")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Pharmacokinetics, pharmacodynamics, and safety of lesinurad, a selective uric acid reabsorption inhibitor, in healthy adult males
Authors Shen Z, Rowlings C, Kerr B, Hingorani V, Manhard K, Quart B, Yeh L, Storgard C
Received 21 March 2015
Accepted for publication 7 May 2015
Published 2 July 2015 Volume 2015:9 Pages 3423—3434
DOI https://doi.org/10.2147/DDDT.S85193
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Zancong Shen, Colin Rowlings, Brad Kerr, Vijay Hingorani, Kimberly Manhard, Barry Quart, Li-Tain Yeh, Chris Storgard
Ardea Biosciences, Inc. (a member of the AstraZeneca group), San Diego, CA, USA
Abstract: Lesinurad is a selective uric acid reabsorption inhibitor under investigation for the treatment of gout. Single and multiple ascending dose studies were conducted to evaluate pharmacokinetics, pharmacodynamics, and safety of lesinurad in healthy males. Lesinurad was administered as an oral solution between 5 mg and 600 mg (single ascending dose; N=34) and as an oral solution or immediate-release capsules once daily (qday) between 100 mg and 400 mg for 10 days under fasted or fed condition (multiple ascending dose; N=32). Following single doses of lesinurad solution, absorption was rapid and exposure (maximum observed plasma concentration and area under the plasma concentration–time curve) increased in a dose-proportional manner. Following multiple qday doses, there was no apparent accumulation of lesinurad. Urinary excretion of unchanged lesinurad was generally between 30% and 40% of dose. Increases in urinary excretion of uric acid and reductions in serum uric acid correlated with dose. Following 400 mg qday dosing, serum uric acid reduction was 35% at 24 hours post-dose, supporting qday dosing. A relative bioavailability study in healthy males (N=8) indicated a nearly identical pharmacokinetic profile following dosing of tablets or capsules. Lesinurad was generally safe and well tolerated.
Keywords: urinary excretion, urate lowering, URAT1, single and multiple doses, food effect, clearance
Introduction
Gout is the most common form of inflammatory arthritis.1 It affects approximately 9 million people in Europe, more than 8 million in the United States, and more than 3 million in Japan.2–6 Gout results from a metabolic disorder, hyperuricemia, where excessive concentrations of uric acid in the blood lead to deposition of monosodium urate crystals in and around the joints and other connective tissues.7 This causes chronic inflammation, leading to acute gout flares and potentially chronic arthritis with joint damage and disfiguring tophi, kidney stones, and chronic kidney disease. Hyperuricemia, typically defined in guidelines as serum uric acid (sUA) >6.8 mg/dL (>400 μmol/L), is caused in most patients by inefficient uric acid excretion.8 Hyperuricemia is also associated with comorbidities such as hypertension, cardiovascular disease, kidney disease, and metabolic syndrome, including diabetes.9
Hyperuricemia is primarily treated with xanthine oxidase inhibitors that block the production of uric acid. In addition, there are uricuretic agents that enhance therapeutic increases in uric acid excretion from the kidney (ie, uricuresis).10 Lesinurad is a selective uric acid reabsorption inhibitor that inhibits the uric acid transporter 1 (URAT1) located in the proximal tubule of the kidney. URAT1 is responsible for the majority of the reabsorption of filtered uric acid from the renal tubular lumen.4–6 By inhibiting URAT1, lesinurad acts as a uricuretic agent, promoting uricuresis and thereby lowering sUA.10–14
The aim of the three current studies was to characterize the pharmacokinetic and pharmacodynamic profile and to evaluate the safety profile of lesinurad in healthy subjects when it is given in pharmacologically active doses. The single ascending dose (SAD) study was conducted using an oral solution of lesinurad, and the multiple ascending dose (MAD) study evaluated the oral solution and an immediate-release capsule. The relative bioavailability study compared the pharmacokinetics between capsules and tablets.
Methods
The protocol and consent form were approved by the Yorkshire Independent Research Ethics Committee (Leeds, UK) prior to the start of the SAD and MAD studies, and approved by the Independent Investigational Review Board Inc. (Plantation, FL, USA) prior to the start of the bioavailability study.
Subjects
All subjects provided written informed consent prior to participation. Only male subjects with body mass index within the range of ≥18 and ≤30 to 32 kg/m2 were eligible for the SAD or MAD studies and ≥18 to 32 kg/m2 for the bioavailability study. Eligible subjects were aged at least 18 years, with an upper limit of 45, 55, and 65 years for the SAD, MAD, and relative bioavailability studies, respectively. For all studies, eligible subjects had sUA concentration ≥5 mg/dL; were considered healthy according to medical history, physical examination findings, 12-lead electrocardiogram findings, and clinical laboratory evaluations; and had a normal (>80 mL/min) estimated creatinine clearance (CrCl).
Exclusion criteria included acute or chronic medical illness; any drug treatment taken in the 14 days (2 months for enzyme-inducing drugs or products) prior to dosing; use of tobacco products within 30 days prior to dosing; heavy caffeine drinker (more than five cups or glasses of caffeinated beverages per day); history or presence of drug addiction or excessive use of alcohol; major surgery within 3 months; blood donation within 12 weeks or plasma donation within 4 weeks prior to screening visit; or subjects in a situation or with a condition that, in the opinion of the investigator, would interfere with optimal participation in the study.
Study design
The SAD and MAD studies were randomized, double-blind, placebo-controlled, sequential, ascending-dose studies. In the SAD study, each dose group included five subjects, of whom one received placebo and four a single dose of lesinurad oral solution from 5 mg to 600 mg in a fasted (5 mg, 25 mg, 100 mg, 200 mg) or fed (100 mg, 400 mg, 600 mg) state. The oral solution was prepared from amorphous lesinurad free acid. Subjects who were dosed in the fed state consumed a standard low-fat diet (17.9% fat; 619 kcal) 30 minutes prior to dosing. Treatment with lesinurad was initiated with the lowest dose group, with an interval of at least a week between each dose level to allow the review of safety and pharmacokinetic data prior to progression to the next dose level. Each subject participated in one dose assessment only and remained in the clinical research unit (CRU) from day −2 (2 days before dosing) until 72 hours post-dose.
In the MAD study, each dose group included eight subjects, of whom two received placebo and six received lesinurad once daily for 10 days as 100 mg in solution (fed), 200 mg and 400 mg as capsules (fasted), and 200 mg as capsules (fed). The capsules contained lesinurad sodium salt. Groups were sequentially enrolled with an interval of at least 14 days between dose levels to allow the review of safety and pharmacokinetic data prior to progression to the next dose group. For subjects administered 100 mg lesinurad in solution in the fed state, a standard low-fat breakfast was consumed 30 minutes prior to dosing, while subjects given 200 mg lesinurad as capsules consumed a moderate-fat diet (36% fat; 985 kcal). Each subject participated in one study period only and remained in the CRU from day −2 until 48 hours after the last dose on day 10.
The relative bioavailability study was an open-label crossover study with four treatment periods. It included eight subjects, and all subjects participated in each treatment period. Subjects consumed a low-fat breakfast (≥23% fat; ≥643 kcal) 30 minutes before being administered a single 400 mg dose of lesinurad capsule (4×100 mg lesinurad sodium salt) followed successively by a single 200 mg dose (1×200 mg), 400 mg dose (1×400 mg), and a 600 mg dose (1×600 mg) of lesinurad as free acid tablets. No one received placebo. Subjects completed a 5-day washout period before administration of study drug in the next treatment period. Each subject remained in the CRU from day −2 until 48 hours post-dose for each of the four treatments.
For all studies, each subject was administered lesinurad with 240 mL of water following an approximately 9-hour fast. During their confinement, subjects abstained from all food and beverages other than the scheduled meals provided by the CRU and water to quench thirst.
Safety assessments for all three studies included adverse events (AEs) based on the medical review of AE reports and the results of vital sign measurements, 12-lead electrocardiograms, physical examinations, and clinical laboratory tests. AEs were recorded throughout the study period and defined as any new medical occurrence or worsening of a preexisting condition after administration of the study drug or placebo. AEs were categorized according to the Medical Dictionary for Regulatory Activities and were reviewed for potential significance and clinical importance. Serious AEs were defined as AEs that resulted in death, hospitalization or prolongation of existing hospitalization; or persistent or significant disability or congenital anomaly; were life threatening; or were considered an important medical event.
Sample collection
For all studies, venous blood samples were taken at predetermined times for determination of plasma concentrations of lesinurad as well as for determination of sUA and serum creatinine concentrations. Urine samples were collected at predetermined intervals for determination of lesinurad, creatinine, and urate concentrations. Blood and urine samples were collected up to 72 hours post-dose for the SAD study, up to 48 hours after the last dose for the MAD study, and up to 48 hours post-dose for the relative bioavailability study.
Sample analysis
Analysis of lesinurad plasma and urine concentrations for all studies was performed using high-performance liquid chromatography (HPLC) with tandem mass spectrometry. All determinations of lesinurad concentrations in human plasma and urine were generated in analytical runs using appropriate calibration curves and quality control samples that met preestablished acceptance criteria and were conducted in compliance with applicable standard operating procedures in place at the time of analysis. Plasma and urine samples were diluted for analysis. The standard curve ranges for lesinurad in plasma were 1 ng/mL to 1,000 ng/mL in the SAD and MAD studies and 5 ng/mL to 2,000 ng/mL in the relative bioavailability study. The standard curve range for lesinurad in urine was 1 ng/mL to 1,000 ng/mL in all studies. The between-run and within-run coefficients of variation for the analytical quality controls in either matrix were ≤15%.
Serum and urine samples were analyzed for uric acid using an HPLC-UV method. Creatinine was analyzed by established enzymatic methods.
Pharmacokinetic analysis
Pharmacokinetic parameters for lesinurad were calculated using noncompartmental analysis. Plasma pharmacokinetic parameters included maximum observed plasma concentration (Cmax), time to Cmax (Tmax), area under the plasma concentration–time curve (AUC) from zero to infinity (AUC[0–inf]) and from zero to 24 hours (AUC[0–24h]), and half-life (t1/2). Cmax and Tmax were obtained from experimental observations. AUC was calculated using the linear trapezoidal rule. The apparent oral clearance (CL/F) was also determined. Urinary pharmacokinetic parameters included the amount of lesinurad excreted unchanged in urine over a time interval (Ae), the fraction of the lesinurad dose (% dose) excreted unchanged in urine following dosing (fe), and the renal clearance of lesinurad (CLR). Ae was calculated as concentration measured × volume, while CLR was calculated as Ae divided by plasma AUC over the same time interval. Pharmacokinetic parameters were derived using WinNonlin Professional, version 5.2 (Pharsight Corporation, St Louis, MO, USA).
Pharmacodynamic analysis
Pharmacodynamic measurements included maximum observed percent changes from baseline (day −1, time-matched) in uric acid (Emax), the amount of urate recovered in urine (AeUR), renal clearance of urate over an interval of time (CLUR), CrCl, and fractional excretion of urate (FEUA). CLUR was calculated as AeUR divided by plasma urate AUC over the same time interval, while FEUA was calculated as (CLUR/CrCl) ×100. The pharmacodynamic parameters were determined using SAS version 8.2 (SAS Institute Inc., Cary, NC, USA).
Statistical analysis
Pharmacokinetics and pharmacodynamics were summarized using descriptive statistics. Data are presented as geometric mean and 95% confidence intervals (CIs) for pharmacokinetic parameters. For pharmacodynamics, least squares means for each dose level and 95% CIs for the mean differences for each pairwise treatment difference were calculated. Placebo was pooled across all groups in the SAD and MAD studies. No adjustments for multiple comparisons were made. Pharmacokinetic–pharmacodynamic relationships were explored graphically.
Dose proportionality assessment for Cmax and AUC was performed using a power model,15 where:
| (1) |
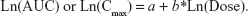
When slope b is close to unity 1 with 95% CI around b within the 0.8 to 1.25 interval, the relationship between dose and the pharmacokinetic parameter was concluded to be dose proportional for the dose range studied.
Results
Subjects
A total of 34 subjects enrolled in and completed the SAD study, while 32 subjects enrolled in and 30 completed the MAD study. There were eight subjects who enrolled in and completed the relative bioavailability study. The demographic and baseline characteristics of subjects in each study are shown in Table S1; the groups in each study had similar demographic and baseline characteristics.
SAD study
The median plasma concentration–time profiles for healthy subjects administered lesinurad oral solution at single doses ranging from 5 mg to 600 mg under fasted or fed conditions is shown in Figure 1A. Lesinurad was rapidly absorbed, with a median Tmax of 0.25 to 1.5 hours (Table 1). Mean estimated terminal t1/2 ranged from 2.7 to 12.7 hours, with the exception of 34.6 hours observed for the 100 mg group with food. This outlier value is likely attributed to individual subjects with anomalously higher concentrations in the terminal phase that led to greater calculated terminal half-lives. Total oral clearance of lesinurad ranged from 92 to 149 mL/min, indicating that lesinurad is a low clearance drug (compared with hepatic blood flow as high as 1.0–1.5 L/min). In urine, unchanged drug ranged from 18.9% to 31.7% (overall, 23.4%) of dose, while renal clearance ranged from 19.8 to 47.7 mL/min (overall, 27.5 mL/min).
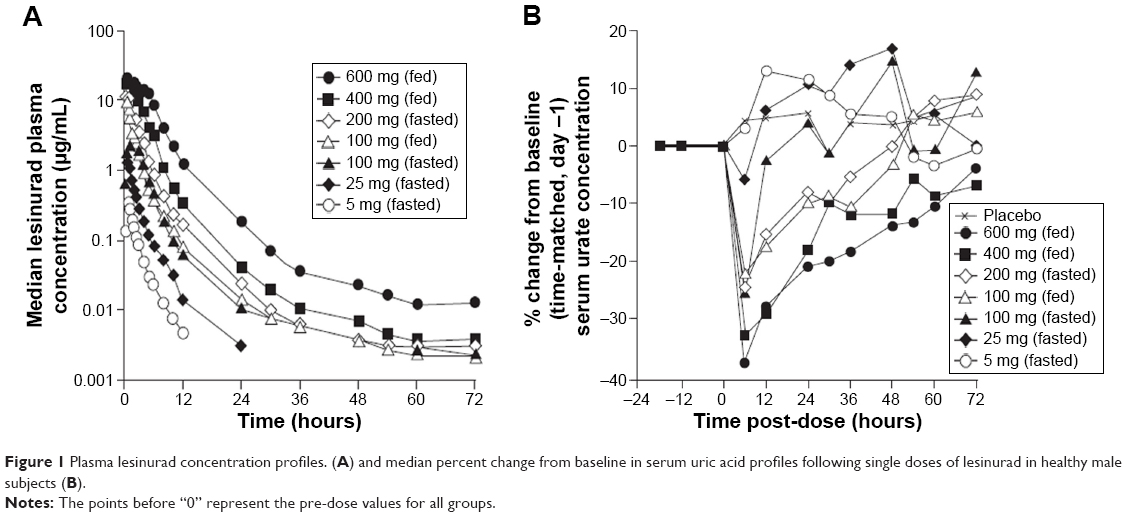 | Figure 1 Plasma lesinurad concentration profiles. (A) and median percent change from baseline in serum uric acid profiles following single doses of lesinurad in healthy male subjects (B). |
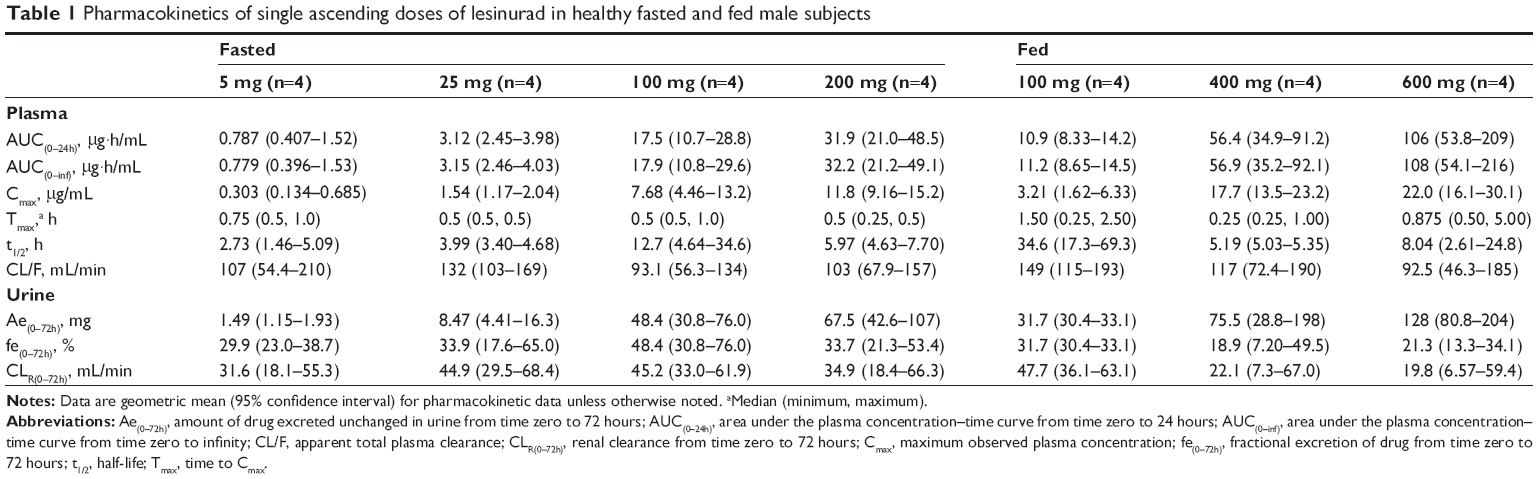 | Table 1 Pharmacokinetics of single ascending doses of lesinurad in healthy fasted and fed male subjects |
sUA concentrations generally showed dose-dependent decreases at lesinurad doses ≥100 mg when assessed over 72 hours post-dose (Figure 1B). At these doses, Emax values, which generally occurred 6 hours post-dose, ranged from −22.3% to −38.6% and were statistically significant when compared with placebo. sUA levels remained lower than baseline for at least 24 hours at doses ≥200 mg. This single dose result suggested that once-daily dosing was appropriate to evaluate in the MAD study.
In general, a dose-dependent increase in the urinary excretion of urate was observed across the 100 mg to 600 mg dose range. Urinary urate levels returned to baseline over 6 to 12 hours after the 100 and 200 mg dose levels and over 12 to 30 hours after the 400 and 600 mg dose levels.
MAD study
When lesinurad was administered once a day for 10 days, total exposure (AUC[0–24h]) increased in a dose-related manner from the 100 mg to the 400 mg dose (Table 2). For all doses, there was little or no difference of systemic exposures between day 1 and day 10 of exposure, suggesting no net changes of absorption and clearance following multiple dosing. The lack of accumulation is consistent with the observation that the majority of lesinurad exposure (>95%) was cleared within 24 hours (Table 1), although terminal half-lives of lesinurad appeared to be variable (2.7–34.6 hours) across dose groups. The fraction of dose excreted into urine as unchanged lesinurad ranged from 34% to 42%, while renal clearance ranged from 28 to 48 mL/min. There was little difference in any urinary pharmacokinetic parameter between day 1 and day 10 of exposure or with dose. A preliminary food effect was explored with lesinurad capsule in this study. The AUC was similar between the 200 mg capsule dose administered in the fed state and in the fasted state. However, Cmax was reduced by half in the fed state, as Tmax occurred 4 hours later than with fasting.
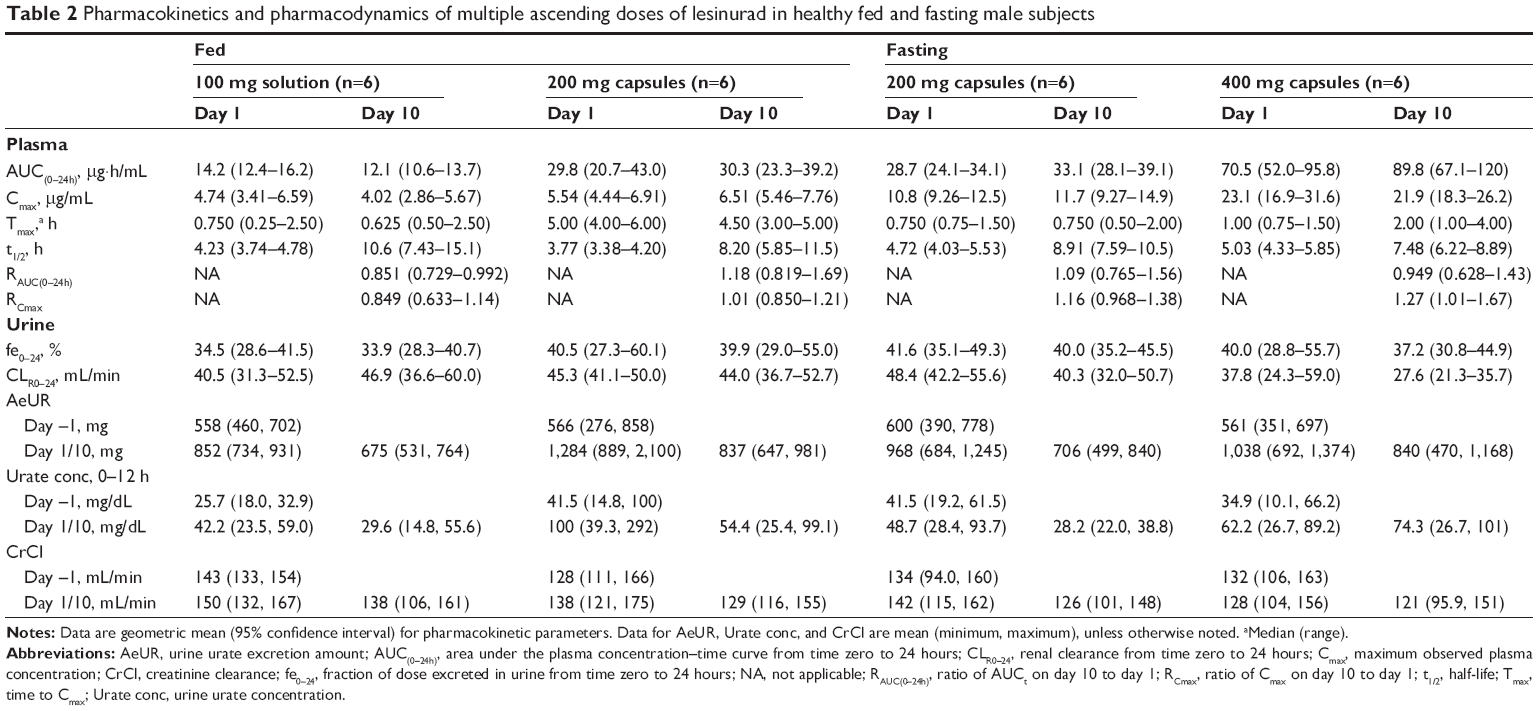 | Table 2 Pharmacokinetics and pharmacodynamics of multiple ascending doses of lesinurad in healthy fed and fasting male subjects |
sUA concentrations decreased significantly over the 100 mg to 400 mg dose range, and the decreases were evident at 12 hours post-dose on day 1 (Figure 2). sUA continued to decline slowly upon repeat dosing, with maximal decreases observed within 6 to 7 days of dosing. Maximum reduction was the least with the 100 mg solution (fed) and greatest with the 200 mg capsule (fed).
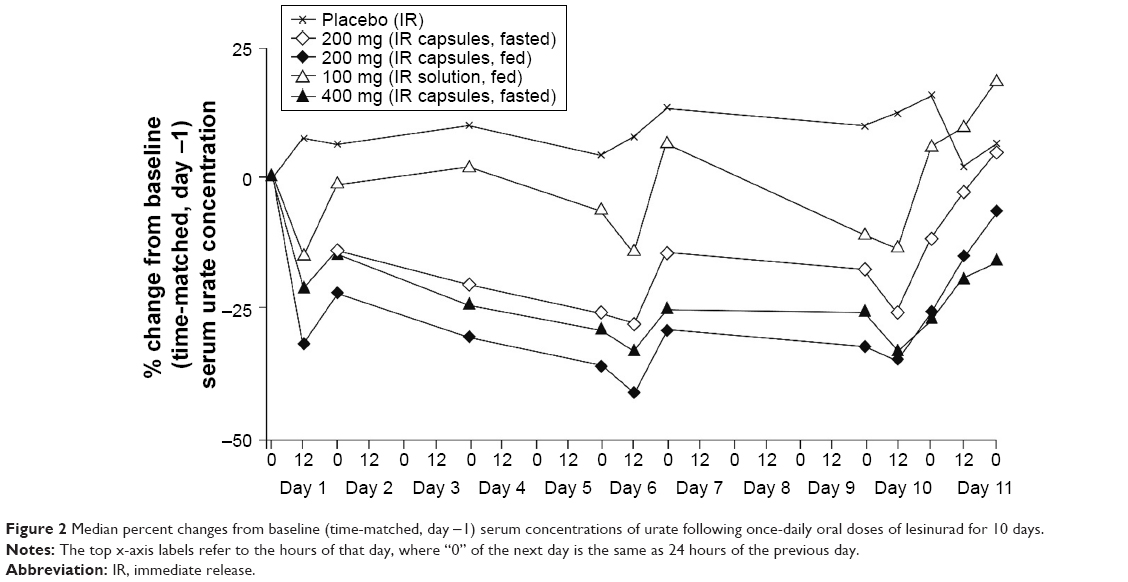 | Figure 2 Median percent changes from baseline (time-matched, day −1) serum concentrations of urate following once-daily oral doses of lesinurad for 10 days. |
The 0- to 24-hour urinary excretion of urate was higher on day 1 and day 10 compared with baseline (day −1) in subjects administered lesinurad (Table 2). However, increases in the amount of urate in urine were generally less following multiple doses on day 10 because urinary urate should parallel the decrease in sUA observed on those days. Urine urate concentrations in the first 12 hours (where the drug effect is mostly exerted) trended similar to the amount excreted. Multiple doses of lesinurad over the 100 to 400 mg dose range had no effect on CrCl.
Relative bioavailability study
The median plasma concentration–time profiles for single oral doses of 400 mg lesinurad capsule and 200, 400, and 600 mg lesinurad tablets are shown in Figure 3. Lesinurad exposure (Cmax and AUC[0–inf]) increased in a dose-related manner from the 200 mg to the 600 mg tablets (Table S2). Systemic exposure of lesinurad was nearly identical following oral administration of the capsules and tablets at the 400 mg dose, with a geometric mean ratio (90% CI) for Cmax of 101 (89.2; 115) and 97.8 (89.6; 107) for AUC(0–inf). The median Tmax was generally similar between the capsule and the tablet. The amount of lesinurad excreted in urine increased in a dose-related manner and was the same with the 400 mg capsule and the 400 mg tablet. Fractional excretion and renal clearance of lesinurad were similar across all doses and both formulations.
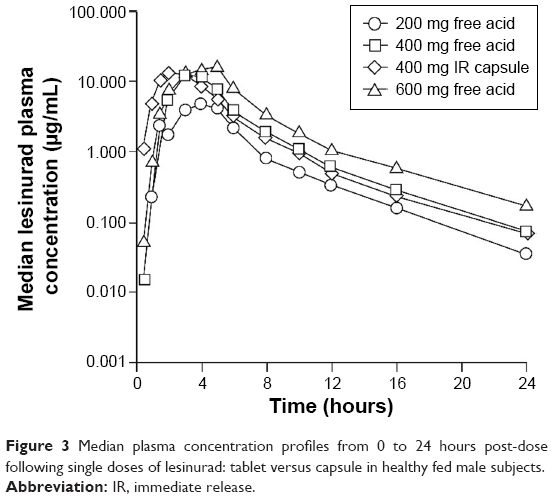 | Figure 3 Median plasma concentration profiles from 0 to 24 hours post-dose following single doses of lesinurad: tablet versus capsule in healthy fed male subjects. |
Dose proportionality
The dose proportionality of lesinurad exposure (Cmax and AUC) following a single oral dose on day 1 was assessed by pooling all available data from the three studies. As shown in Figure 4A, both Cmax and AUC exposures showed a dose-proportional increase at doses of 5 to 400 mg under fasted conditions with power model slope b wholly within the 0.8 to 1.25 interval. Under fed conditions, the increase in lesinurad Cmax and AUC was considered dose proportional at doses of 100 to 600 mg despite the slope b (1.13, 95% CI: 1.00–1.26) for AUC being marginally outside the 0.8 to 1.25 interval, likely due to small group size and variability between studies, subjects, and formulation. Dose proportionality was not explored in multiple doses due to the narrow dose range evaluated in the fed condition.
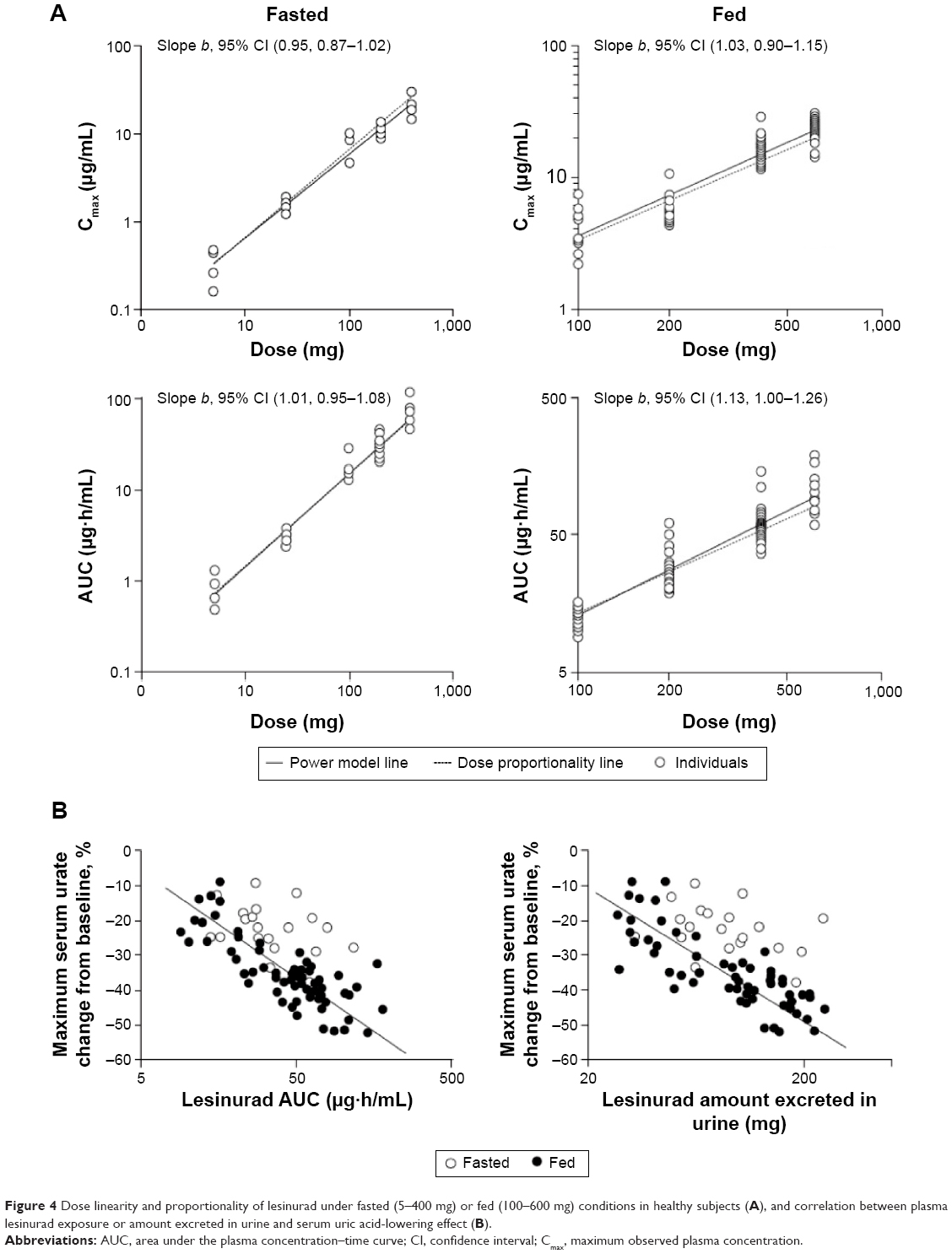 | Figure 4 Dose linearity and proportionality of lesinurad under fasted (5–400 mg) or fed (100–600 mg) conditions in healthy subjects (A), and correlation between plasma lesinurad exposure or amount excreted in urine and serum uric acid-lowering effect (B). |
Pharmacokinetic and pharmacodynamic relationship of lesinurad
The pharmacokinetic and pharmacodynamic relationship of lesinurad following a single oral dose on day 1 was evaluated by pooling all available data from the three studies. Figure 4B shows that the maximum sUA-lowering effect is well correlated with lesinurad plasma AUC exposure where data from the fed groups clearly delineate the relationship. The data also suggest a somewhat greater reduction in sUA may be associated with food despite the same drug exposure. As shown in Figure 4B, a good correlation between sUA lowering and lesinurad amount excreted in the urine was also established, consistent with lesinurad action on the URAT1 transporter in the kidney.
Safety and tolerability
Lesinurad was well tolerated in both the SAD and MAD studies even at the highest dose administered (600 mg). The incidence of AEs in SAD was low and generally similar over the entire dose range and between placebo and active groups. All AEs were mild in severity and resolved without treatment. One subject in the placebo group in the SAD study experienced ventricular tachyarrhythmia. No subjects discontinued because of AEs.
The overall incidence of AEs in MAD was low, with the highest number of AEs reported with the 200 mg lesinurad (fed) dose. The number of events in this group was higher (n=16) than the other active dose groups, but was similar to placebo (n=19). The number of subjects reporting at least one AE was similar among the dose groups and the placebo group. The majority of AEs reported were mild and resolved without treatment, with the most frequently reported treatment-related AE being diarrhea (Table S3). Diarrhea was also reported by subjects receiving placebo. No severe or serious treatment-emergent AEs were reported. One subject in the placebo group was withdrawn due to renal colic. The number of AEs possibly related to study drug was higher in the fed state (n=13) compared with the fasted (n=6) for the 200 mg treatment, although the number of subjects with at least one AE was similar (n=5 versus 4). For all treatments, AEs were reported throughout the dosing period with no evidence of a greater number at the start or end.
Single oral doses of lesinurad were well tolerated in the bioavailability study as well, with AEs only reported with the 400 mg tablet in the fasted state. Most AEs were mild in severity and resolved without treatment; no severe or serious AEs were reported.
There were no significant findings on laboratory examination in the three studies, other than the decrease in sUA. The results for some parameters, including serum creatinine, were outside the appropriate reference ranges, but the findings were generally transient and occurred at isolated time points only. No findings were considered to be of clinical importance at any dose level. Change from baseline in QTc >30 ms was observed in one subject who received lesinurad 5 mg in SAD; no changes were observed in the bioavailability study. Increases in QTc >30 ms were infrequent in MAD, occurring in subjects who received placebo (two subjects), lesinurad 200 mg (three fed subjects, three fasted) and lesinurad 400 mg (two subjects).
Discussion
The results of these studies in healthy subjects are consistent with other published data showing that inhibition by lesinurad of URAT1 in the proximal tubule of the kidney increases urinary urate excretion and lowers sUA concentrations.11
Lesinurad exposure increased in a dose-proportional manner over the range of doses examined in both the fed and fasting states following a single oral dose. The exploratory food effect evaluation in a limited number of subjects indicated that food had no effect on the extent of absorption but reduced the rate of absorption of lesinurad. Multiple once-daily doses resulted in no accumulation in plasma.
Serum urate concentrations significantly decreased following the multiple once-daily dosing of lesinurad over the dose range of 100 mg to 400 mg. The long duration of the effect of lesinurad on serum urate, more than 24 hours after a single dose and up to more than 48 hours after the last dose on day 10, supports the viability of a once-daily dosing regimen. The steady-state sUA-lowering effect was achieved at approximately 6 to 7 days.
Lesinurad appeared to be safe and well tolerated by healthy male subjects when given either as single or multiple daily doses in either the fed or the fasting state. The incidence of AEs was low after single doses and did not appear to be dose related. Single daily oral doses of lesinurad over 10 days had no apparent effect on the safety and laboratory parameters measured. Safety, tolerability, pharmacokinetics, and pharmacodynamics were similar following administration of the sodium salt capsules and free acid tablet, supporting the use of either the sodium salt or free acid formulations in the Phase II and III clinical trials.
In summary, these studies indicate that the selective uric acid reabsorption inhibitor lesinurad induces sustained, dose-dependent decreases in sUA concentration, with concomitant increases in urinary urate concentrations in healthy subjects. Phase III studies utilizing the tablet formulation will evaluate whether changes in sUA will lead to clinically meaningful changes in patients with this metabolic disorder.
Acknowledgments
Funding was provided by Ardea Biosciences/AstraZeneca. Editorial support was provided by Tom Claus, PhD, of PAREXEL and funded by AstraZeneca. Ardea Biosciences, Inc. is a member of the AstraZeneca group.
Disclosure
Z Shen, C Rowlings, K Manhard, and C Storgard are employees of Ardea Biosciences, Inc. B Quart and LT Yeh are former employees of Ardea Biosciences, Inc. B Kerr and V Hingorani are paid consultants of Ardea Biosciences, Inc. The authors report no other conflicts of interest in this work.
References
Doghramji PP, Wortmann RL. Hyperuricemia and gout: new concepts in diagnosis and management. Postgrad Med. 2012;124(6):98–109. 2 de Oliveira EP, Burini RC. High plasma uric acid concentration: causes and consequences. Diabetol Metab Syndr. 2012;4:12. | ||
Gout: Novel Biologics Rev Up A Slow Market. Burlington: Decision Resources; 2010. Available from: http://decisionresources.com/Products-and-Services/Report?r=dbaspd1810. Accessed March 10, 2015. | ||
Kuo CF, Grainge MJ, Mallen C, Zhang W, Doherty M. Rising burden of gout in the UK but continuing suboptimal management: a nationwide population study. Ann Rheum Dis. 2015;74(4):661–667. | ||
Zhang W, Doherty M, Pascual E, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part I: Diagnosis. Report of a task force of the Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65(10):1301–1311. | ||
Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65(10):1312–1324. | ||
Bieber JD, Terkeltaub RA. Gout: on the brink of novel therapeutic options for an ancient disease. Arthritis Rheum. 2004;50(8):2400–2414. | ||
Boss GR, Seegmiller JE. Hyperuricemia and gout. Classification, complications and management. N Engl J Med. 1979;300(26):1459–1468. | ||
Becker MA, Jolly M. Hyperuricemia and associated diseases. Rheum Dis Clin North Am. 2006;32(2):275–293, v–vi. | ||
Girardet J-L, Miner JN. Urate crystal deposition disease and gout – new therapies for an old problem. Annual Reports in Medicinal Chemistry. Academic Press; 2014;49:151–164. | ||
Fleischmann R, Kerr B, Yeh LT, et al; RDEA594-111 Study Group. Pharmacodynamic, pharmacokinetic and tolerability evaluation of concomitant administration of lesinurad and febuxostat in gout patients with hyperuricaemia. Rheumatology (Oxford). 2014;53(12):2167–2174. | ||
Perez-Ruiz F, Hingorani V, Welp J, et al. Efficacy and safety of a range of doses of rdea594, a novel uricosuric agent, as a single agent in hyperuricemic gout patients: multicenter, randomized, double-blind, placebo-controlled, phase 2 experience. Ann Rheum Dis. 2010;69(Suppl 3):121. | ||
Perez-Ruiz F, Sundy J, Krishnan E, et al. Efficacy and safety of lesinurad (RDEA594), a novel URAT1 inhibitor, in combination with allopurinol in allopurinol-refractory gout patients: results from a randomized, blinded, placebo-controlled, phase 2b extension study. Ann Rheum Dis. 2013;71(Suppl 3):439. | ||
Yeh LT, Shen Z, Kerr B. RDEA594: a potent URAT1 inhibitor without affecting other renal transporters OAT 1 and OAT 3. Ann Rheum Dis. 2009;68(Suppl 3):320. | ||
Peng B, Hayes M, Resta D, et al. Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J Clin Oncol. 2004;22(5):935–942. |
Supplementary materials
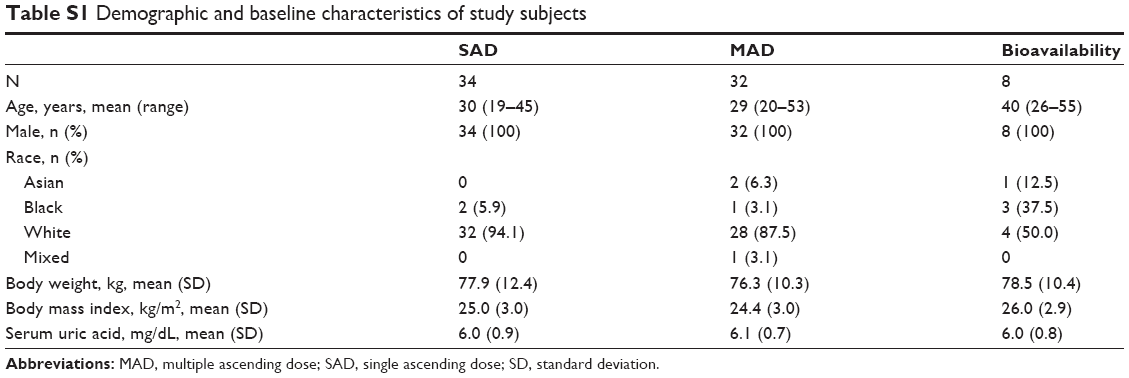 | Table S1 Demographic and baseline characteristics of study subjects |
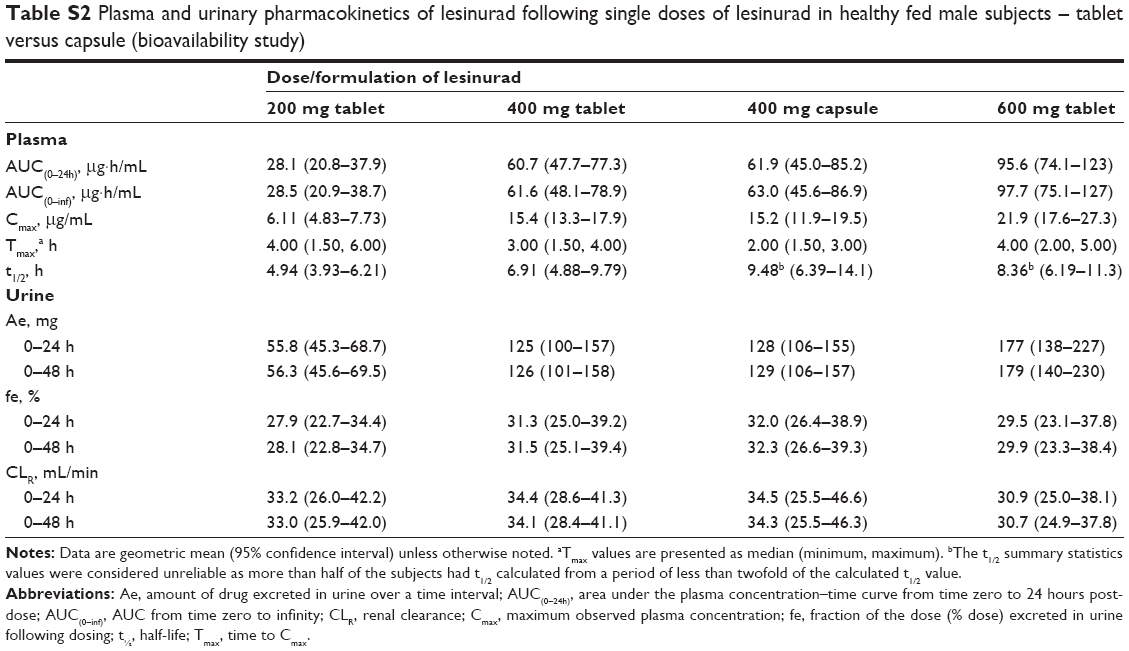 | Table S2 Plasma and urinary pharmacokinetics of lesinurad following single doses of lesinurad in healthy fed male subjects – tablet versus capsule (bioavailability study) |
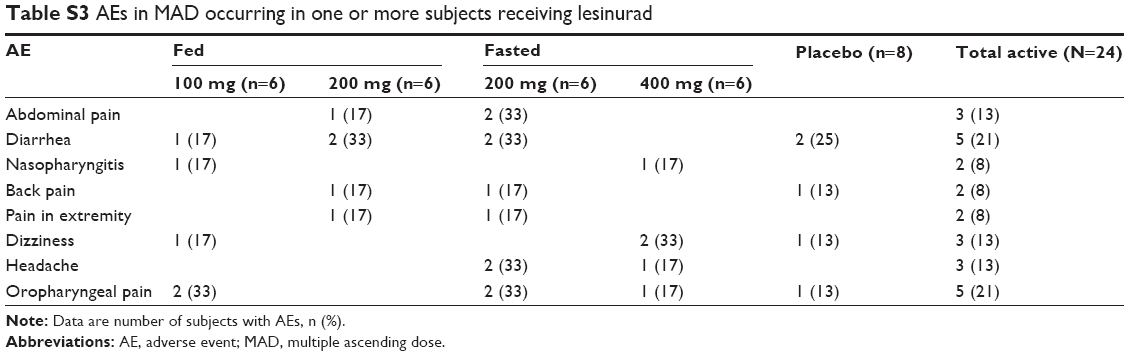 | Table S3 AEs in MAD occurring in one or more subjects receiving lesinurad |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.