Back to Journals » Drug Design, Development and Therapy » Volume 16
Pharmacokinetics of a Fixed-Dose Combination of Teneligliptin Hydrochloride Hydrate and Modified-Release Metformin Under Fasting and Fed Conditions in Healthy Subjects
Authors Goak IS ![]() , Lee JA
, Lee JA ![]() , Jeong MH, Moon SJ
, Jeong MH, Moon SJ ![]() , Kim MG
, Kim MG ![]()
Received 27 October 2022
Accepted for publication 15 December 2022
Published 28 December 2022 Volume 2022:16 Pages 4439—4448
DOI https://doi.org/10.2147/DDDT.S393675
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
In Sun Goak,1,2 Jin A Lee,1,3 Min Ho Jeong,4 Seol Ju Moon,1,3 Min Gul Kim1,3,5
1Department of Pharmacology, School of Medicine, Jeonbuk National University, Jeonju, Republic of Korea; 2Division of Endocrinology and Metabolism, Department of Internal Medicine, Jeonbuk National University Hospital, Jeonju, Republic of Korea; 3Center for Clinical Pharmacology and Biomedical Research Institute, Jeonbuk National University Hospital, Jeonju, Republic of Korea; 4Division of Clinical Team, Kingdom Pharmaceutical Co., Ltd, Seoul, Republic of Korea; 5Research Institute of Clinical Medicine of Jeonbuk National University, Jeonju, Republic of Korea
Correspondence: Min Gul Kim, Department of Pharmacology, School of Medicine, Jeonbuk National University, Center for Clinical Pharmacology and Biomedical Research Institute, Jeonbuk National University Hospital, Geonji road #20, Deokjin-gu, Jeonju, 54907, Republic of Korea, Tel +82-63-250-3458, Fax +82-63-259-3483, Email [email protected]
Purpose: This study was performed to compare the pharmacokinetics of two fixed-dose combination (FDC) formulations of teneligliptin combined with modified-release metformin in healthy Korean subjects under fasting and fed conditions.
Patients and Methods: The study was a single-center, open-label, single-dose, 2-way, 2-period, crossover trial. A total of 72 eligible subjects (40 subjects in the fasting state study and 32 subjects in the fed study) were enrolled in the study and were randomized to treatment. After the administration of a single FDC tablet of the investigational products, blood samples were collected at specific time intervals from 0 to 96 hours. The plasma concentrations of teneligliptin and metformin were measured by ultra performance liquid chromatography-tandem mass spectrometry (UPLC‒MS/MS). Pharmacokinetic parameters were calculated, and 90% confidence intervals (CIs) of the geometric mean ratios (test/reference) of the parameters were obtained through analysis of variance of the logarithmically transformed data.
Results: The corresponding 90% CIs of area under the plasma concentration-time curve from time zero to the time of last measurable concentration (AUCt) and maximum plasma drug concentration (Cmax) for the test/reference geometric mean ratio (GMR) of teneligliptin were 94.81– 101.32% and 86.03– 97.63%, respectively, under fasting conditions. The corresponding 90% CIs of AUCt and Cmax for the test/reference GMR of metformin were 95.01– 108.36% and 94.69– 108.40%, respectively, under the fasting state and 98.82– 107.56% and 97.25– 106.99%, respectively, after feeding. All adverse events were of mild intensity, and the subjects recovered spontaneously without sequelae.
Conclusion: The test FDC drug is equivalent to the reference FDC drug in subjects under fasting and fed conditions within the Korean regulatory bioequivalence criteria. Both formulations were safe and well tolerated, and there were no differences in the safety profiles between the two single FDC formulation drugs.
Trial Registration No: Clinicaltrials.gov. KCT0007757, KCT0007759.
Keywords: type 2 diabetes mellitus, dipeptidyl peptidase-4 inhibitors, modified release metformin, fixed dose combination
Introduction
Type 2 diabetes mellitus (T2DM) causes a variety of acute and chronic complications, such as diabetic ketoacidosis, hyperosmolar hyperglycemic coma, unexpected or uncontrolled infection, diabetic polyneuropathy, retinopathy and nephropathy.1 Poorly controlled diabetic patients do not simply present a problem with hyperglycemia but, rather, one involving insulin resistance or a relative lack of insulin, which leads to severe systemic complications.1 According to several diabetes guidelines, it has been reported that dual combination therapy is more effective than monotherapy for reaching the therapeutic goal when beginning treatment in patients with high glycated hemoglobin at the time of diagnosis.2 The major mechanisms of controlling glucose levels are different in metformin and dipeptidyl peptidase-4 (DPP-4) inhibitors. Metformin primarily lowers glucose by improving hepatic insulin resistance and reducing hepatic gluconeogenesis. Incretin hormones are released from enteroendocrine cells in a rapid response to a meal and are rapidly inactivated by the enzyme DPP-4. DPP-4 inhibitors improve hyperglycemia in glucose-dependent action by increasing active incretin levels, increasing serum insulin levels, and decreasing serum glucagon levels.3–5 Therefore, incretin-related agents such as DPP-4 inhibitors have the advantage of reducing glycemic fluctuations and minimizing the risk of hypoglycemia and weight gain.5 Moreover, several clinical trials have proven the additive or synergistic action of these two drugs when used in combination therapy.6–9
Fixed-dose combinations (FDCs) can simplify treatment compared with 2-pill administration and can potentially improve drug adherence and reduce medication errors, especially in diabetic patients, because such patients often take multiple medications after meals. The first FDC for the treatment of diabetes, glucovance, a combination of sulfonylurea and biguanide, was approved by the US Food and Drug Administration (FDA) in the early 2000s. Since then, several combinations have been created because of the advantages of FDCs. Metformin was previously the first-line treatment in patients with T2DM. Therefore, metformin is one of the most common FDC drugs. However, the most common side effects of metformin, which are also the most important factors in lowering drug adherence, include diarrhea, nausea, and vomiting. Modified-release metformin (extended release—XR formulation, or sustained release—SR formulation) may be better tolerated than comparable doses of the immediate-release metformin.10 Based on this understanding, a novel FDC formulation of teneligliptin hydrochloride hydrate 20 mg and metformin hydrochloride (HCl) XR 1000 mg will not only increase the overall efficacy of the treatment of T2DM but also increase compliance and reduce medication errors in patients who are prescribed long-term polypharmacy.
The objective of this study was to evaluate the pharmacokinetics of the drug teneligliptin combined with modified-release metformin and administered as the teneligliptin hydrochloride hydrate 20 mg plus metformin HCl XR 1000 mg FDC formulation or combined with other FDC drugs consisting of the same corresponding dose. In addition, this study aimed to evaluate the effect of food on the pharmacokinetics of modified-release metformin.
Materials and Methods
Study Population
Healthy adult volunteers aged over 19 years with a body mass index ranging from 17.5 to 30.5 kg/m2 were enrolled in the study at the Jeonbuk National University Hospital Clinical Trial Center (Jeonju, Korea). The subjects’ health was confirmed by medical history, physical examination, measurement of vital signs, 12-lead electrocardiography (ECG) and clinical laboratory tests.
Specific exclusion criteria included type 1 diabetes mellitus, lactic acidosis, acute or chronic metabolic acidosis including diabetic ketoacidosis with or without coma, and a history of hypersensitivity to biguanides. Additionally, subjects who had moderate (stage 3b) and severe renal impairment (eGFR <45 mL/min/1.73 m2), congestive heart failure requiring drug treatment, and involvement in strenuous exercise were excluded.
The volunteers were informed about the details, including the risks and benefits of the study, and they provided their written informed consent before participating in the study. They were free to withdraw from the study at any time.
Study Design
The study was a single-center, randomized, open-label, single-dose, 2-way, 2-period, crossover trial. Each patient was randomly assigned to 1 of 2 treatment groups in a 1:1 ratio (Figure 1). This clinical trial consisted of 2 independent studies. A total of 72 subjects were hospitalized at the clinical trial center, and each patient was assigned to one of two parts: 40 subjects participated in a fasting study (Study 1), and the other 32 subjects participated in a fed study (Study 2). There were 14 days of washout between the 2 study periods to allow for sufficient excretion time, the washout period being more than 5 times the half-life (t1/2) of teneligliptin and metformin. In both studies, eligible subjects were admitted to Jeonbuk National University Hospital a day before administration of the investigational product. Because the fasted state was defined as having no food or liquid except water for at least 10 hours, the subjects were provided with the dinner and made to fast until administration. Water was also restricted 1 hour before and 1 hour after administration of the investigational product. Lunch was provided after pharmacokinetic blood sampling approximately 4 hours after administration. Grapefruit or grapefruit-containing food was restricted from 7 days before the first administration of the investigational drug to the final pharmacokinetic blood sample collection. During the study period, drinking alcohol, smoking, and imbibing caffeine-containing products were not allowed.
 |
Figure 1 Schematic diagram of the study design and dosing schedules. |
In the fasting study (Study 1), 40 subjects were randomized to 1 of 2 treatment sequences, in which the treatments consisted of a single FDC of test drug (KD-4002) comprising teneligliptin hydrochloride hydrate 20 mg combined with metformin HCl XR 1000 mg (Kyung Dong Pharmaceutical Co., Ltd, Republic of Korea) or a single FDC of reference drug comprising teneligliptin hydrobromide hydrate 20 mg combined with metformin HCl SR 1000 mg (Tenelia M SR Table 20/1000 mg tablet; Han Dok Pharmaceutical Corp, Republic of Korea). Subjects received the test or reference drug with 150 mL of water after a 10-hour overnight fast and then fasted for 4 hours postdose.
In the fed study (Study 2), 32 subjects were served a high-fat meal containing approximately 900 kcal (29.5% carbohydrate, 17.5% protein, and 53.0% fat) of breakfast following a 10-hour overnight fast. The subjects were administered the same test drug or reference drug with 150 mL of water and fasted for 4 hours post-dose.
In consideration of hypoglycemia, dizziness, and expected adverse reactions during administration of the clinical trial drug, blood was collected in bed for up to 4 hours after administration. If blood sugar was less than 70 mg/dL when measured 2 hours and 4 hours after administration of the clinical trial drug, an immediate break was given for the patient to take sugar (sucrose) at once regardless of symptoms. After administration of the clinical trial drug, if lactic acidosis was suspected by the investigator, arterial blood gas analysis was performed, and the patient was transferred to the emergency room.
Pharmacokinetic blood samples were collected by direct venipuncture or through an indwelling peripheral venous heparin lock catheter into ethylenediaminetetraacetic acid (EDTA) tubes and were centrifuged at 1800 g within 60 minutes of collection at 4 °C for 10 minutes. Then, separated plasma was aliquoted into polypropylene tubes and stored at –80 °C ~ −60 °C until further analysis. Blood samples for determination of teneligliptin concentration were obtained at 0 hours (predose) and 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, 12, 24, 48, 72 and 96 hours after drug administration. For determination of the metformin concentration, blood samples were obtained 0 hours (predose) and 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 10, 12 and 24 hours after drug administration.
The study protocol was approved by the Institutional Review Board of Jeonbuk National University Hospital, and the study was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice. In the fasting study (Study 1), subjects’ first enrollment date was October 15, 2020, and the last observation date was November 24, 2020. In the fed study (Study 2), subjects’ first enrollment date was September 24, 2020, and the last observation date was October 29, 2020.
Plasma Drug Concentration Analysis
The plasma concentrations of teneligliptin and metformin were determined using a validated methodology that included ultra performance liquid chromatography (UPLC) with tandem mass spectrometry. Each analytical run included appropriate standards and quality-control samples.
The lower limit of quantitation (LLOQ) for teneligliptin was 1.00 ng/mL. Chromatographic separation was performed using an ACQUITY UPLC BEH C18 column (2.1 × 50 mm, 1.7 µm, Waters) at a flow rate of 0.3 mL/min and column temperature of 30 ± 5 °C, and the injection volume was 10 µL. The mobile phase consisted of a mixture of injector wash solution strong [acetonitrile: distilled water: formate (70:30:0.1 v/v)] and injector wash solution weak [0.1% (v/v) formate in acetonitrile:0.1% (w/v) ammonium formate and 0.1% (v/v) formate in distilled water (25:75 v/v)]. Ionization in the positive ion electrospray was used for detection and quantitation. Ion pairs from m/z 427.22 → 243.17 for teneligliptin and from m/z 435.29 → 251.24 for the internal standard (IS) were selected for quantitation. Teneligliptin-d8 was used as the IS for analytes, and drug-to-IS ratios were used to create a linear calibration curve using 1/χ2-weighted linear regression analysis. The validated quantification range was 1.00–1000 ng/mL for teneligliptin. Intra- and interassay accuracy and precision for the analyses were within 15% of the theoretical values, and stability was confirmed according to standard operating procedures and Ministry of Food and Drug Safety guidelines for bioanalytical methods. A calibration curve covering the range of 1.00–1000 ng/mL was constructed, which was linear over the concentration range (correlation coefficient r2 ≥ 0.9971).
The lower limit of quantitation (LLOQ) for metformin was 20.0 ng/mL. Chromatographic separation was performed using an ACQUITY UPLC BEH HILIC C18 column (2.1 × 50 mm, 1.7 µm, Waters) at a flow rate of 0.4 mL/min and column temperature of 30 ± 5 °C, and the injection volume was 5 µL. The mobile phase consisted of a mixture of injector wash solution strong [acetonitrile: distilled water (70:30 v/v)] and injector wash solution weak [acetonitrile:0.1% (w/v) ammonium formate in distilled water (90:10 v/v)]. Ionization in the positive ion electrospray was used for detection and quantification. Ion pairs from m/z 130.18 → 60.24 for metformin and from m/z 136.25 → 60.24 for the internal standard (IS) were selected for quantitation. Metformin-d6 was used as the IS for analytes, and drug-to-IS ratios were used to create a linear calibration curve using 1/χ2-weighted linear regression analysis. The validated quantification range was 20.0–5000 ng/mL for metformin. Intra- and interassay accuracy and precision for the analyses were within 15% of the theoretical values, and stability was confirmed according to standard operating procedures and Ministry of Food and Drug Safety guidelines for bioanalytical methods. A calibration curve covering the range of 5–1000 ng/mL was constructed, and it was linear over the concentration range (correlation coefficient r2 ≥ 0.9950).
Pharmacokinetic Analysis
The pharmacokinetic analysis included all subjects who had completed pharmacokinetic blood sampling according to the protocol. The pharmacokinetic parameters were assessed using a noncompartmental method provided by MassLynx software (version 4.1, Waters Inc. USA). The maximum plasma drug concentration (Cmax) and time to Cmax (Tmax) were directly obtained from the plasma concentration-time profiles. The terminal elimination half-life (t1/2) was calculated as ln 2/λz, where λz reflects the slope of the apparent elimination phase of the natural logarithmic (ln) transformation of the plasma drug concentration-time profiles. The area under the plasma concentration-time curve from time zero to the time of last measurable concentration (AUCt) was calculated according to the linear trapezoidal method. The AUC from time zero to infinity (AUCinf) was estimated as AUCt + Ct/λz, where Ct is the plasma concentration of the last measurable sample. Apparent total plasma clearance (CL/F) was calculated as Dose/AUCinf.
Statistical Analysis
Pharmacokinetic equivalence was assessed for the principal parameters of systemic exposure (AUCt and Cmax). The log-transformed AUCt and Cmax were analyzed in SAS version 9.4 (SAS Institute, Inc., Cary, NC, USA) using a mixed-effects analysis of variance (ANOVA) with sequence, period, and treatment as fixed effects and subject within sequence as a random effect. The results were reported in 90% confidence intervals (CIs) surrounding the ratio of the geometric least-square mean of the pharmacokinetic parameters after retransformation. The products were considered bioequivalent when the 90% CIs for these parameters were within the range of 0.8–1.25.
Sample Size
From pharmacokinetic studies of teneligliptin and modified-release metformin in fasting state, the intra-subject coefficients of variation (CV) of AUCt and Cmax were estimated the following values; teneligliptin AUCt 12.1%, teneligliptin Cmax 18.0%, metformin AUCt 27.5% and metformin Cmax 25.2%.11 Assuming that the CV within the subject was 0.27 and the equivalence range is 0.8 to 1.25, a total of 34 subjects were required to have a test power of about 80% at the significance level of 0.05 in Study 1. Considering the dropout rate, the number of subjects was set to 40. Following pharmacokinetic study of modified-release metformin in fed state, the intra-subject CV of AUCt and Cmax were estimated the 14.6% and 16.1%, respectively.12 Assuming that the coefficient of variation within the subject is 0.16 and the equivalence range is 0.8 to 1.25, a total of 26 subjects were required to have a test power of about 80% at the significance level of 0.05 in Study 2. The number of subjects was set similar to Study 1.
Safety Analysis
The safety analysis included all 40 subjects who received at least 1 dose of any of the investigational drugs. Safety measurements included physical examination, clinical laboratory test results (including hematology, serum chemistry, and urinalysis), vital signs, 12-lead ECGs, and assessment of adverse events (AEs). Descriptive statistics were used to summarize any clinically significant findings from the clinical laboratory test results, vital signs, and ECGs in each treatment arm.
Results
Subjects
From 92 volunteers screened for enrollment in the study, 72 healthy volunteers were enrolled and randomized to treatment. Baseline demography and characteristics for the study populations are listed in Table 1. In Study 1, a total of 40 healthy Korean subjects were enrolled, and 38 subjects completed the study according to the protocol (pharmacokinetic population); two subjects withdrew consent. The mean ± standard deviation (SD) values of the subjects’ age, height, weight, and BMI were 27.68 ± 7.43 years, 170.33 ± 7.38 cm, 69.53 ± 11.49 kg, and 23.79 ± 2.72 kg/m2, respectively. In Study 2, a total of 32 healthy Korean subjects were enrolled, and 27 subjects completed the study according to the protocol (pharmacokinetic population); four subjects withdrew consent and one subject dropped out due to not completing a high-fat meal within 20 minutes prior to drug administration. The mean ± SD values of the subject’s age, height, weight, and BMI were 24.19 ± 2.58 years, 172.91 ± 5.46 cm, 73.51 ± 10.38 kg, and 24.46 ± 2.68 kg/m2, respectively.
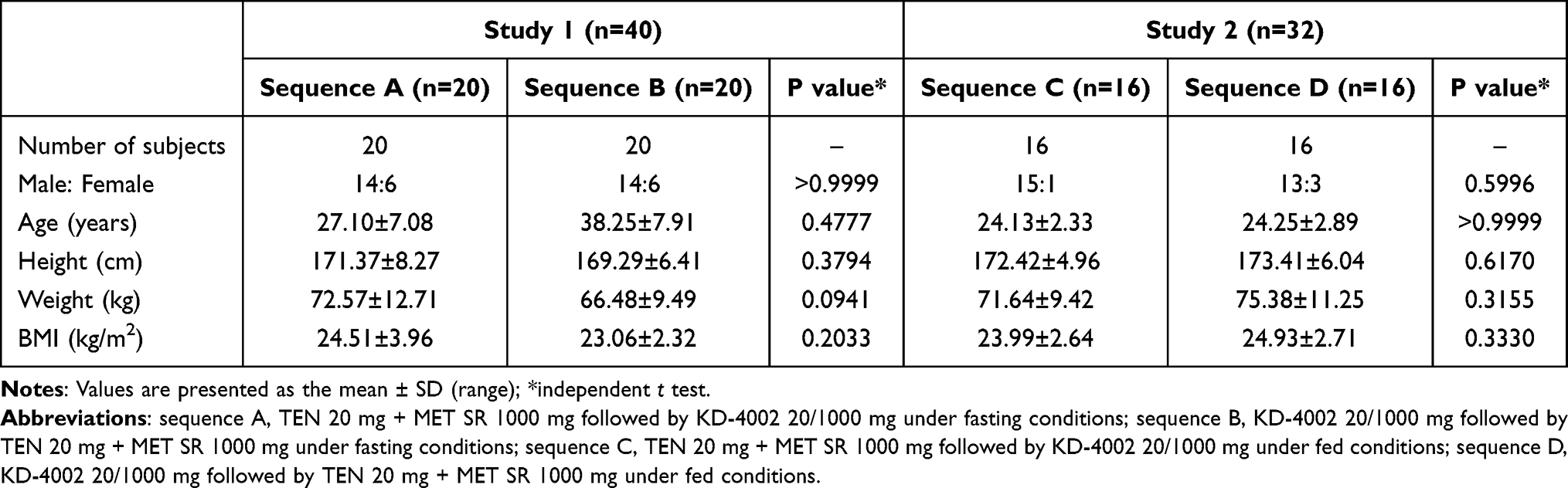 |
Table 1 Baseline Characteristics of the Study Population of Study 1 (Fasting) and Study 2 (Fed) (Total n = 72) |
Pharmacokinetics of Teneligliptin
The mean plasma concentration-time curve and arrhythmic mean of pharmacokinetic parameters of teneligliptin, including AUCt, AUCinf, Cmax, Tmax, t1/2, Vd/F and CL/F, following oral administration under fasting conditions are shown in Figure 2 and Table 2, respectively. The point estimate and 90% CIs for the geometric mean ratios (test/reference) of log-transformed AUCt and Cmax of teneligliptin for the fasting study were assessed by ANOVA. The corresponding 90% CIs for the geometric mean ratio of the AUCt and Cmax were 94.81–101.32% and 86.03–97.63%, respectively, in the fasting state study (Table 3). These results were within the acceptance range of 80–125%, indicating that the test product was equivalent to the reference product in subjects under both the fasting and fed conditions.
 |
Table 2 Pharmacokinetic Parameters of Teneligliptin Under Fasting Conditions (Study 1) |
 |
Table 3 Primary Pharmacokinetic Parameters of Teneligliptin Under Fasting Conditions (Study 1) |
 |
Figure 2 Mean plasma concentration-time curve. (A) Teneligliptin after a single FDC administration of the test or reference drug under fasting conditions (Study 1). (B) Modified-release metformin after a single FDC administration of the test or reference drug under fasting conditions (Study 1). (C) Modified-release metformin after a single FDC administration of the test or reference drug under fed conditions (Study 2). |
Pharmacokinetics of Modified-Release Metformin
The mean plasma concentration-time curve and geometric mean of pharmacokinetic parameters of modified-release metformin, including AUCt, AUCinf, Cmax, Tmax, t1/2, Vd/F and CL/F, following oral administration under fasting and fed conditions are shown in Figure 2 and Table 4, respectively. The point estimate and 90% CIs for the geometric mean ratios (test/reference) of log-transformed AUCt and Cmax of modified-release metformin for the fasting and fed state studies were assessed by ANOVA. The corresponding 90% CIs for the geometric mean ratio of the AUCt and Cmax were 95.01–108.36% and 94.69–108.40% in the fasting state study, AUCt and Cmax were 98.82–107.56% and 97.25–106.99% in the fed state study (Table 5). These results were within the acceptance range of 80–125%, indicating that the test product was equivalent to the reference product in subjects under both the fasting and fed conditions.
 |
Table 4 Pharmacokinetic Parameters of Metformin Under Fasting and Fed Conditions (Studies 1 and 2) |
 |
Table 5 Primary Pharmacokinetic Parameters of Metformin Under Fasting and Fed Conditions (Studies 1 and 2) |
Safety and Tolerability of Teneligliptin and Modified-Release Metformin
In Study 1, a total of 17 AEs were reported by 14 subjects who were administered the investigational products at least once. There were 2 cases of dizziness, 3 cases of headache, 2 cases of increased creatinine, 1 case of abdominal pain, 5 cases of pyuria, 3 cases of increased blood pressure and 1 case of changed echocardiogram. As a result of confirming causal relationship with the clinical trial drug, 11 cases were related, and 4 cases were not related.
In Study 2, a total of 9 AEs were reported by 7 subjects. There were 3 cases of increased alanine aminotransferase, 1 case of increased creatinine, 3 cases of pyuria, 1 case of abdominal tenderness and 1 case of diarrhea. After confirming causal relationship with the clinical trial drug, 3 cases were related, and 6 cases were not related (Table 6).
 |
Table 6 Safety and Tolerability of Teneligliptin and Modified-Release Metformin (Studies 1 and 2) |
All AEs were of mild intensity, and the subjects recovered spontaneously without sequelae. There was no statistically significant difference between the treatment groups in the incidence of adverse events and adverse drug reactions.
Discussion
According to the American Diabetes Association guidelines, it is recommended that patients who are taking hypoglycemic drugs for the first time should start with a combination therapy to avoid treatment failure if their blood glucose is high.2 When deciding to start treatment with a combination therapy, it is recommended that drugs with different mechanisms of action be chosen to enhance the hypoglycemic effect. Teneligliptin and metformin have different mechanisms of action among the many ways to lower glucose. Teneligliptin, a DPP4 inhibitor, showed glucose-dependent glucose-lowering effects and activated incretin action. Metformin, a biguanide, increased insulin sensitivity in tissue and decreased gluconeogenesis in the liver. The efficacy and safety of teneligliptin and metformin dual combination therapy was recently confirmed once again in Asian patients in a Chinese clinical trial.13 This trial compared teneligliptin versus placebo for type 2 diabetes patients who were inadequately controlled with metformin and lifestyle modification. The inclusion criteria were almost the same as those in a previous study conducted in Europe. At 24 weeks, both clinical trials showed that the number of patients with glycated hemoglobin less than 7% was over 40%, and the glycated hemoglobin decreased by approximately 0.8%.13,14 Providing a stabilized FDC formulation for a drug whose efficacy has been proven as described above has the advantage of possibly reducing the burdens of administration and improving low medication compliance in patients with type 2 diabetes.
In the present study, we designed the results to demonstrate pharmacokinetics between the administration newly developed FDC formulation KD-4002 (ie, teneligliptin hydrochloride hydrate 20 mg combined with modified-release metformin HCl 1000 mg tablet) and the coadministration of a reference FDC drug (teneligliptin hydrobromide hydrate 20 mg combined with modified-release metformin HCl 1000 mg tablet) under fasted or fed conditions.
In Study 1, we verified that all pharmacokinetic parameters for both teneligliptin and modified-release metformin were similar in patients who received the test FDC drug (KD-4002) and those who received the reference FDC drug under fasting conditions, with GMR and 90% CI values that fell entirely within 80%–125% for both Cmax and AUCt. In pharmacokinetic studies in healthy adults, teneligliptin was well tolerated and did not significantly affect the pharmacokinetics of metformin. Throughout the study, the administration of teneligliptin combined with the modified-release metformin FDC formulation was well tolerated by all subjects.
In Study 2, we verified that all pharmacokinetic parameters for modified-release metformin were similar in patients under fed conditions. This is because US FDA guidance published in 2002 recommends that all modified-release drugs be subjected to postprandial bioavailability and bioequivalence studies.15 There was an increase in metformin AUCt of approximately 43% and a decrease in metformin Cmax of approximately 19% when patients were given a high-fat meal. The high-fat meal prolonged the Tmax by approximately 3 hours compared with the fasted state, whereas the Cmax was not affected under fasted conditions compared with fed conditions. In a previous bioequivalence study under fed conditions, the AUCt of metformin increased by 36%–60% compared to the fasted condition, whereas the Cmax of metformin was not significantly affected following administration of the FDC formulation.16 Therefore, the results of increased metformin pharmacokinetics after a high-fat diet are unlikely to be clinically meaningful.
No serious adverse events occurred in the present study, and the AEs observed were mild in nature and resolved without sequelae. A total of six subjects who dropped out in Study 1 and 2 were also unrelated to the cause of AEs.
Conclusion
This pharmacokinetic study compared administration of a newly developed FDC formulation, KD-4002, containing teneligliptin hydrochloride hydrate 20 mg and metformin HCl XR 1000 mg, with that of the other FDC formulation, teneligliptin hydrobromide hydrate 20 mg combined metformin HCl SR 1000 mg under fasted and fed conditions. This study suggests that the test drug (KD-4002) treatment has similar exposure and absorption rates as the reference drug treatment.
A single FDC drug of teneligliptin hydrochloride hydrate 20 mg combined metformin HCl XR 1000 mg (KD-4002) was well tolerated under both fasted and fed conditions.
Data Sharing Statement
The authors will not share de-identified individual participant data. All data contained in the manuscript is shareable.
Ethics Approval and Informed Consent
This study was approved by the Ministry of Food and Drug Safety of the Republic of Korea and the Institutional Review Board of Jeonbuk National University Hospital. This study was registered at ClinicalTrials.gov (identifier number KCT0007757, KCT0007759). It was conducted according to the Declaration of Helsinki and the Guidelines for Good Clinical Practice at Jeonbuk National University Hospital, Center of Clinical Pharmacology, Republic of Korea. All subjects provided written informed consent prior to screening.
Acknowledgments
The authors wish to thank the investigators, the participating subjects, and Kyung Dong Pharmaceutical Co., Ltd for their assistance in conducting the study.
Funding
This study was supported by the Kyung Dong Pharmaceutical Co., Ltd, Seoul, Republic of Korea.
Disclosure
The research article is original, has not already been published in any other journal (medical, or otherwise) or is not currently under consideration for publication by another journal, and does not infringe any existing copyright or any other rights prescribed by law. Min Ho Jeong is paid employees of Kyung Dong Pharmaceutical Co., Ltd, Seoul, Republic of Korea. The other authors report no conflicts of interest in this work. The sponsor did not participate in the study design, study conduct, or data analysis.
References
1. American Diabetes Association. Introduction: Standards of Medical Care in Diabetes. Vol. 45. Virginia, US: American Diabetes Association; 2022.
2. Draznin B, Aroda VR, Bakris G, et al. 9. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes. Diabetes Care. 2022;45(Supplement_1):S125–S143.
3. Liu Y, Hong T. Combination therapy of dipeptidyl peptidase-4 inhibitors and metformin in type 2 diabetes: rationale and evidence. Diabetes Obes Metab. 2014;16(2):111–117. doi:10.1111/dom.12128
4. Meier JJ, Nauck MA. Incretins and the development of type 2 diabetes. Curr Diabetes Rep. 2006;6(3):194–201. doi:10.1007/s11892-006-0034-7
5. Moore KB, Saudek CD. Therapeutic potential of dipeptidyl peptidase-IV inhibitors in patients with diabetes mellitus. Am J Ther. 2008;15(5):484–491. doi:10.1097/MJT.0b013e3180ed42dc
6. Williams-Herman D, Johnson J, Teng R, et al. Efficacy and safety of sitagliptin and metformin as initial combination therapy and as monotherapy over 2 years in patients with type 2 diabetes. Diabetes Obes Metab. 2010;12(5):442–451. doi:10.1111/j.1463-1326.2010.01204.x
7. Pérez‐Monteverde A, Seck T, Xu L, et al. Efficacy and safety of sitagliptin and the fixed-dose combination of sitagliptin and metformin vs. pioglitazone in drug-naive patients with type 2 diabetes. Int J Clin Pract. 2011;65(9):930–938. doi:10.1111/j.1742-1241.2011.02749.x
8. Bosi E, Dotta F, Jia Y, Goodman M. Vildagliptin plus metformin combination therapy provides superior glycaemic control to individual monotherapy in treatment-naive patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2009;11(5):506–515. doi:10.1111/j.1463-1326.2009.01040.x
9. Haak T, Meinicke T, Jones R, Weber S, Eynatten M, Woerle HJ. Initial combination of linagliptin and metformin improves glycaemic control in type 2 diabetes: a randomized, double‐blind, placebo‐controlled study. Diabetes Obes Metab. 2012;14(6):565–574. doi:10.1111/j.1463-1326.2012.01590.x
10. Blonde L, Dailey GE, Jabbour SA, Reasner CA, Mills DJ. Gastrointestinal tolerability of extended-release metformin tablets compared to immediate-release metformin tablets: results of a retrospective cohort study. Curr Med Res Opin. 2004;20(4):565–572. doi:10.1185/030079904125003278
11. Idkaidek N, Arafat T, Melhim M, Alawneh J, Hakooz N. Metformin IR versus XR pharmacokinetics in humans. J Bioequiv Availab. 2011;3(10):
12. Batolar L, Iqbal M, Monif T, Khuroo A, Sharma P. Bioequivalence and pharmacokinetic comparison of 3 metformin extended/sustained release tablets in healthy Indian male volunteers. Arzneimittelforschung. 2012;62(01):22–26. doi:10.1055/s-0031-1295428
13. Ji L, Li L, Ma J, et al. Efficacy and safety of teneligliptin added to metformin in Chinese patients with type 2 diabetes mellitus inadequately controlled with metformin: a Phase 3, randomized, double‐blind, placebo‐controlled study. Endocrinol Diabetes Metab. 2021;4(2):e00222. doi:10.1002/edm2.222
14. Bryson A, Jennings PE, Deak L, Paveliu FS, Lawson M. The efficacy and safety of teneligliptin added to ongoing metformin monotherapy in patients with type 2 diabetes: a randomized study with open label extension. Expert Opin Pharmacother. 2016;17(10):1309–1316. doi:10.1080/14656566.2016.1190334
15. Food Drug Administration. Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies. Maryland, US: Food Drug Administration; 2002.
16. Rhee S-J, Lee S, Yoon SH, Cho J-Y, Jang I-J, Yu K-S. Pharmacokinetics of the evogliptin/metformin extended-release (5/1000 mg) fixed-dose combination formulation compared to the corresponding loose combination, and food effect in healthy subjects. Drug Des Dev Ther. 2016;10:1411.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.