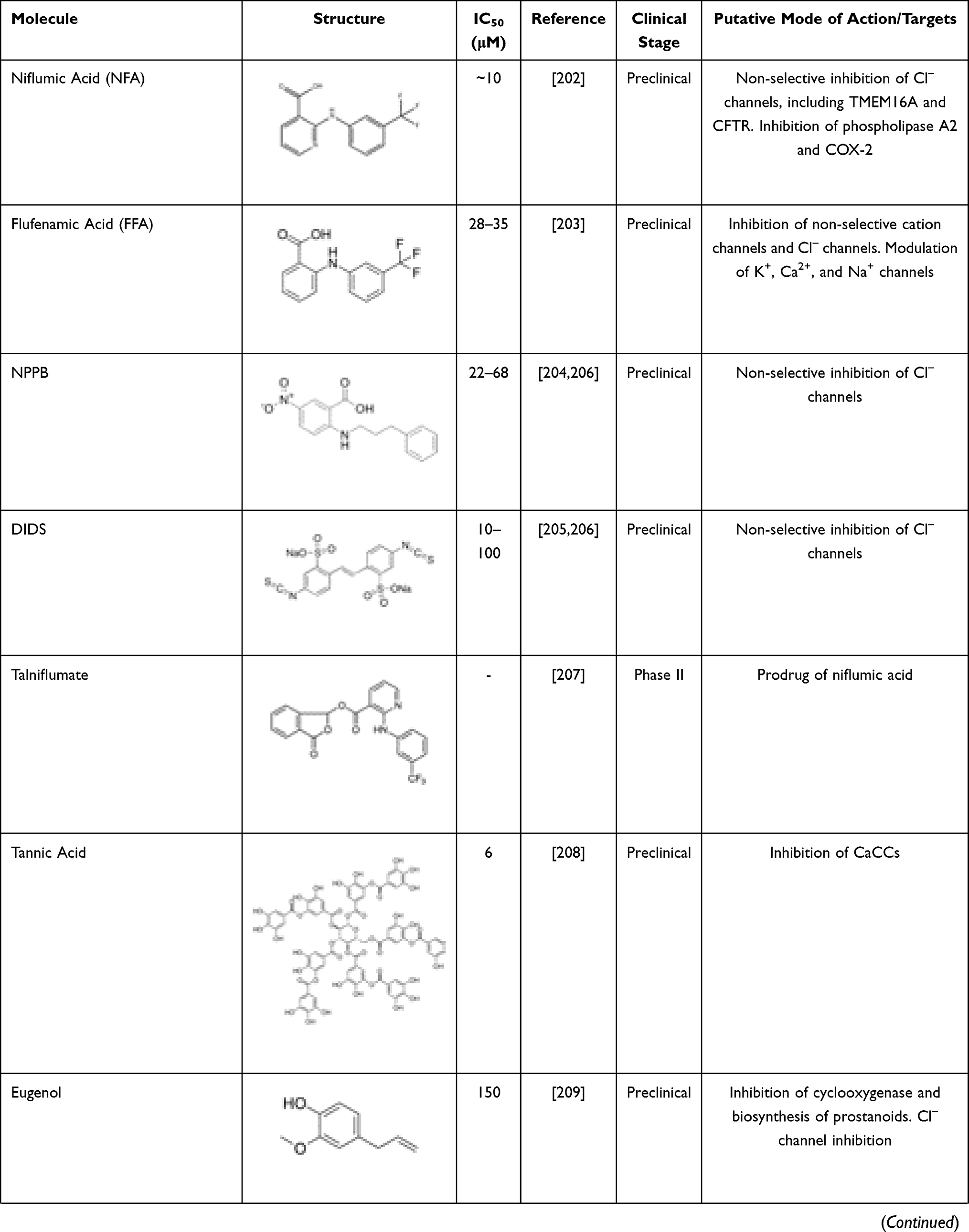 |
 |
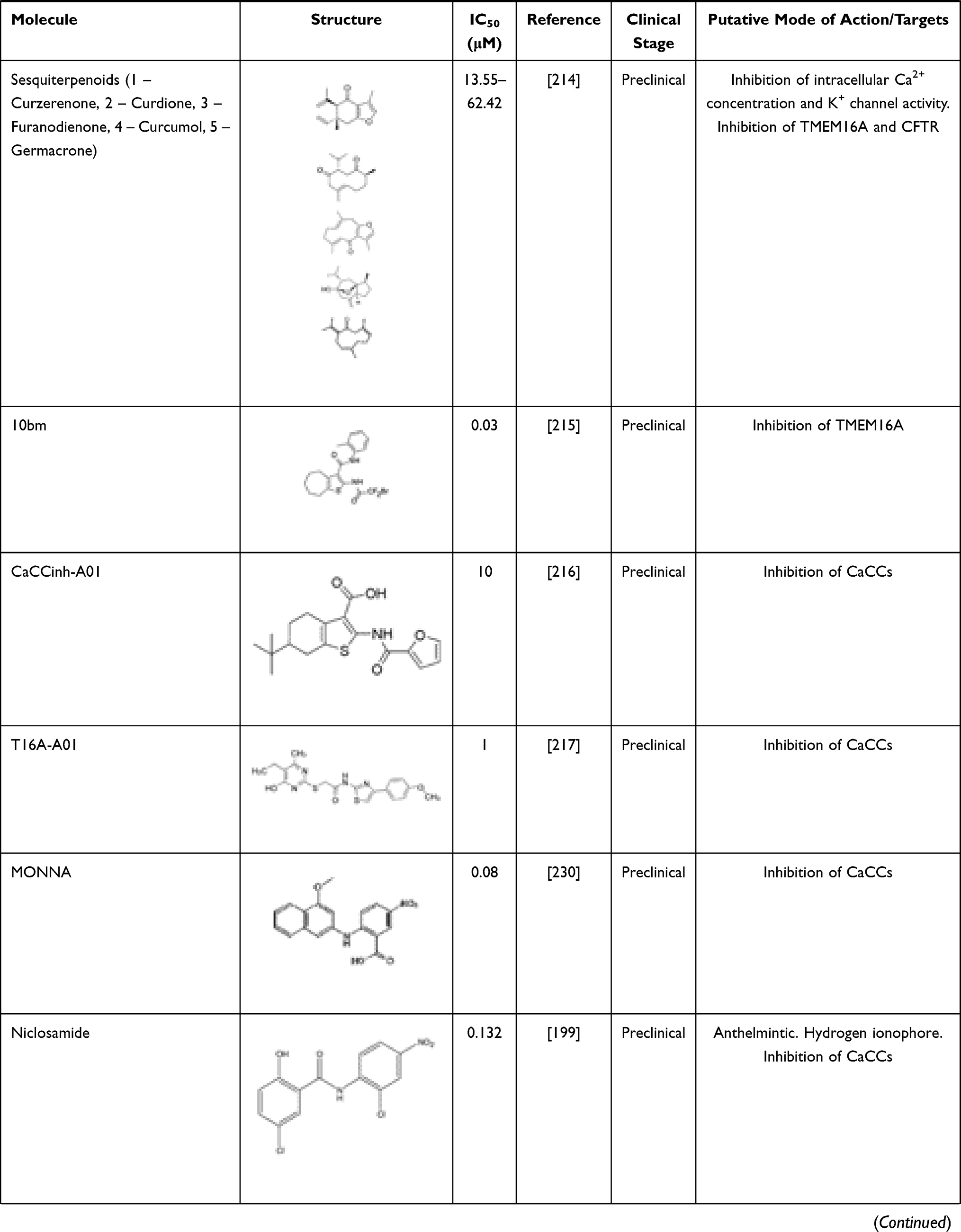 |
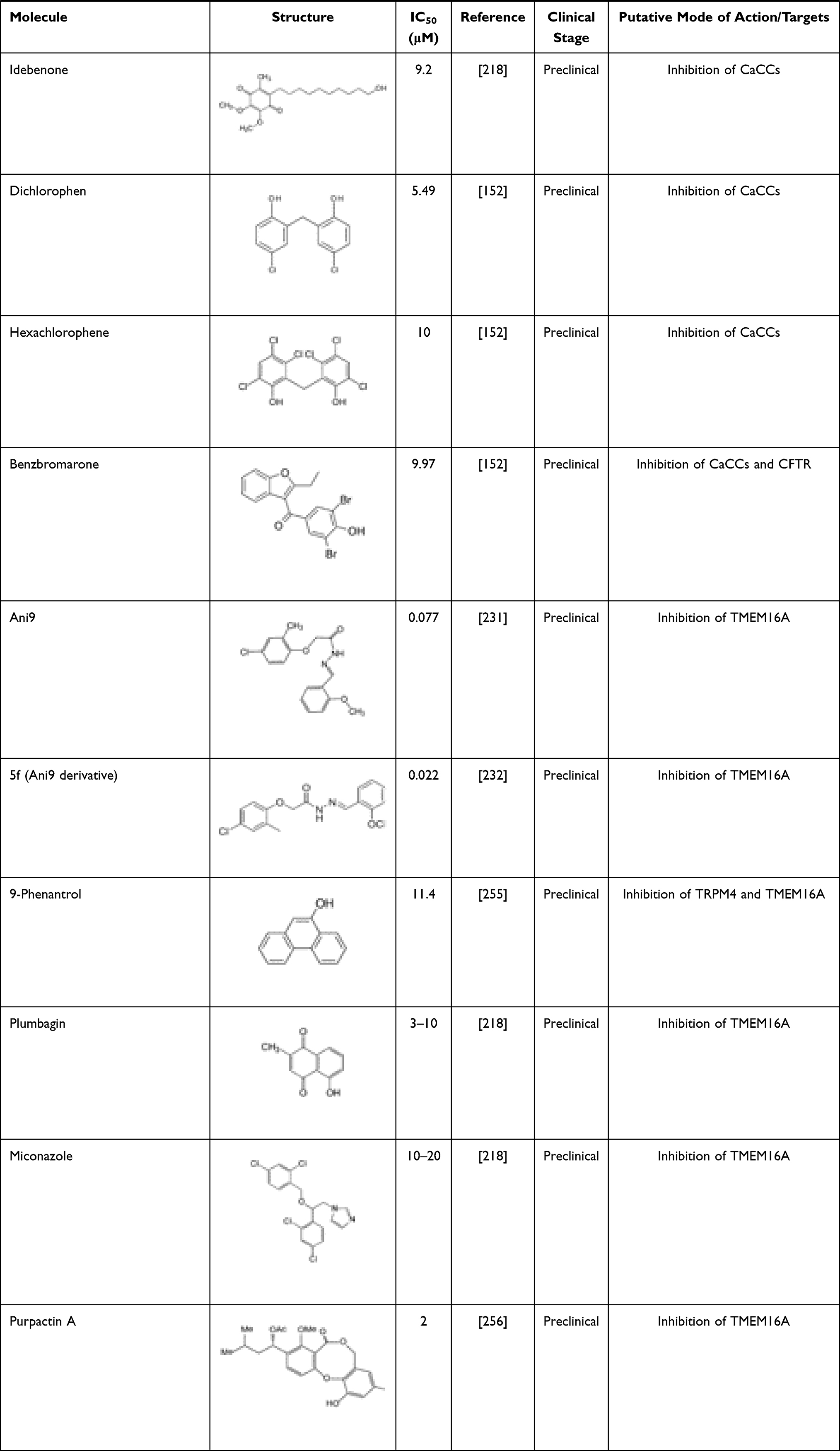 |
Table 3 TMEM16A Inhibitors |
Some natural products have also been identified as TMEM16A inhibitors, including tannic acid and other gallotannins, which are known to inhibit smooth muscle contraction and intestinal Cl– secretion.208 Other natural compounds like eugenol209 and shikonin210 have antidiarrheal effects, and flavonoids (galangin, luteolin, quercetin and fisetin),210 matrine,211 dehydroandrographolide,212 avermectins213 and sesquiterpenoids (curzerenone, curdione, curcumol, furanodienone and germacrone)214 demonstrated anti-proliferative and anti-cancer effects.
Several TMEM16A inhibitors have been identified by HTS, such as cycloalkylthiophene 10bm,215 CaCCinh-A01,216 T16inh-A01,217 MONNA,199 niclosamide,218 idebenone, dichlorophem, hexachlorophenom and benzbromarone.152 The latter is a uricosuric agent that was initially approved for the treatment of gout. Since the discovery of its role on TMEM16A inhibition, it has been suggested that it might be repurposed for TMEM16A-related disorders. A good example has been the repurposing of benzbromarone for the treatment of idiopathic pulmonary arterial hypertension, a disease in which TMEM16A was found to be upregulated.219,220 However, it is important to note that benzbromarone is not a TMEM16A-specific inhibitor, thus also inhibiting other channels, such as CFTR.221 Some safety concerns have also been raised for the long-term use of benzbromarone due to reports of hepatotoxicity.222
Niclosamide is an antihelminthic drug that has also demonstrated anti-cancer effects on several types of human cancer cells.223–225 It has been more extensively investigated in experimental and clinical studies for individuals suffering from prostate and colorectal cancer.226,227 Apart from its anti-neoplastic effects, niclosamide also demonstrated to inhibit TMEM16A and other members of the TMEM16 family.228 Other studies also suggested that niclosamide reduces bronchoconstriction199 by relaxing airway smooth muscles and mitigates mucus hypersecretion.229 Interestingly, the potential antiviral efficacy of niclosamide is under clinical investigation in combination with an established therapeutic regimen for SARS-CoV-2 infection (NCT04558021).
T16inh-A01,217 MONNA,230 and Ani9231 were initially characterized as the most potent and selective TMEM16A inhibitors. However, T16inh-A01 and MONNA were found to also inhibit other members of this family, such as TMEM16B, in a dose-dependent manner.231 Ani9 and its derivative 5f232 are, therefore, presented as the most powerful and apparently selective TMEM16A inhibitors to date. Both Ani9 and 5f demonstrated high selectivity for TMEM16A without affecting intracellular Ca2+ concentration or other channels, such as CFTR. Nevertheless, recent studies demonstrated that Ani9 may affect TMEM16F whole-cell currents, indicating that this inhibitor is not completely selective for TMEM16A.228 Overall, despite the existence of various TMEM16A inhibitors, the potency and selectivity of these compounds are a considerable limitation. The development of potent and more selective molecules that directly target TMEM16A may provide invaluable information on the role of this channel in CF and TMEM16A-related disorders.
Solute Carrier Family 26 Member 9 (SLC26A9)
SLC26A9 has been proposed to function as a constitutively active Cl– transporter,233,234 predominantly expressed in epithelial cells of the respiratory tract, stomach, duodenum, ileum and pancreas.31,235 In human airways, SLC26A9 contributes to constitutive Cl– secretion and MCC,236 and, in contrast to CFTR and TMEM16A, it is described as being spontaneously active once inserted into the PM of airway epithelial cells.237
SLC26A9 was found to be mostly cytoplasmic or to localize closer to tight junctions.238 Nevertheless, it was proposed to functionally interact with CFTR, which positively regulates its transport, function and targeting to the PM.31 In this context, the presence of F508del-CFTR reduced SLC26A9 expression by promoting its retention in the endoplasmic reticulum and premature degradation by the proteasome, similar to F508del-CFTR itself.238
The importance of investigating SLC26A9 in the context of CF has been highlighted due to its striking effects on the pathogenesis of organs affected in CF, as well as other disorders. SLC26A9 has been described as a modifier gene of lung function and of responses to CFTR modulator drugs.121,122 SLC26A9 polymorphisms have been associated with the risk of developing meconium ileus and early exocrine pancreatic disease in individuals with CF.239–241 Interestingly, SLC26A9 polymorphisms have also been linked to asthma in children, namely due to a reduction in SLC26A9 protein expression.236 As SLC26A9 contributes to epithelial Cl– secretion and prevents mucus obstruction under inflammatory conditions,236 it possibly also plays a role in the regulation of the ASL, similar to CFTR. Furthermore, a recent study demonstrated that increased SLC26A9 expression in pancreatic ductal cells delays the age of onset of CF-related diabetes.242
Taken together, these data support a clinically relevant role of SLC26A9 as both a CF disease modifier and a promising therapeutic target to circumvent deficient Cl– secretion in CF. In contrast to TMEM16A, SLC26A9 modulation as a therapeutic target for CF is unanimous – ie, all data point toward the need to increase SLC26A9 expression and function. Nevertheless, the study of SLC26A9 has been limited by the lack of sensitive and selective pharmacological modulators. Some compounds have demonstrated to inhibit SLC26A9, namely non-selective Cl– channel inhibitors NFA, NPPB, DIDS and GlyH101243 with different degrees of potency. The use of inhibitors that are virtually specific for other channels, such as Ani9 for TMEM16A and CFTRinh172 for CFTR, for which SLC26A9 is probably not sensitive,244 allows only for the indirect study of SLC26A9-mediated Cl– currents. Accordingly, identification of selective activators of SLC26A9 is necessary to further understand whether and how it can be used as a therapeutic target. Due to its spontaneous activation once inserted into the PM,237 molecules that increase or stabilize SLC26A9 PM expression may contribute to increase epithelial Cl– secretion.
Anionophores
Besides the aforementioned channels/transporters, small artificial transmembrane ion transporters (termed as ionophores) have emerged as therapeutic candidates for CF.245 Among these, anionophores, which selectively facilitate the transport of anions, have the potential to compensate for the faulty CFTR transport activity in CF.246
Studies have identified promising molecules, including derivatives of natural products, such as prodiginines247,248 and tambjamines,249 capable of facilitating Cl– and HCO3– transport in mammalian cells. More recently, these compounds were also reported to effectively correct abnormal ASL parameters in primary cultures from individuals with CF.250 Interestingly, the activity of some anionophores is additive to F508del-CFTR rescue by the modulators VX-809 and VX-770, while the activity of others is additive to TMEM16A activation.251 These data reinforce the idea that anionophores, either by themselves or in combination with other modulator drugs, may offer promising therapeutic strategies for CF.
Amphotericin B (AmB) is a channel-forming molecule capable of creating non-selective ion channels. Recent studies with AmB showed that it can increase Cl– secretion, ASL height and pH, reduce ASL viscosity, and restore antibacterial activity in primary cultures of airway epithelia from individuals with CF caused by different mutations, including the ones that produce little to no CFTR.252,253 Additionally, a small clinical study involving people with CF not treated with CFTR modulators showed that intranasal application of AmB caused a significant change in nasal potential difference, a commonly used biomarker to assess experimental therapies in people with CF.253 These data combined with the fact that AmB is an already clinically approved drug used to treat fungal infections, further emphasize the need to carry out additional studies to assess if inhaled AmB can in fact benefit people with CF, regardless of their CFTR mutations.
Similar to the use of alternative channels/transporters mentioned here, taking advantage of ionophores to replace defective CFTR has the potential to provide an alternative therapy for all individuals with CF, that would work independently of their CF genotype. Nevertheless, further studies are needed to determine the efficacy and safety profiles of these molecules to enable their clinical use.
Outlook and Conclusions
Numerous milestones have been achieved in CF research since the very first pathological description of this disease in 1938. The development of novel therapies for CF has been a success story with a real transformation in the disease prognosis, quality of life and life expectancy. In fact, in the 1960s, CF was a deadly and untreatable disease in early childhood and nowadays many individuals with CF achieve the adulthood, although they still face a high therapeutic burden and several disease-related complications. While the use of symptomatic therapies remains of great importance, the introduction of CFTR modulator drugs in the clinic, namely highly effective CFTR modulator therapies, has demonstrated significant benefit by targeting the root cause of CF and thus prevent several pathological consequences downstream of CFTR dysfunction. Accordingly, it is expected that these drugs may induce a profound alteration on the course of disease, especially for individuals who initiate their use in the early disease onset. Despite such progress, not all individuals with CF are eligible for the currently available CFTR modulator drugs, and novel and more potent CFTR modulators are still under development. Novel cell models and assays using ex vivo individual-derived specimens have also been optimized to comparatively evaluate drug efficacies and determine which may provide the greatest therapeutic benefits in a precision medicine approach, ie, theranostics. Furthermore, a better understanding of the complexity of CF epithelial cell physiology and ion transport has enabled the pursue for alternative targets to compensate for the absence of CFTR function. Modulation of alternative ion channels/transporters, such as ENaC, TMEM16A or SLC26A9, as therapeutic targets in CF is promising but still challenging. It is still critical to develop selective and potent modulators of these targets and understand how (and whether) they may be used in the clinical scenario. Nevertheless, such approaches, if successful, may be beneficial for all individuals with CF, regardless of their CFTR genotypes.
Acknowledgments
Work of authors is supported by UIDB/04046/2020 and UIDP/04046/2020 center grants (to BioISI) and research grants: Research Scholar Program in CF (2018) from Gilead Sciences (to MLP), “HIT-CF” (H2020-SC1-2017-755021) from EU (to MDA), and SRC 013 from CF Trust/UK (to MDA). MFF is supported by the CF Foundation Postdoctoral Grant (FIGUEI18F0).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science (80-). 1989;245(4922):1059–1065. doi:10.1126/science.2772657
2. Riordan JR, Rommens JM, Kerem BS, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science (80-). 1989;245(4922):1066–1073. doi:10.1126/science.2475911
3. Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science (80-). 1989;245(4922):1073–1080. doi:10.1126/science.2570460
4. Lopes-Pacheco M. CFTR modulators: shedding light on precision medicine for cystic fibrosis. Front Pharmacol. 2016;7:275. doi:10.3389/fphar.2016.00275
5. Saint-Criq V, Gray MA. Role of CFTR in epithelial physiology. Cell Mol Life Sci. 2017;74(1):93–115. doi:10.1007/s00018-016-2391-y
6. Liu F, Zhang Z, Csanády L, Gadsby DC, Chen J. Molecular structure of the human CFTR Ion channel. Cell. 2017;169(1):85–95. doi:10.1016/j.cell.2017.02.024
7. Zhang Z, Liu F, Chen J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc Natl Acad Sci U S A. 2018;115(50):12757–12762. doi:10.1073/pnas.1815287115
8. Chappe V, Hinkson DA, Zhu T, Chang XB, Riordan JR, Hanrahan JW. Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR channel activation by PKA. J Physiol. 2003;548(Pt 1):39–52. doi:10.1113/jphysiol.2002.035790
9. Du K, Lukacs GL. Cooperative assembly and misfolding of CFTR domains in vivo. Mol Biol Cell. 2009;20(7):2673–2683. doi:10.1091/mbc.e08-09-0950
10. Kim SJ, Skach WR. Mechanisms of CFTR folding at the endoplasmic reticulum. Front Pharmacol. 2012;3:201. doi:10.3389/fphar.2012.00201
11. Lopes-Pacheco M. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol. 2020;10:1662. doi:10.3389/fphar.2019.01662
12. De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med Respir Med. 2016;4(8):662–674. doi:10.1016/S2213-2600(16)00023-0
13. Lopes-Pacheco M, Kitoko JZ, Morales MM, Petrs-Silva H, Rocco PRM. Self-complementary and tyrosine-mutant rAAV vectors enhance transduction in cystic fibrosis bronchial epithelial cells. Exp Cell Res. 2018;372(2):99–107. doi:10.1016/j.yexcr.2018.09.015
14. Castellani C, Duff AJA, Bell SC, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros. 2018;17(2):153–178. doi:10.1016/j.jcf.2018.02.006
15. Bell SC, Mall MA, Gutierrez H, et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med. 2020;8(1):65–124. doi:10.1016/S2213-2600(19)30337-6
16. Lopes-Pacheco M, Pedemonte N, Kicic A. Editorial: emerging therapeutic approaches for cystic fibrosis. Front Pharmacol. 2019;10(November):1–4. doi:10.3389/fphar.2019.01440
17. Lopes-Pacheco M, Pedemonte N, Veit G. Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin Drug Discov. 2021;1–17. doi:10.1080/17460441.2021.1912732
18. Ramsey BW, Davies JC, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–1672. doi:10.1056/NEJMoa1105185
19. Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–231. doi:10.1056/nejmoa1409547
20. Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med. 2017;377(21):2013–2023. doi:10.1056/nejmoa1709846
21. Rowe SM, Daines C, Ringshausen FC, et al. Tezacaftor–ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med. 2017;377(21):2024–2035. doi:10.1056/nejmoa1709847
22. Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del Allele. N Engl J Med. 2019;381(19):1809–1819. doi:10.1056/nejmoa1908639
23. Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, Phase 3 trial. Lancet. 2019;394(10212):1940–1948. doi:10.1016/S0140-6736(19)32597-8
24. Jih KY, Hwang TC. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc Natl Acad Sci U S A. 2013;110(11):4404–4409. doi:10.1073/pnas.1215982110
25. Farinha CM, King-Underwood J, Sousa M, et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem Biol. 2013;20(7):943–955. doi:10.1016/j.chembiol.2013.06.004
26. Eckford PDW, Ramjeesingh M, Molinski S, et al. VX-809 and related corrector compounds exhibit secondary activity stabilizing active F508del-CFTR after its partial rescue to the cell surface. Chem Biol. 2014;21(5):666–678. doi:10.1016/j.chembiol.2014.02.021
27. Lopes-Pacheco M, Boinot C, Sabirzhanova I, Morales MM, Guggino WB, Cebotaru L. Combination of correctors rescue ΔF508-CFTR by reducing its association with Hsp40 and Hsp27. J Biol Chem. 2015;290(42):25636–25645. doi:10.1074/jbc.m115.671925
28. Phuan PW, Tan JA, Rivera AA, et al. Nanomolar-potency ‘co-potentiator’ therapy for cystic fibrosis caused by a defined subset of minimal function CFTR mutants. Sci Rep. 2019;9(1):1–12. doi:10.1038/s41598-019-54158-2
29. Veit G, Vaccarin C, Lukacs GL. Elexacaftor co-potentiates the activity of F508del and gating mutants of CFTR. J Cyst Fibros. 2021;3–6. doi:10.1016/j.jcf.2021.03.011
30. Amaral MD. How to determine the mechanism of action of CFTR modulator compounds: a gateway to theranostics. Eur J Med Chem. 2021;210(xxxx):112989. doi:10.1016/j.ejmech.2020.112989
31. Balázs A, Mall MA. Role of the SLC26A9 chloride channel as disease modifier and potential therapeutic target in cystic fibrosis. Front Pharmacol. 2018;9:1112. doi:10.3389/fphar.2018.01112
32. Moore PJ, Tarran R. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis lung disease. Expert Opin Ther Targets. 2018;22(8):687–701. doi:10.1080/14728222.2018.1501361
33. Kunzelmann K, Ousingsawat J, Cabrita I, et al. TMEM16A in cystic fibrosis: activating or inhibiting? Front Pharmacol. 2019;10(3). doi:10.3389/fphar.2019.00003
34. Morais P, Adachi H, Yu YT. Suppression of nonsense mutations by new emerging technologies. Int J Mol Sci. 2020;21(12):4394. doi:10.3390/ijms21124394
35. Pranke I, Bidou L, Martin N, et al. Factors influencing readthrough therapy for frequent cystic fibrosis premature termination codons. ERJ Open Res. 2018;4(1):00080–02017. doi:10.1183/23120541.00080-2017
36. Xue X, Mutyam V, Thakerar A, et al. Identification of the amino acids inserted during suppression of CFTR nonsense mutations and determination of their functional consequences. Hum Mol Genet. 2017;26(16):3116–3129. doi:10.1093/hmg/ddx196
37. Welch EM, Barton ER, Zhuo J, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87–91. doi:10.1038/nature05756
38. Kerem E, Konstan MW, De Boeck K, et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2(7):539–547. doi:10.1016/S2213-2600(14)70100-6
39. Konstan MW, VanDevanter DR, Rowe SM, et al. Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: the international, randomized, double-blind, placebo-controlled Ataluren Confirmatory Trial in Cystic Fibrosis (ACT CF). J Cyst Fibros. 2020;19(4):595–601. doi:10.1016/j.jcf.2020.01.007
40. Pibiri I, Lentini L, Melfi R, et al. Enhancement of premature stop codon readthrough in the CFTR gene by Ataluren (PTC124) derivatives. Eur J Med Chem. 2015;101:236–244. doi:10.1016/j.ejmech.2015.06.038
41. Pibiri I, Lentini L, Melfi R, et al. Rescuing the CFTR protein function: introducing 1,3,4-oxadiazoles as translational readthrough inducing drugs. Eur J Med Chem. 2018;159:126–142. doi:10.1016/j.ejmech.2018.09.057
42. Pibiri I, Melfi R, Tutone M, Di Leonardo A, Pace A, Lentini L. Targeting nonsense: optimization of 1,2,4-oxadiazole trids to rescue cftr expression and functionality in cystic fibrosis cell model systems. Int J Mol Sci. 2020;21(17):6420. doi:10.3390/ijms21176420
43. Gopalsamy A, Bennett EM, Shi M, Zhang WG, Bard J, Yu K. Identification of pyrimidine derivatives as hSMG-1 inhibitors. Bioorganic Med Chem Lett. 2012;22(21):6636–6641. doi:10.1016/j.bmcl.2012.08.107
44. Valley HC, Bukis KM, Bell A, et al. Isogenic cell models of cystic fibrosis-causing variants in natively expressing pulmonary epithelial cells. J Cyst Fibros. 2019;18(4):476–483. doi:10.1016/j.jcf.2018.12.001
45. Santos L, Mention K, Cavusoglu-doran K, et al. Comparison of Cas9 and Cas12a CRISPR editing methods to correct the W1282X-CFTR mutation. J Cyst Fibros. 2021. doi:10.1016/j.jcf.2021.05.014
46. Laselva O, Eckford PD, Bartlett C, et al. Functional rescue of c.3846G>A (W1282X) in patient-derived nasal cultures achieved by inhibition of nonsense mediated decay and protein modulators with complementary mechanisms of action. J Cyst Fibros. 2020;19(5):717–727. doi:10.1016/j.jcf.2019.12.001
47. McHugh DR, Cotton CU, Hodges CA. Synergy between readthrough and nonsense mediated decay inhibition in a murine model of cystic fibrosis nonsense mutations. Int J Mol Sci. 2021;22(1):334. doi:10.3390/ijms22010344
48. Kerem E. ELX-02: an investigational read-through agent for the treatment of nonsense mutation-related genetic disease. Expert Opin Investig Drugs. 2020;29(12):1347–1354. doi:10.1080/13543784.2020.1828862
49. Leubitz A, Vanhoutte F, Hu M, et al. A randomized, double-blind, placebo-controlled, multiple dose escalation study to evaluate the safety and pharmacokinetics of ELX-02 in Healthy Subjects. Clin Pharmacol Drug Dev. 2021. doi:10.1002/cpdd.914
50. Crawford DK, Mullenders J, Pott J, Boj SF, Landskroner-Eiger S, Goddeeris MM. Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J Cyst Fibros. 2021;20(3):436–442. doi:10.1016/j.jcf.2021.01.009
51. Liang F, Shang H, Jordan NJ, et al. High-throughput screening for readthrough modulators of CFTR PTC mutations. SLAS Technol. 2017;22(3):315–324. doi:10.1177/2472630317692561
52. Smith E, Dukovski D, Shumate J, Scampavia L, Miller JP, Spicer TP. Identification of compounds that promote readthrough of premature termination codons in the CFTR. SLAS Discov. 2021;26(2):205–215. doi:10.1177/2472555220962001
53. Sharma N, Evans TA, Pellicore MJ, et al. Capitalizing on the heterogeneous effects of CFTR nonsense and frameshift variants to inform therapeutic strategy for cystic fibrosis. PLoS Genet. 14;2018. doi:10.1371/journal.pgen.1007723
54. Pedemonte N, Galietta LJV. Pharmacological correctors of mutant CFTR mistrafficking. Front Pharmacol. 2012;3:175. doi:10.3389/fphar.2012.00175
55. Cheng SH, Gregory RJ, Marshall J, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;83(1):129–135. doi:10.1016/0092-8674(95)90241-4
56. Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83(1):129–135. doi:10.1016/0092-8674(95)90241-4
57. Boyle MP, Bell SC, Konstan MW, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a Phase 2 randomised controlled trial. Lancet Respir Med. 2014;2(7):527–538. doi:10.1016/S2213-2600(14)70132-8
58. Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor/ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med. 2018;197(2):214–224. doi:10.1164/rccm.201704-0717OC
59. Pranke IM, Hatton A, Simonin J, et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci Rep. 2017;7(1):375. doi:10.1038/s41598-017-07504-1
60. Van Goor F, Hadida S, Grootenhuis PDJ, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108(46):18843–18848. doi:10.1073/pnas.1105787108
61. Capurro V, Tomati V, Sondo E, et al. Partial rescue of f508del-cftr stability and trafficking defects by double corrector treatment. Int J Mol Sci. 2021;22(10):5262. doi:10.3390/ijms22105262
62. Awatade NT, Uliyakina I, Farinha CM, et al. Measurements of functional responses in human primary lung cells as a basis for personalized therapy for cystic fibrosis. EBioMedicine. 2015;2(2):147–153. doi:10.1016/j.ebiom.2014.12.005
63. Awatade NT, Ramalho S, Silva IAL, et al. R560S: a class II CFTR mutation that is not rescued by current modulators. J Cyst Fibros. 2019;18(2):182–189. doi:10.1016/j.jcf.2018.07.001
64. Lopes-Pacheco M, Sabirzhanova I, Rapino D, Morales MM, Guggino WB, Cebotaru L. Correctors rescue CFTR mutations in Nucleotide-Binding Domain 1 (NBD1) by modulating proteostasis. ChemBioChem. 2016;17(6):493–505. doi:10.1002/cbic.201500620
65. Lopes-Pacheco M, Boinot C, Sabirzhanova I, Rapino D, Cebotaru L. Combination of Correctors rescues CFTR transmembrane-domain mutants by mitigating their interactions with proteostasis. Cell Physiol Biochem. 2017;41(6):2194–2210. doi:10.1159/000475578
66. Rapino D, Sabirzhanova I, Lopes-Pacheco M, Grover R, Guggino WB, Cebotaru L. Rescue of NBD2 mutants N1303K and S1235R of CFTR by Small-molecule correctors and transcomplementation. PLoS One. 2015;10(3):e0119796. doi:10.1371/journal.pone.0119796
67. Van Willigen M, Vonk AM, Yeoh HY, et al. Folding-function relationship of the most common cystic fibrosis-causing CFTR conductance mutants. Life Sci Alliance. 2019;2(1):2201800172. doi:10.26508/lsa.201800172
68. Veit G, Roldan A, Hancock MA, et al. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight. 2020;5(18):e139983. doi:10.1172/JCI.INSIGHT.139983
69. da Silva Filho LVRF, Zampoli M, Cohen-Cymberknoh M, Kabra SK. Cystic fibrosis in low and middle-income countries (LMIC): a view from four different regions of the world. Paediatr Respir Rev. 2020;38:37–44. doi:10.1016/j.prrv.2020.07.004
70. Wang X, Liu B, Searle X, et al. Discovery of 4-[(2R,4R)-4-({[1-(2,2-Difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-7-(difluoromethoxy)-3,4-dihydro-2H-chromen-2-yl]benzoic Acid (ABBV/GLPG-2222), a potent Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Corrector for. J Med Chem. 2018;61(4):1436–1449. doi:10.1021/acs.jmedchem.7b01339
71. Singh AK, Fan Y, Balut C, et al. Biological characterization of F508DelCFTR protein processing by the CFTR corrector ABBV-2222/GLPG2222. J Pharmacol Exp Ther. 2020;372(1):107–118. doi:10.1124/jpet.119.261800
72. Bell SC, Barry PJ, De Boeck K, et al. CFTR activity is enhanced by the novel corrector GLPG2222, given with and without ivacaftor in two randomized trials. J Cyst Fibros. 2019;18(5):700–707. doi:10.1016/j.jcf.2019.04.014
73. Gees M, Musch S, Van Der Plas S, et al. Identification and characterization of novel CFTR potentiators. Front Pharmacol. 2018;9:1221. doi:10.3389/fphar.2018.01221
74. De Wilde G, Gees M, Musch S, et al. Identification of GLPG/ABBV-2737, a novel class of corrector, which exerts functional synergy with other CFTR modulators. Front Pharmacol. 2019;10:514. doi:10.3389/fphar.2019.00514
75. van Koningsbruggen-rietschel S, Conrath K, Fischer R, et al. GLPG2737 in lumacaftor/ivacaftor-treated CF subjects homozygous for the F508del mutation: a randomized phase 2A trial (PELICAN). J Cyst Fibros. 2020;19(2):292–298. doi:10.1016/j.jcf.2019.09.006
76. Scanio MJC, Searle XB, Liu B, et al. Discovery of ABBV/GLPG-3221, a potent corrector of CFTR for the treatment of cystic fibrosis. ACS Med Chem Lett. 2019;10(11):1543–1548. doi:10.1021/acsmedchemlett.9b00377
77. Laselva O, Bartlett C, Popa A, et al. Emerging preclinical modulators developed for F508del-CFTR have the potential to be effective for ORKAMBI resistant processing mutants. J Cyst Fibros. 2021;20(1):106–119. doi:10.1016/j.jcf.2020.07.015
78. Lopes-Pacheco M, Silva IAL, Turner MJ, et al. Characterization of the mechanism of action of RDR01752, a novel corrector of F508del-CFTR. Biochem Pharmacol. 2020;180:114133. doi:10.1016/j.bcp.2020.114133
79. Veit G, Xu H, Dreano E, et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat Med. 2018;24(11):1732–1742. doi:10.1038/s41591-018-0200-x
80. Pedemonte N, Bertozzi F, Caci E, et al. Discovery of a picomolar potency pharmacological corrector of the mutant CFTR chloride channel. Sci Adv. 2020;6(8):eaay9669. doi:10.1126/sciadv.aay9669
81. Centko RM, Carlile GW, Barne I, et al. Combination of selective PARP3 and PARP16 inhibitory analogues of latonduine a corrects F508del-CFTR Trafficking. ACS Omega. 2020;5(40):25593–25604. doi:10.1021/acsomega.0c02467
82. Spanò V, Barreca M, Cilibrasi V, et al. Evaluation of fused pyrrolothiazole systems as correctors of mutant CFTR protein. Molecules. 2021;26(5):1275. doi:10.3390/molecules26051275
83. Moran O. The gating of the CFTR channel. Cell Mol Life Sci. 2017;74(1):85–92. doi:10.1007/s00018-016-2390-z
84. Van Goor F, Hadida S, Grootenhuis PDJ, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106(44):18825–18830. doi:10.1073/pnas.0904709106
85. McKone EF, Borowitz D, Drevinek P, et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir Med. 2014;2(11):902–910. doi:10.1016/S2213-2600(14)70218-8
86. De Boeck K, Munck A, Walker S, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13(6):674–680. doi:10.1016/j.jcf.2014.09.005
87. Guimbellot J, Solomon GM, Baines A, et al. Effectiveness of ivacaftor in cystic fibrosis patients with non-G551D gating mutations. J Cyst Fibros. 2019;18(1):102–109. doi:10.1016/j.jcf.2018.04.004
88. Avramescu RG, Kai Y, Xu H, et al. Mutation-specific downregulation of CFTR2 variants by gating potentiators. Hum Mol Genet. 2017;26(24):4873–4885. doi:10.1093/hmg/ddx367
89. Dekkers JF, Van Mourik P, Vonk AM, et al. Potentiator synergy in rectal organoids carrying S1251N, G551D, or F508del CFTR mutations. J Cyst Fibros. 2016;15(5):568–578. doi:10.1016/j.jcf.2016.04.007
90. Laselva O, Bartlett C, Gunawardena TNA, et al. Rescue of multiple class II CFTR mutations by elexacaftor + tezacaftor + ivacaftor mediated in part by the dual activities of Elexacaftor as both corrector and potentiator. Eur Respir J. 2020:2002774. doi:10.1183/13993003.02774-2020.
91. Harbeson SL, Morgan AJ, Liu JF, et al. Altering metabolic profiles of drugs by precision deuteration 2: discovery of a deuterated analog of ivacaftor with differentiated pharmacokinetics for clinical development. J Pharmacol Exp Ther. 2017;362(2):359–367. doi:10.1124/jpet.117.241497
92. Van Der Plas SE, Kelgtermans H, De munck T, et al. Discovery of N-(3-Carbamoyl-5,5,7,7-tetramethyl-5,7-dihydro-4H-thieno[2,3-c]pyran-2-yl)-lH-pyrazole-5-carboxamide (GLPG1837), a novel potentiator which can open Class III mutant Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Channels to a High. J Med Chem. 2018;61(4):1425–1435. doi:10.1021/acs.jmedchem.7b01288
93. Van Der Plas SE, Kelgtermans H, Mammoliti O, et al. Discovery of GLPG2451, a novel once daily potentiator for the treatment of cystic fibrosis. J Med Chem. 2021;64(1):343–353. doi:10.1021/acs.jmedchem.0c01796
94. Laselva O, McCormack J, Bartlett C, et al. Preclinical studies of a rare cf-causing mutation in the second nucleotide binding domain (C.3700a>g) show robust functional rescue in primary nasal cultures by novel cftr modulators. J Pers Med. 2020;10(4):209. doi:10.3390/jpm10040209
95. Kazani S, Rowlands DJ, Bottoli I, et al. Safety and efficacy of the cystic fibrosis transmembrane conductance regulator potentiator icenticaftor (QBW251). J Cyst Fibros. 2021;20(2):250–256. doi:10.1016/j.jcf.2020.11.002
96. Rowe SM, Jones I, Dransfield MT, et al. Efficacy and safety of the cftr potentiator icenticaftor (QBW251) in copd: results from a phase 2 randomized trial. Int J COPD. 2020;15:2399–2409. doi:10.2147/COPD.S257474
97. Pesce E, Bellotti M, Liessi N, et al. Synthesis and structure-activity relationship of aminoarylthiazole derivatives as correctors of the chloride transport defect in cystic fibrosis. Eur J Med Chem. 2015;99:14–35. doi:10.1016/j.ejmech.2015.05.030
98. Pesce E, Pedemonte N, Leoni A, Locatelli A, Morigi R. Synthesis and biological evaluation of thiazole derivatives on basic defects underlying cystic fibrosis. Bioorganic Med Chem Lett. 2020;30(21):127473. doi:10.1016/j.bmcl.2020.127473
99. Brindani N, Gianotti A, Giovani S, et al. Identification, structure-activity relationship, and biological characterization of 2,3,4,5-Tetrahydro-1 H-pyrido[4,3- b]indoles as a Novel Class of CFTR Potentiators. J Med Chem. 2020;63(19):11169–11194. doi:10.1021/acs.jmedchem.0c01050
100. Son J, Phuan P, Zhu JS, et al. 1-Benzylspiro[Piperidine-4,1ʹ-Pyrido[3,4-b]indole] “co-potentiators” for minimal function CFTR mutants. Eur J Med Chem. 2021;209:112888. doi:10.1016/j.ejmech.2020.112888
101. Haggie PM, Phuan PW, Tan JA, et al. Correctors and potentiators rescue function of the truncated W1282X-Cystic Fibrosis Transmembrane Regulator (CFTR) translation product. J Biol Chem. 2017;292(3):771–785. doi:10.1074/jbc.M116.764720
102. Giuliano KA, Wachi S, Drew L, et al. Use of a high-throughput phenotypic screening strategy to identify amplifiers, a novel pharmacological class of small molecules that exhibit functional synergy with potentiators and correctors. SLAS Discov. 2018;23(2):111–121. doi:10.1177/2472555217729790
103. Dukovski D, Villella A, Bastos C, et al. Amplifiers co-translationally enhance CFTR biosynthesis via PCBP1-mediated regulation of CFTR mRNA. J Cyst Fibros. 2020;19(5):733–741. doi:10.1016/j.jcf.2020.02.006
104. Molinski SV, Ahmadi S, Ip W, et al. Orkambi® and amplifier co‐therapy improves function from a rare CFTR mutation in gene‐edited cells and patient tissue. EMBO Mol Med. 2017;9(9):1224–1243. doi:10.15252/emmm.201607137
105. Swiatecka-Urban A, Brown A, Moreau-Marquis S, et al. The short apical membrane half-life of rescued ΔF508-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) results from accelerated endocytosis of ΔF508-CFTR in polarized human airway epithelial cells. J Biol Chem. 2005:36762–36772. doi:10.1074/jbc.M508944200
106. Sharma M, Pampinella F, Nemes C, et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol. 2004;164(6):923–933. doi:10.1083/jcb.200312018
107. Sharma M, Benharouga M, Hu W, Lukacs GL. Conformational and temperature-sensitive stability defects of the ΔF508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J Biol Chem. 2001;276(12):8942–8950. doi:10.1074/jbc.M009172200
108. He L, Kota P, Aleksandrov AA, et al. Correctors of F508 CFTR restore global conformational maturation without thermally stabilizing the. FASEB J. 2013(27):536–545. doi:10.1096/fj.12-216119
109. Okiyoneda T, Barrière H, Bagdány M, et al. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science (80-). 2010;329(5993):805–810. doi:10.1126/science.1191542
110. Donaldson SH, Solomon GM, Zeitlin PL, et al. Pharmacokinetics and safety of cavosonstat (N91115) in healthy and cystic fibrosis adults homozygous for F508DEL-CFTR. J Cyst Fibros. 2017;16(3):371–379. doi:10.1016/j.jcf.2017.01.009
111. Zaman K, Sawczak V, Zaidi A, et al. Augmentation of cftr maturation by s-nitrosoglutathione reductase. Am J Physiol - Lung Cell Mol Physiol. 2016;310(3):L263–L270. doi:10.1152/ajplung.00269.2014
112. Moniz S, Sousa M, Moraes BJ, et al. HGF stimulation of Rac1 signaling enhances pharmacological correction of the most prevalent cystic fibrosis mutant F508del-CFTR. ACS Chem Biol. 2013;8(2):432–442. doi:10.1021/cb300484r
113. Loureiro CA, Matos AM, Â D-A, et al. A molecular switch in the scaffold NHERF1 enables misfolded CFTR to evade the peripheral quality control checkpoint. Sci Signal. 2015;8(377):ra48. doi:10.1126/scisignal.aaa1580
114. Matos AM, Gomes-Duarte A, Faria M, et al. Prolonged co-treatment with HGF sustains epithelial integrity and improves pharmacological rescue of Phe508del-CFTR. Sci Rep. 2018;8(1):13026. doi:10.1038/s41598-018-31514-2
115. Matos AM, Pinto FR, Barros P, Amaral MD, Pepperkok R, Matos P. Inhibition of calpain 1 restores plasma membrane stability to pharmacologically rescued Phe508del-CFTR variant. J Biol Chem. 2019;294(36):13396–13410. doi:10.1074/jbc.RA119.008738
116. Mancini G, Loberto N, Olioso D, et al. GM1 as adjuvant of innovative therapies for cystic fibrosis disease. Int J Mol Sci. 2020;21(12):4486. doi:10.3390/ijms21124486
117. Cheng J, Moyer BD, Milewski M, et al. A golgi-associated PDZ domain protein modulates cystic fibrosis transmembrane regulator plasma membrane expression. J Biol Chem. 2002;277(5):3520–3529. doi:10.1074/jbc.M110177200
118. Cushing PR, Vouilleme L, Pellegrini M, Boisguerin P, Madden DR. A stabilizing influence CAL PDZ inhibition extends the half-life of ΔF508-CFTR. Angew Chem Int Ed Engl. 2010:9907–9911. doi:10.1002/anie.201005585
119. Dougherty PG, Wellmerling JH, Koley A, et al. Cyclic peptidyl inhibitors against CAL/CFTR interaction for treatment of cystic fibrosis. J Med Chem. 2020;63(24):15773–15784. doi:10.1021/acs.jmedchem.0c01528
120. Sosnay PR, Siklosi KR, Van Goor F, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet. 2013;45(10):1160–1167. doi:10.1038/ng.2745
121. Strug LJ, Gonska T, He G, et al. Cystic fibrosis gene modifier SLC26A9 modulates airway response to CFTR-directed therapeutics. Hum Mol Genet. 2016;25(20):4590–4600. doi:10.1093/hmg/ddw290
122. Corvol H, Mésinèle J, Douksieh IH, Strug LJ, Boëlle PY, Guillot L. SLC26A9 gene is associated with lung function response to ivacaftor in patients with cystic fibrosis. Front Pharmacol. 2018;9:828. doi:10.3389/fphar.2018.00828
123. O’Neal WK, Knowles MR. Cystic fibrosis disease modifiers: complex genetics defines the phenotypic diversity in a monogenic disease. Annu Rev Genomics Hum Genet. 2018;19(1):201–222. doi:10.1146/annurev-genom-083117-021329
124. Amaral MD, de Boeck K. CF’ ESPTF on ’Speeding up access to new drugs for. Theranostics by testing CFTR modulators in patient-derived materials: the current status and a proposal for subjects with rare CFTR mutations. J Cyst Fibros. 2019;18(5):685–692. doi:10.1016/j.jcf.2019.06.010
125. Mou H, Vinarsky V, Tata PR, et al. Dual SMAD signaling inhibition enables long-term expansion of diverse epithelial basal cells. Cell Stem Cell. 2016;19(2):217–231. doi:10.1016/j.stem.2016.05.012
126. Gianotti A, Delpiano L, Caci E. In vitro methods for the development and analysis of human primary airway epithelia. Front Pharmacol. 2018;9:1176. doi:10.3389/fphar.2018.01176
127. Sachs N, Papaspyropoulos A, Zomer‐van Ommen DD, et al. Long‐term expanding human airway organoids for disease modeling. EMBO J. 2019;38(4):e100300. doi:10.15252/embj.2018100300
128. Vonk AM, van Mourik P, Ramalho AS, et al. Protocol for application, standardization and validation of the forskolin-induced swelling assay in cystic fibrosis human colon organoids. STAR Protoc. 2020;1(1):100019. doi:10.1016/j.xpro.2020.100019
129. Dekkers JF, Wiegerinck CL, De Jonge HR, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19(7):939–945. doi:10.1038/nm.3201
130. de Courcey F, Zholos AV, Atherton-Watson H, et al. Development of primary human nasal epithelial cell cultures for the study of cystic fibrosis pathophysiology. Am J Physiol - Cell Physiol. 2012;303(11):C1173–C1179. doi:10.1152/ajpcell.00384.2011
131. McCravy MS, Quinney NL, Cholon DM, et al. Personalised medicine for non-classic cystic fibrosis resulting from rare CFTR mutations. Eur Respir J. 2020;56(1):2000062. doi:10.1183/13993003.00062-2020
132. Merkert S, Bednarski C, Göhring G, Cathomen T, Martin U. Generation of a gene-corrected isogenic control iPSC line from cystic fibrosis patient-specific iPSCs homozygous for p.Phe508del mutation mediated by TALENs and ssODN. Stem Cell Res. 2017;23:95–97. doi:10.1016/j.scr.2017.07.010
133. Mithal A, Capilla A, Heinze D, et al. Generation of mesenchyme free intestinal organoids from human induced pluripotent stem cells. Nat Commun. 2020;11(1):215. doi:10.1038/s41467-019-13916-6
134. Hawkins FJ, Suzuki S, L. beermann M, et al. Derivation of airway basal stem cells from human pluripotent stem cells. Cell Stem Cell. 2021;28(1):79–95. doi:10.1016/j.stem.2020.09.017
135. Berkers G, van Mourik P, Vonk AM, et al. Rectal organoids enable personalized treatment of cystic fibrosis. Cell Rep. 2019;26(7):1701–1708. doi:10.1016/j.celrep.2019.01.068
136. de Winter – de Groot KM, Berkers G, Marck – van der Wilt REP, et al. Forskolin-induced swelling of intestinal organoids correlates with disease severity in adults with cystic fibrosis and homozygous F508del mutations. J Cyst Fibros. 2020;19(4):614–619. doi:10.1016/j.jcf.2019.10.022
137. Ramalho AS, Förstová E, Vonk AM, et al. Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis. Eur Respir J. 2021;57(1):1902426. doi:10.1183/13993003.02426-2019
138. Collawn JF, Lazrak A, Bebok Z, Matalon S. The CFTR and ENaC debate: how important is ENaC in CF lung disease? Am J Physiol - Lung Cell Mol Physiol. 2012;302(11):L1141–L1146. doi:10.1152/ajplung.00036.2012
139. Figueira MF, Webster MJ, Tarran R. CrossTalk proposal: mucosal acidification drives early progressive lung disease in cystic fibrosis. J Physiol. 2018;596(16):3433–3437. doi:10.1113/JP275425
140. Althaus M. ENaC Inhibitors and airway re-hydration in cystic fibrosis: state of the art. Curr Mol Pharmacol. 2013;6(1):3–12. doi:10.2174/18744672112059990025
141. Butler R, Hunt T, Smith NJ. ENaC inhibitors for the treatment of cystic fibrosis. Pharm Pat Anal. 2015;4(1):17–27. doi:10.4155/ppa.14.51
142. Couroux P, Farias P, Rizvi L, et al. First clinical trials of novel ENaC targeting therapy, SPX-101, in healthy volunteers and adults with cystic fibrosis. Pulm Pharmacol Ther. 2019;58:101819. doi:10.1016/j.pupt.2019.101819
143. Mackie A, Rascher J, Schmid M, Endriss V, Brand T, Seibold W. First clinical trials of the inhaled epithelial sodium channel inhibitor BI 1265162 in healthy volunteers. ERJ Open Res. 2021;7(1):00447–02020. doi:10.1183/23120541.00447-2020
144. Nickolaus P, Jung B, Sabater J, Constant S, Gupta A. Preclinical evaluation of the epithelial sodium channel inhibitor BI 1265162 for treatment of cystic fibrosis. ERJ Open Res. 2020;6(4):00429–02020. doi:10.1183/23120541.00429-2020
145. Sesma JI, Wu B, Stuhlmiller TJ, Scott DW. SPX-101 is stable in and retains function after exposure to cystic fibrosis sputum. J Cyst Fibros. 2019;18(2):244–250. doi:10.1016/j.jcf.2018.06.002
146. Scott DW, Walker MP, Sesma J, et al. SPX-101 is a novel epithelial sodium channel–targeted therapeutic for cystic fibrosis that restores mucus transport. Am J Respir Crit Care Med. 2017;196(6):734–744. doi:10.1164/rccm.201612-2445OC
147. Mukherjee A, MacDonald KD, Kim J, Henderson MI, Eygeris Y, Sahay G. Engineered mutant α-ENaC subunit mRNA delivered by lipid nanoparticles reduces amiloride currents in cystic fibrosis–based cell and mice models. Sci Adv. 2021;6(47):eabc5911. doi:10.1126/sciadv.abc5911
148. Almaça J, Faria D, Sousa M, et al. High-content siRNA screen reveals global ENaC regulators and potential cystic fibrosis therapy targets. Cell. 2013;154(6):1390–1400. doi:10.1016/j.cell.2013.08.045
149. Kunzelmann K, Tian Y, Martins JR, et al. Anoctamins. Pflugers Arch Eur J Physiol. 2011;462(2):195–208. doi:10.1007/s00424-011-0975-9
150. Cho H, Yang YD, Lee J, et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat Neurosci. 2012;15(7):1015–1021. doi:10.1038/nn.3111
151. Hong GS, Lee SH, Lee B, et al. ANO1/TMEM16A regulates process maturation in radial glial cells in the developing brain. Proc Natl Acad Sci U S A. 2019;116(25):12494–12499. doi:10.1073/pnas.1901067116
152. Huang F, Zhang H, Wu M, et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc Natl Acad Sci U S A. 2012;109(40):16354–16359. doi:10.1073/pnas.1214596109
153. Crottès D, Jan LY. The multifaceted role of TMEM16A in cancer. Cell Calcium. 2019;82:102050. doi:10.1016/j.ceca.2019.06.004
154. Shi S, Pang C, Guo S, et al. Recent progress in structural studies on TMEM16A channel. Comput Struct Biotechnol J. 2020;18:714–722. doi:10.1016/j.csbj.2020.03.015
155. West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol. 2004;165(1):107–113. doi:10.1016/S0002-9440(10)63279-8
156. Carles A, Millon R, Cromer A, et al. Head and neck squamous cell carcinoma transcriptome analysis by comprehensive validated differential display. Oncogene. 2006;25(12):1821–1831. doi:10.1038/sj.onc.1209203
157. Britschgi A, Bill A, Brinkhaus H, et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci U S A. 2013;110(11):E1026–E1034. doi:10.1073/pnas.1217072110
158. Jia L, Liu W, Guan L, Lu M, Wang KW. Inhibition of calcium-activated chloride channel ANO1/TMEM16A suppresses tumor growth and invasion in human lung cancer. PLoS One. 2015;10(8):e0136584. doi:10.1371/journal.pone.0136584
159. Duvvuri U, Shiwarski DJ, Xiao D, et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012;72(13)):3270–3281. doi:10.1158/0008-5472.CAN-12-0475-T
160. Ousingsawat J, Martins JR, Schreiber R, Rock JR, Harfe BD, Kunzelmann K. Loss of TMEM16A causes a defect in epithelial Ca2+ -dependent chloride transport. J Biol Chem. 2009;284(42):28698–28703. doi:10.1074/jbc.M109.012120
161. Rock JR, O’Neal WK, Gabriel SE, et al. Transmembrane protein 16A (TMEM16A) is a Ca2+ -regulated Cl- secretory channel in mouse airways. J Biol Chem. 2009;284(22):14875–14880. doi:10.1074/jbc.C109.000869
162. He M, Wu B, Ye W, et al. Chloride channels regulate differentiation and barrier functions of the mammalian airway. Elife. 2020;9:e53085. doi:10.7554/eLife.53085
163. Lee B, Hong GS, Lee SH, et al. Anoctamin 1/TMEM16A controls intestinal Cl− secretion induced by carbachol and cholera toxin. Exp Mol Med. 2019;51(8):1–14. doi:10.1038/s12276-019-0287-2
164. Simões FB, Quaresma MC, Clarke LA, et al. TMEM16A chloride channel does not drive mucus production. Life Sci Alliance. 2019;2(6):1–13. doi:10.26508/LSA.201900462
165. Benedetto R, Ousingsawat J, Wanitchakool P, et al. Epithelial chloride transport by CFTR requires TMEM16A. Sci Rep. 2017;7(1):12397. doi:10.1038/s41598-017-10910-0
166. Sondo E, Caci E, Galietta LJV. The TMEM16A chloride channel as an alternative therapeutic target in cystic fibrosis. Int J Biochem Cell Biol. 2014;52:73–76. doi:10.1016/j.biocel.2014.03.022
167. Le SC, Jia Z, Chen J, Yang H. Molecular basis of PIP2-dependent regulation of the Ca2+-activated chloride channel TMEM16A. Nat Commun. 2019;10(1):3769. doi:10.1038/s41467-019-11784-8
168. Tembo M, Wozniak KL, Bainbridge RE, Carlson AE. Phosphatidylinositol 4,5-bisphosphate (PIP2) and Ca2 are both required to open the Cl channel TMEM16A. J Biol Chem. 2019;294(33):12556–12564. doi:10.1074/jbc.RA118.007128
169. Tian Y, Schreiber R, Wanitchakool P, et al. Control of TMEM16A by INO-4995 and other inositolphosphates. Br J Pharmacol. 2013;168(1):253–265. doi:10.1111/j.1476-5381.2012.02193.x
170. Lérias JR, Pinto MC, Botelho HM, et al. A novel microscopy-based assay identifies extended synaptotagmin-1 (ESYT1) as a positive regulator of anoctamin 1 traffic. Biochim Biophys Acta Mol Cell Res. 2018;1865(2):421–431. doi:10.1016/j.bbamcr.2017.11.009
171. Kellerman D, Rossi Mospan A, Engels J, Schaberg A, Gorden JA, Smiley L. Denufosol: a review of studies with inhaled P2Y2 agonists that led to Phase 3. Pulm Pharmacol Ther. 2008;21(4):600–607. doi:10.1016/j.pupt.2007.12.003
172. Ratjen F, Durham T, Navratil T, et al. Long term effects of denufosol tetrasodium in patients with cystic fibrosis. J Cyst Fibros. 2012;11(6):539–549. doi:10.1016/j.jcf.2012.05.003
173. Moss RB. Pitfalls of drug development: lessons learned from trials of denufosol in cystic fibrosis. J Pediatr. 2013;162(4):676–680. doi:10.1016/j.jpeds.2012.11.034
174. Ousingsawat J, Kongsuphol P, Schreiber R, Kunzelmann K. CFTR and TMEM16A are separate but functionally related Cl - channels. Cell Physiol Biochem. 2011;28(4):715–724. doi:10.1159/000335765
175. Kester MB, Sokolove PM. The effect of adriamycin and duramycin on calcium translocation in liposome systems modeled on the inner mitochondrial membrane. Arch Biochem Biophys. 1990;280(2):405–411. doi:10.1016/0003-9861(90)90349-4
176. Cloutier MM, Guernsey L, Sha’afi RI. Duramycin increases intracellular calcium in airway epithelium. Mol Membr Biol. 1993;10(2):107–118. doi:10.3109/09687689309150258
177. Zeitlin PL, Boyle MP, Guggino WB, Molina L. A phase I trial of intranasal Moli1901 for cystic fibrosis. Chest. 2004;125(1):143–149. doi:10.1378/chest.125.1.143
178. Steiner I, Errhalt P, Kubesch K, et al. Pulmonary pharmacokinetics and safety of nebulized duramycin in healthy male volunteers. Naunyn Schmiedebergs Arch Pharmacol. 2008;378(3):323–333. doi:10.1007/s00210-008-0293-8
179. Grasemann H, Stehling F, Brunar H, et al. Inhalation of Moli1901 in patients with cystic fibrosis. Chest. 2007;131(5):1461–1466. doi:10.1378/chest.06-2085
180. Gold EP, Jacocks HM, Bourdelais AJ, Baden DG. Brevenal, a brevetoxin antagonist from Karenia brevis, binds to a previously unreported site on mammalian sodium channels. Harmful Algae. 2013;26:12–19. doi:10.1016/j.hal.2013.03.001
181. Abraham WM, Bourdelais AJ, Sabater JR, et al. Airway responses to aerosolized brevetoxins in an animal model of asthma. Am J Respir Crit Care Med. 2005;171(1):26–34. doi:10.1164/rccm.200406-735OC
182. Keeler DM, Grandal MK, McCall JR. Brevenal, a marine natural product, is anti-inflammatory and an immunomodulator of macrophage and lung epithelial cells. Mar Drugs. 2019;17(3):184. doi:10.3390/md17030184
183. Namkung W, Yao Z, Finkbeiner WE, Verkman AS. Small‐molecule activators of TMEM16A, a calcium‐activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J. 2011;25(11):4048–4062. doi:10.1096/fj.11-191627
184. Liu S, Feng J, Luo J, Yang P, Brett TJ, Hu H. Eact, a small molecule activator of TMEM16A, activates TRPV1 and elicits pain- and itch-related behaviours. Br J Pharmacol. 2016;173(7):1208–1218. doi:10.1111/bph.13420
185. Genovese M, Borrelli A, Venturini A, et al. TRPV4 and purinergic receptor signalling pathways are separately linked in airway epithelia to CFTR and TMEM16A chloride channels. J Physiol. 2019;597(24):5859–5878. doi:10.1113/JP278784
186. Guo S, Wang H, Pang C, et al. Entering the spotlight: chitosan oligosaccharides as novel activators of CaCCs/TMEM16A. Pharmacol Res. 2019;146:104323. doi:10.1016/j.phrs.2019.104323
187. Guo S, Chen YF, Shi S, et al. The molecular mechanism of ginsenoside analogs activating TMEM16A. Biophys J. 2019;118(1):262–272. doi:10.1016/j.bpj.2019.11.015
188. Chai R, Chen Y, Yuan H, et al. Identification of resveratrol, an herbal compound, as an activator of the calcium-activated chloride channel, TMEM16A. J Membr Biol. 2017;250(5):483–492. doi:10.1007/s00232-017-9975-9
189. Ji Q, Shi S, Guo S, et al. Activation of TMEM16A by natural product canthaxanthin promotes gastrointestinal contraction. FASEB J. 2020;34(10):13430–13444. doi:10.1096/fj.202000443RR
190. Huang Y, Guo S, Ren S, Chen Y, Zhan Y, An H. The natural compound cinnamaldehyde is a novel activator of calcium-activated chloride channel. J Membr Biol. 2018;251(5–6):747–756. doi:10.1007/s00232-018-0052-9
191. Lee G, Bae H. Anti-inflammatory applications of melittin, a major component of bee venom: detailed mechanism of action and adverse effects. Molecules. 2016;21(5):616. doi:10.3390/molecules21050616
192. Schreiber R, Ousingsawat J, Wanitchakool P, et al. Regulation of TMEM16A/ANO1 and TMEM16F/ANO6 ion currents and phospholipid scrambling by Ca 2+ and plasma membrane lipid. J Physiol. 2018;596(2):217–229. doi:10.1113/JP275175
193. Oršolić N. Bee venom in cancer therapy. Cancer Metastasis Rev. 2012;31(1–2):173–194. doi:10.1007/s10555-011-9339-3
194. Kunzelmann K, Ousingsawat J, Benedetto R, Cabrita I, Schreiber R. Contribution of anoctamins to cell survival and cell death. Cancers (Basel). 2019;11(3):382. doi:10.3390/cancers11030382
195. Danahay HL, Lilley S, Fox R, et al. TMEM16A potentiation: a novel therapeutic approach for the treatment of cystic fibrosis. Am J Respir Crit Care Med. 2020;201(8):946–954. doi:10.1164/RCCM.201908-1641OC
196. Danahay H, Fox R, Lilley S, et al. Potentiating TMEM16A does not stimulate airway mucus secretion or bronchial and pulmonary arterial smooth muscle contraction. FASEB Bio Adv. 2020;2(8):464–477. doi:10.1096/fba.2020-00035
197. Lin J, Jiang Y, Li L, Liu Y, Tang H, Jiang D. TMEM16A mediates the hypersecretion of mucus induced by Interleukin-13. Exp Cell Res. 2015;334(2):260–269. doi:10.1016/j.yexcr.2015.02.026
198. Benedetto R, Cabrita I, Schreiber R, Kunzelmann K. TMEM16A is indispensable for basal mucus secretion in airways and intestine. FASEB J. 2019;33(3):4502–4512. doi:10.1096/fj.201801333RRR
199. Miner K, Labitzke K, Liu B, et al. Drug repurposing: the anthelmintics niclosamide and nitazoxanide are potent TMEM16A antagonists that fully bronchodilate airways. Front Pharmacol. 2019;10:51. doi:10.3389/fphar.2019.00051
200. Singh RD, Gibbons SJ, Saravanaperumal SA, et al. Ano1, a Ca2+-activated Cl- channel, coordinates contractility in mouse intestine by Ca2+ transient coordination between interstitial cells of Cajal. J Physiol. 2014;592(18):4051–4068. doi:10.1113/jphysiol.2014.277152
201. Jiang Y, Yu B, Yang H, Ma T. Shikonin inhibits intestinal calcium-activated chloride channels and prevents rotaviral diarrhea. Front Pharmacol. 2016;7:
202. Hogg RC, Wang Q, Large WA. Action of niflumic acid on evoked and spontaneous calcium‐activated chloride and potassium currents in smooth muscle cells from rabbit portal vein. Br J Pharmacol. 1994;112(3):977–984. doi:10.1111/j.1476-5381.1994.tb13177.x
203. Guinamard R, Simard C, Del Negro C. Flufenamic acid as an ion channel modulator. Pharmacol Ther. 2013;138(2):272–284. doi:10.1016/j.pharmthera.2013.01.012
204. Wu G, Hamill OP. NPPB block of Ca++-activated Cl- currents in Xenopus oocytes. Pflügers Arch Eur J Physiol. 1992;420(2):227–229. doi:10.1007/BF00374996
205. Kubitz R, Warth R, Allert N, Kunzelmann K, Greger R. Small-conductance chloride channels induced by cAMP, Ca2+, and hypotonicity in HT29 cells: ion selectivity, additivity and stilbene sensitivity. Pflügers Arch Eur J Physiol. 1992;421(5):447–454. doi:10.1007/BF00370255
206. Frings S, Reuter D, Kleene SJ. Neuronal Ca2+-activated Cl- channels - Homing in on an elusive channel species. Prog Neurobiol. 2000;60(3):247–289. doi:10.1016/S0301-0082(99)00027-1
207. Walker NM, Simpson JE, Levitt RC, Boyle KT, Clarke LL. Talniflumate increases survival in a cystic fibrosis mouse model of distal intestinal obstructive syndrome. J Pharmacol Exp Ther. 2006;317(1):275–283. doi:10.1124/jpet.105.094847
208. Namkung W, Thiagarajah JR, Phuan P, Verkman AS. Inhibition of Ca 2+ ‐activated Cl − channels by gallotannins as a possible molecular basis for health benefits of red wine and green tea. FASEB J. 2010;24(11):4178–4186. doi:10.1096/fj.10-160648
209. Yao Z, Namkung W, Ko EA, Park J, Tradtrantip L, Verkman AS. Fractionation of a herbal antidiarrheal medicine reveals Eugenol as an inhibitor of Ca2+-activated Cl- channel TMEM16A. PLoS One. 2012;7(5):e38030. doi:10.1371/journal.pone.0038030
210. Zhang X, Li H, Zhang H, et al. Inhibition of transmembrane member 16A calcium-activated chloride channels by natural flavonoids contributes to flavonoid anticancer effects. Br J Pharmacol. 2017;174(14):2334–2345. doi:10.1111/bph.13841
211. Guo S, Chen Y, Pang C, et al. Matrine is a novel inhibitor of the TMEM16A chloride channel with antilung adenocarcinoma effects. J Cell Physiol. 2019;234(6):8698–8708. doi:10.1002/jcp.27529
212. Sui Y, Wu F, Lv J, et al. Identification of the novel TMEM16A inhibitor dehydroandrographolide and its anticancer activity on SW620 cells. PLoS One. 2015;10(12):e0144715. doi:10.1371/journal.pone.0144715
213. Zhang X, Zhang G, Zhai W, Zhao Z, Wang S, Yi J. Inhibition of TMEM16A Ca2+-activated Cl− channels by avermectins is essential for their anticancer effects. Pharmacol Res. 2020;156:104763. doi:10.1016/j.phrs.2020.104763
214. Zhu X, Zhang W, Jin L, Zhang G, Yang H, Yu B. Inhibitory activities of curzerenone, curdione, furanodienone, curcumol and germacrone on Ca2+-activated chloride channels. Fitoterapia. 2020;147:104736. doi:10.1016/j.fitote.2020.104736
215. Truong EC, Phuan PW, Reggi AL, et al. Substituted 2-Acylaminocycloalkylthiophene-3-carboxylic acid arylamides as inhibitors of the calcium-activated chloride channel transmembrane protein 16A (TMEM16A). J Med Chem. 2017;60(11):4626–4635. doi:10.1021/acs.jmedchem.7b00020
216. De La Fuente R, Namkung W, Mills A, Verkman AS. Small-molecule screen identifies inhibitors of a human intestinal calcium-activated chloride channel. Mol Pharmacol. 2008;73(3):758–768. doi:10.1124/mol.107.043208
217. Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286(3):2365–2374. doi:10.1074/jbc.M110.175109
218. Seo Y, Park J, Kim M, et al. Inhibition of ANO1/TMEM16A chloride channel by idebenone and its cytotoxicity to cancer cell lines. PLoS One. 2015;10(7):e0133656. doi:10.1371/journal.pone.0133656
219. Papp R, Nagaraj C, Zabini D, et al. Targeting TMEM16A to reverse vasoconstriction and remodelling in idiopathic pulmonary arterial hypertension. Eur Respir J. 2019;53(6):1800965. doi:10.1183/13993003.00965-2018
220. Theilmann AL, Ormiston ML. Repurposing benzbromarone for pulmonary arterial hypertension: can channelling the past deliver the therapy of the future? Eur Respir J. 2019;53(6):1900583. doi:10.1183/13993003.00583-2019
221. Diena T, Melani R, Caci E, et al. Block of CFTR-dependent chloride currents by inhibitors of multidrug resistance-associated proteins. Eur J Pharmacol. 2007;560(2–3):127–131. doi:10.1016/j.ejphar.2007.01.051
222. Felser A, Lindinger PW, Schnell D, et al. Hepatocellular toxicity of benzbromarone: effects on mitochondrial function and structure. Toxicology. 2014;324:136–146. doi:10.1016/j.tox.2014.08.002
223. Jin Y, Lu Z, Ding K, et al. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: inactivation of the NF-κB pathway and generation of reactive oxygen species. Cancer Res. 2010;70(6):2516–2527. doi:10.1158/0008-5472.CAN-09-3950
224. Ahn SY, Yang JH, Kim NH, et al. Anti-helminthic niclosamide inhibits Ras-driven oncogenic transformation via activation of GSK-3. Oncotarget. 2017;8(19):31856–31863. doi:10.18632/oncotarget.16255
225. Lee MC, Chen YK, Hsu YJ, Lin BR. Niclosamide inhibits the cell proliferation and enhances the responsiveness of esophageal cancer cells to chemotherapeutic agents. Oncol Rep. 2020;43(2):549–561. doi:10.3892/or.2019.7449
226. Schweizer MT, Haugk K, McKiernan JS, et al. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PLoS One. 2018;13(6):e0198389. doi:10.1371/journal.pone.0198389
227. Burock S, Daum S, Keilholz U, Neumann K, Walther W, Stein SU. Phase II trial to investigate the safety and efficacy of orally applied niclosamide in patients with metachronous or sychronous metastases of a colorectal cancer progressing after therapy: the NIKOLO trial. BMC Cancer. 2018;18(1):297. doi:10.1186/s12885-018-4197-9
228. Centeio R, Cabrita I, Benedetto R, et al. Pharmacological inhibition and activation of the Ca2+activated Cl− channel TMEM16A. Int J Mol Sci. 2020;21(7):2557. doi:10.3390/ijms21072557
229. Cabrita I, Benedetto R, Schreiber R, Kunzelmann K. Niclosamide repurposed for the treatment of inflammatory airway disease. JCI Insight. 2019;4(15):15. doi:10.1172/jci.insight.128414
230. Oh SJ, Hwang SJ, Jung J, et al. Monna, a potent and selective blocker for transmembrane protein with unknown function 16/anoctamin-1. Mol Pharmacol. 2013;84(5):726–735. doi:10.1124/mol.113.087502
231. Seo Y, Lee HK, Park J, et al. Ani9, a novel potent small-molecule ANO1 inhibitor with negligible effect on ANO2. PLoS One. 2016;11(5):e01557711. doi:10.1371/journal.pone.0155771
232. Seo Y, Kim J, Chang J, Kim SS, Namkung W, Kim I. Synthesis and biological evaluation of novel Ani9 derivatives as potent and selective ANO1 inhibitors. Eur J Med Chem. 2018;160:245–255. doi:10.1016/j.ejmech.2018.10.002
233. Quesada R, Dutzler R. Alternative chloride transport pathways as pharmacological targets for the treatment of cystic fibrosis. J Cyst Fibros. 2020;19(Suppl 1):S37–S41. doi:10.1016/j.jcf.2019.10.020
234. Walter JD, Sawicka M, Dutzler R. Cryo-EM structures and functional characterization of murine Slc26a9 reveal mechanism of uncoupled chloride transport. Elife. 2019;8:e46986. doi:10.7554/eLife.46986
235. Dorwart MR, Shcheynikov N, Yang D, Muallem S. The solute carrier 26 family of proteins in epithelial ion transport. Physiology. 2008;23(2):104–114. doi:10.1152/physiol.00037.2007
236. Anagnostopoulou P, Riederer B, Duerr J, et al. SLC26A9-mediated chloride secretion prevents mucus obstruction in airway inflammation. J Clin Invest. 2012;122(10):3629–3634. doi:10.1172/JCI60429
237. Bertrand CA, Zhang R, Pilewski JM, Frizzell RA. SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J Gen Physiol. 2009;133(4):421–438. doi:10.1085/jgp.200810097
238. Sato Y, Thomas DY, Hanrahan JW. The anion transporter SLC26A9 localizes to tight junctions and is degraded by the proteasome when co-expressed with F508del-CFTR. J Biol Chem. 2019;294(48):18269–18284. doi:10.1074/jbc.RA119.010192
239. Li W, Soave D, Miller MR, et al. Unraveling the complex genetic model for cystic fibrosis: pleiotropic effects of modifier genes on early cystic fibrosis-related morbidities. Hum Genet. 2014;133(2):151–161. doi:10.1007/s00439-013-1363-7
240. Sun L, Rommens JM, Corvol H, et al. Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nat Genet. 2012;44(5):562–569. doi:10.1038/ng.2221
241. Miller MR, Soave D, Li W, et al. Variants in solute carrier SLC26A9 modify prenatal exocrine pancreatic damage in cystic fibrosis. J Pediatr. 2015;166(5):1152–1157. doi:10.1016/j.jpeds.2015.01.044
242. Lam ATN, Aksit MA, Vecchio-Pagan B, et al. Increased expression of anion transporter SLC26A9 delays diabetes onset in cystic fibrosis. J Clin Invest. 2020;130(1):272–286. doi:10.1172/JCI129833
243. Salomon JJ, Spahn S, Wang X, Füllekrug J, Bertrand CA, Mall MA. Generation and functional characterization of epithelial cells with stable expression of SLC26A9 Cl- channels. Am J Physiol - Lung Cell Mol Physiol. 2016;310(7):L593–L602. doi:10.1152/ajplung.00321.2015
244. Loriol C, Dulong S, Avella M, et al. Characterization of SLC26A9, facilitation of Cl - transport by bicarbonate. Cell Physiol Biochem. 2008;22(1–4):15–30. doi:10.1159/000149780
245. Davis JT, Okunola O, Quesada R. Recent advances in the transmembrane transport of anions. Chem Soc Rev. 2010;39(10):3843–3862. doi:10.1039/b926164h
246. Li H, Salomon JJ, Sheppard DN, Mall MA, Galietta LJ. Bypassing CFTR dysfunction in cystic fibrosis with alternative pathways for anion transport. Curr Opin Pharmacol. 2017;34:91–97. doi:10.1016/j.coph.2017.10.002
247. Hernando E, Capurro V, Cossu C, et al. Small molecule anionophores promote transmembrane anion permeation matching CFTR activity. Sci Rep. 2018;8(1):2608. doi:10.1038/s41598-018-20708-3
248. Cossu C, Fiore M, Baroni D, et al. Anion-transport mechanism of a triazole-bearing derivative of prodigiosine: a candidate for cystic fibrosis therapy. Front Pharmacol. 2018;9:852. doi:10.3389/fphar.2018.00852
249. Fiore M, Cossu C, Capurro V, et al. Small molecule-facilitated anion transporters in cells for a novel therapeutic approach to cystic fibrosis. Br J Pharmacol. 2019;176(11):1764–1779. doi:10.1111/bph.14649
250. Gianotti A, Capurro V, Delpiano L, et al. Small molecule anion carriers correct abnormal airway surface liquid properties in cystic fibrosis airway epithelia. Int J Mol Sci. 2020;21(4):1488. doi:10.3390/ijms21041488
251. Li H, Valkenier H, Thorne AG, et al. Anion carriers as potential treatments for cystic fibrosis: transport in cystic fibrosis cells, and additivity to channel-targeting drugs. Chem Sci. 2019;10(42):9663–9672. doi:10.1039/c9sc04242c
252. Muraglia KA, Chorghade RS, Kim BR, et al. Small-molecule ion channels increase host defences in cystic fibrosis airway epithelia. Nature. 2019;567(7748):405–408. doi:10.1038/s41586-019-1018-5
253. Chorghade RS, Kim BR, Launspach JL, Karp PH, Welsh MJ, Burke MD. Amphotericin B induces epithelial voltage responses in people with cystic fibrosis. J Cyst Fibros. 2021;20(3):540–550. doi:10.1016/j.jcf.2020.11.018
254. Guo S, Chen Y, Pang C, et al. Ginsenoside Rb1, a novel activator of the TMEM16A chloride channel, augments the contraction of guinea pig ileum. Pflugers Arch Eur J Physiol. 2017;469(5–6):681–692. doi:10.1007/s00424-017-1934-x
255. Burris SK, Wang Q, Bulley S, Neeb ZP, Jaggar JH. 9-Phenanthrol inhibits recombinant and arterial myocyte TMEM16A channels. Br J Pharmacol. 2015;172(10):2459–2468. doi:10.1111/bph.13077
256. Yimnual C, Satitsri S, Ningsih BNS, Rukachaisirikul V, Muanprasat C. A fungus-derived purpactin A as an inhibitor of TMEM16A chloride channels and mucin secretion in airway epithelial cells. Biomed Pharmacother. 2021;139:111583. doi:10.1016/j.biopha.2021.111583
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.