Back to Journals » Drug Design, Development and Therapy » Volume 14
Pharmacokinetics and Tolerability of Single and Multiple Intravenous Doses of Cefotetan Disodium in Healthy Chinese Volunteers
Authors Liu J ![]() , Zhai Y
, Zhai Y ![]() , Wu L
, Wu L ![]() , Wu G, Zheng Y, Hu X, Shentu J
, Wu G, Zheng Y, Hu X, Shentu J
Received 16 October 2019
Accepted for publication 29 January 2020
Published 13 February 2020 Volume 2020:14 Pages 613—620
DOI https://doi.org/10.2147/DDDT.S234619
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Jian Liu,1,2 You Zhai,1,2 Lihua Wu,1,2 Guolan Wu,1,2 Yunliang Zheng,1,2 Xingjiang Hu,1,2 Jianzhong Shentu1– 3
1Research Center for Clinical Pharmacy, State Key Laboratory for Diagnosis and Treatment of Infectious Disease, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, People’s Republic of China; 2Zhejiang Provincial Key Laboratory for Drug Evaluation and Clinical Research, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, People’s Republic of China; 3College of Medicine, Zhejiang University, Hangzhou, People’s Republic of China
Correspondence: Jianzhong Shentu
Research Center for Clinical Pharmacy, State Key Laboratory for Diagnosis and Treatment of Infectious Disease, The First Affiliated Hospital, College of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou 310003, People’s Republic of China
Tel +86 571 87236560
Fax +86 571 87214223
Email [email protected]
Background: Cefotetan is highly stable to penicillinase and cephalosporin produced by gram-negative bacteria, and it has strong antimicrobial activity against most gram-negative bacteria, some anaerobic bacteria and streptococcus. The objective of this study was to evaluate the pharmacokinetic profile and tolerability of single and multiple intravenous doses of cefotetan disodium in healthy Chinese volunteers.
Methods: In this single-center, open-label, dose-escalating study, subjects were randomized to receive a single dose of cefotetan disodium 0.5, 1.0, or 2.0 g administered as a 1 h intravenous infusion. After completion of the single-dose phase, subjects continued into the multiple-dose phase, in which they received 1.0 g cefotetan disodium BID for 7 consecutive days. Plasma samples were assayed by a validated high-performance liquid chromatography-tandem mass spectrometry method. Pharmacokinetic parameters were calculated and analyzed statistically. Tolerability was assessed based on physical examinations, vital signs, laboratory tests, and subject interviews.
Results: After intravenous administration of single doses of 0.5, 1.0, and 2.0 g cefotetan disodium, the pharmacokinetics of cefotetan were as follows: Cmax was 69.49± 12.10 μg·mL− 1, 132.03± 22.56 μg·mL− 1 and 237.75± 42.12 μg·mL− 1, respectively; AUClast was 278.29± 51.13 μg·mL− 1·h, 543.25± 92.44 μg·mL− 1·h and 1003.8± 172.39 μg·mL− 1·h, respectively; AUC∞ was 284.42± 50.76 μg·mL− 1·h, 551.38± 95.83 μg·mL− 1·h and 1020.18± 181.19 μg·mL− 1·h, respectively; t1/2 was 4.21± 0.83 h, 4.39± 0.53 h and 4.27± 0.74 h, respectively; CL was 1.81± 0.33 L·h− 1, 1.86± 0.32 L·h− 1 and 2.02± 0.38 L·h− 1, respectively; Vd was 10.80± 1.89L, 11.78± 2.20L and 12.25± 1.99L, respectively. In the multiple-dose study, the pharmacokinetics of cefotetan were as follows: Cmax,ss was 147.58± 22.71 μg·mL− 1; Cmin,ss was 12.92± 3.70 μg·mL− 1; Cavg was 45.10± 7.78 μg·mL− 1; AUCτ,ss was 541.15± 93.36 μg·mL− 1·h; AUC∞ was 612.06± 114.23 μg·mL− 1·h; t1/2 was 4.30± 0.63 h; CL was 1.90± 0.35L·h− 1; Vd was 8.91± 1.57L; DF was 300.92± 33.28%; Accumulation Index was 1.17± 0.05. No serious adverse events were reported. Adverse events were generally mild.
Conclusion: Cefotetan disodium showed favorable tolerability in this study. The Cmax and AUCs of cefotetan disodium demonstrated dose-dependent pharmacokinetic characteristics after single dose over a dose range (0.5– 2.0 g) in healthy subjects, whereas the t1/2 was independent of dose. Except for Vd, there was no difference in other pharmacokinetic parameters between multiple and single administration.
Keywords: cefotetan disodium, pharmacokinetics, healthy volunteers, plasma, single-dose, multiple-dose
Introduction
With the wide application of the third-generation cephalosporins, gram-negative bacterial infections have been effectively treated and controlled. However, in the 1980s, extended-spectrum beta-lactamases (ESBLs), which can hydrolyze penicillins, cephalosporins and other beta-lactam antibiotics at the same time, were found in some gram-negative bacteria. These enzymes are mediated by plasmids and can be transferred and spread among strains, resulting in the propagation of drug-resistant bacteria, which brings great difficulties to clinical anti–infective treatment. Therefore, it has become a hot spot to find effective and broad-spectrum antibiotics which are stable to ESBLs in the research of antibiotics.
The introduction of methoxyl group into the 7-alpha carbon atom in the mother nucleus of cefotetan can make it highly stable to the penicillinase and cephalosporinase produced by gram-negative bacteria. It can inhibit the synthesis of bacterial cell wall by binding with one or more penicillin-binding proteins, thus exerting short-term strong bactericidal activity. It has a good antimicrobial effect on gram-negative bacteria and anaerobic bacteria, especially for Coliform bacteria, Serratia, Proteus, Morgan, Providence and Pseudomonas.1,2 Meanwhile, cefotetan has good pharmacokinetic characteristics. The average half-life of the elimination of cefotetan is about 3–4 h. Thus, people need to use this drug only twice a day, which is in accord with the habit of clinical administration.3
Cefotetan and its injection have been loaded into a basic catalog of antibiotics in Japan. It is the second generation of cephalosporin antibiotics initiated by Yamanouchi Pharmaceutical Co., Ltd. in Japan and was approved to be on the market in Japan in 1993. At present, cefotetan disodium for injection has been listed in the United States and Europe. The manufacturer is Astra Zeneca Group, and the trade name is “Cefotan.” Cefoxitin is the representative cephalomycin antibiotics on the market in China. However, its antimicrobial activity, safety and pharmacokinetic characteristics are slightly inferior to cefotetan.
Cefotetan disodium for injection of the present study was developed by Zhejiang Jianfeng Pharmaceutical Co., Ltd. Its raw materials and its injections had been on the market abroad, and did not change its dosage form and its administration route. Thus, it belongs to Class 3.1 in Chemicals Registration Classification, and its pharmacokinetic studies in human are needed, according to the relevant regulations of “Registration Classification of Chemicals” in the “Drug Registration Regulations” (No. 28 of the Bureau Order).4
The present pharmacokinetic studies of cefotetan disodium for injection were carried out in healthy Chinese volunteers. The concentrations of cefotetan in blood were determined, and the main pharmacokinetic parameters were calculated. These results will facilitate the understanding of pharmacological characteristics of cefotetan disodium, as well as its absorption, distribution, metabolism and excretion. This study will provide a basis for better clinical use of cefotetan disodium for injection in Chinese people in the future.
Materials and Methods
Subjects
Healthy male and female subjects, aged 18–45 years, with body mass index (BMI) between 18.0 and 25.0 kg·m−2, were eligible to participate in this study. The health status of subjects was determined by medical history, complete physical examination, 12-lead ECG, and laboratory tests during the screening period. All female subjects were demanded to have a negative pregnancy test result during the screening period and expected to use appropriate contraceptives throughout the whole study. Subjects with clinically significant abnormalities at screening were excluded from the study. Other exclusions included a history of clinically significant cardiovascular, renal, hepatic, pulmonary, gastrointestinal, endocrine, hematologic, vascular, or collagen disease; a history of nervous system disease, muscle disease, seizure, or a psychiatric disorder that might hinder compliance with the study protocol; a history of alcohol or drug abuse; allergy or hypersensitivity to study drug; positive tests for human immunodeficiency virus, hepatitis B virus surface antigen, anti-hepatitis C virus antibody, or syphilis antibody; blood donation within the previous 3 months; participation in a study of another investigational drug within the previous 3 months; and any drug treatment in the previous 2 weeks. Women who were breastfeeding or menstruating, or receiving hormone replacement therapy, were excluded.
Study Design
This was a single-center, open-label, randomized cross-over, two-part (part 1 and part2) study conducted in healthy adult Chinese subjects. The study protocol was approved by the independent the Ethical Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University. The study was conducted in accordance with Guideline for Good Clinical Practice,5 and the Guideline for Pharmacokinetics studies recommended by the China Food and Drug Administration6 and International Conference on Harmonisation Good Clinical Practiceguidelines7 and the Declaration of Helsinki (as revised, Fortaleza, 2013).8 All subjects were fully informed about the procedure and risk associated with this study and have signed informed consent before undergoing any study procedures. This study has been registered at www.chictr.org.cn under registration No ChiCTR-TTRCC-14004652.
Part 1 was a randomized 3-way crossover, open-label, single-dose study in which 12 subjects received cefotetan disodium each of the following 3 treatments in a randomized sequence: 0.5–1.0–2.0 g, 1.0-2.0-0.5 g and 2.0-0.5-1.0 g (administered as 1 h intravenous infusion), according to a computer-generated randomization schedule (SAS 9.1, SAS Institute Inc., Cary, NC, USA). Treatments were separated by 7-day washout periods. All subjects fasted (ie, no food and liquid except water) for at least 10 h prior to drug administration. Water was allowed as needed for up to 2 h after drug intake. During the 24 h after drug administration, no strenuous physical activity was permitted. Smoking and consumption of alcohol or caffeine-containing beverages were prohibited throughout the study.
In Part 2, 12 subjects (6 males and 6 females) who completed the single-dose phase (Part 1) were enrolled in the multiple-dose phase (Part 2). They were received a 1 h infusion of cefotetan disodium 1.0 g every 12 h for 7 times. Subjects were confined to the study center, received standard meals daily (1 h before administration, 4 h after administration, and 10 h after administration) for 3 consecutive days. At approximately 7:30 am on day 4, subjects received the last dose of their assigned treatment. Standard meals were provided to all subjects at 4 and 10 h after study drug administration.
For both parts of this study, cefotetan disodium for injection (1.0 g per ampoule; lot 130702) was supplied by Zhejiang Jianfeng Pharmaceutical Co., Ltd. Strenuous physical activity, smoking and consumption of alcohol, caffeine-containing beverages carbonated drinks, or grapefruit (or other citrus fruit) were prohibited throughout the study.
Safety Assessment
Safety was assessed by the investigators based on subject interviews, spontaneous reporting, vital signs, physical examination, and clinical laboratory tests (urinalysis, hematology, and blood chemistry) during the whole study period. All AEs were considered serious when led to death, life-threatening, required hospitalization, led to disability, or required intervention to prevent permanent impairment or damage. All AEs were recorded in the source data record and on case report forms by investigators.
Pharmacokinetic Parameters and Assessment
In Part 1 of the study (single dosing phase), blood samples (5 mL) were collected from an indwelling venous catheter into heparinized tubes in three stages: 1) before each administration (0 h); 2) at 0.25, 0.5, 0.75 and 1 h (the end of the infusion) after each start of administration; 3) at 0.25, 0.5, 1, 2, 3, 5, 7, 9, 12, 15 and 24 h after each end of administration. The blood samples were centrifuged at 4,000 rpm for 10 mins at 4°C, and the acquired plasma samples were stored at −70°C until analyzed.
In Part 2 of the study (multiple dosing phase, 1.0 g q12h for a total of 7 doses), blood samples were collected in four stages: 1) at 48, 36, 24 and 12h before the last administration; 2) before the last administration (0 h); 3) at 0.25, 0.5, 0.75 and 1 h (the end of the infusion) after the start of the last administration; 4) at 0.25, 0.5, 1, 2, 3, 5, 7, 9, 12, 15 and 24 h after the end of the last administration. Other experimental conditions were the same as in the single-dose phase.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) was used to measure the concentrations of cefotetan in plasma samples. The LC-MS/MS system included a Spark Pico high-performance liquid chromatography system (Spark Holland, Emmen, Netherlands) and API4000 triple quadrupole mass spectrometer (AB Sciex, Ontario, Canada). Chromatography was performed with an Agilent ZORBAX SB-C18 column (4.6 mm×250 mm, 5 μm) and the mobile phase was water with 0.5% formic acid (A)–acetonitrile(B) at a flow rate of 1.0 mL·min−1.The gradient elution program: 0–4 min 30 s, 70%A/30%B; 4 min 30 s-4 min 40 s, 70%~5%A/30%~95%B; 4 min 40 s-6 min, 5%A/95%B; 6 min-6 min 10 s, 5%~70%A/95%~30%B; 6 min 10 s- 9 min, 70%A/30%B. The column temperature was set at 25°C, and the injection volume was 5 μL.
All detections were carried out with the mass spectrometer in negative electrospray ionization mode. The multiple reaction monitoring transitions were m/z 574.1/458.1 for cefotetan and m/z 452.1/282.9 for cefixime (internal standard). The optimized instrumental parameters for mass spectral acquisition were collision gas at 10 psi, curtain gas at 15 psi, ion source gas 1 (nitrogen) at 20 psi, ion source gas 2 (nitrogen) at 30 psi, dwell time of 200 msec, ion spray voltage of −4500 V, and temperature of 350°C.
The method was validated in terms of specificity, calibration curve, sensitivity, matrix effect, accuracy, precision, and stability. The relative standard deviation (RSD) of quality control should be within 15% of the actual value, except at the lower limit of quantitation, where it should not deviate by >20%. Calibration curves were constructed at concentrations of 0.5105–510.5 μg·mL−1 (r>0.999). Blood samples above the limit of quantitation were diluted and reanalyzed to yield results within the calibrated range. Stability tests showed that plasma sample decomposition was<10% for all conditions examined (5 h at room temperature, 3 freeze-thaw cycles at or below −70°C, and 175 days after storage at or below −70°C). Separate sets of analytical quality control samples (for concentrations of 0.5105, 0.9320, 18.64 and 372.8 μg·mL−1) were used to assess assay precision and accuracy. The intra- and inter-day precisions (relative standard deviation, %RSD) were ≤4.78% and ≤5.81%, respectively. The intra- and inter-day recovery rates were 94.55–95.83% and 97.11–97.96%, respectively. The matrix effect was 104.61%, 103.53%, and 104.15% with RSD≤4.40% for cefotetan with the concentrations of 0.9320, 18.64 and 372.8 μg·mL−1.
The blood sample analysis and determination method of cefotetan disodium refer to foreign references.9–11 The methodology of this study meets the requirements of pharmacokinetics. The whole research was strictly operated according to SOPs to ensure that the research data were true, accurate and complete, and the research results were scientific and reliable.
Single- and multiple-dose pharmacokinetic parameters were calculated from the plasma concentration-time data by non-compartmental methods using WinNonlin 6.3 software (Pharsight Corporation, Sunnyvale, CA, USA).
The Cmax (the maximum plasma concentration at single dose), Cmax,ss and Cmin,ss (the maximum and minimum plasma concentration at the steady state) were obtained from the observed data. The AUClast was calculated using the linear trapezoidal rule. The AUC∞ (AUC∞,ss) was calculated as AUClast + Clast/λz, where Clast is the last measurable concentration and λz is the slope of the log-linear regression of the terminal concentration data points. The t1/2 was calculated as (ln2)/λz. CL after administration of single doses was calculated as Dose/AUC∞; after multiple dosing, CL was calculated as Dose/AUCτ,ss (area under the concentration–time curve at the steady state), where τ is the dosing interval (12 h). The mean concentration at steady state (Cavg) was calculated as AUCτ,ss/τ. Vd was based on the terminal elimination phase (CL/λz).The accumulation index (AI) was calculated as the ratio of AUCτ,ss to AUCτ (single dose), and the degree of fluctuation (DF) was calculated by (Cmax,ss-Cmin,ss)/Cavg. Individual pharmacokinetic values were calculated and averaged.
Statistical Analysis
Pharmacokinetic parameters were expressed by using arithmetic means and standard deviations. For single-dose and multiple-dose studies, descriptive statistical analyses for pharmacokinetic parameters were performed by using SPSS (Version 16.0, SPSS Inc., Chicago, IL, USA). To test dose proportionality in the single-dose range studied, log-transformed pharmacokinetic parameters AUC and Cmax were analyzed by using the power model, PK=A×(Dose)β, where PK is the pharmacokinetic parameter, A is the intercept, and β is the dose-proportionality coefficient. A 95% confidence interval (CI) for the dose-proportionality parameter β was then calculated. Note that a slope of 1 would correspond to perfect dose proportionality. A t-test was used to determine whether the multiple-dose pharmacokinetic parameters were consistent with those in the single-dose phase. The results for Tmax were evaluated using the Wilcoxon rank-sum analysis. Data were categorized by sex to determine statistical differences in PK parameters between males and females. P<0.05 was considered statistically significant.
Results
Study Participants
In the single-dose and multiple-dose study, a total of 12 healthy subjects (6 males and 6 females) were enrolled in and completed both two studies. The mean age [SD] of subjects was 25.67 [2.67] years (range, 20–29 years), mean body weight [SD] was 62.00 [9.98] kg (range, 47.5–85.0 kg), mean height [SD] was 1.70 [0.09] m (range, 1.58–1.85 m), and mean BMI [SD] was 21.43 [1.80] kg·m−2 (range, 19.0–24.8 kg·m−2). Demographic data for the subjects receiving cefotetan disodium are shown in Table 1.
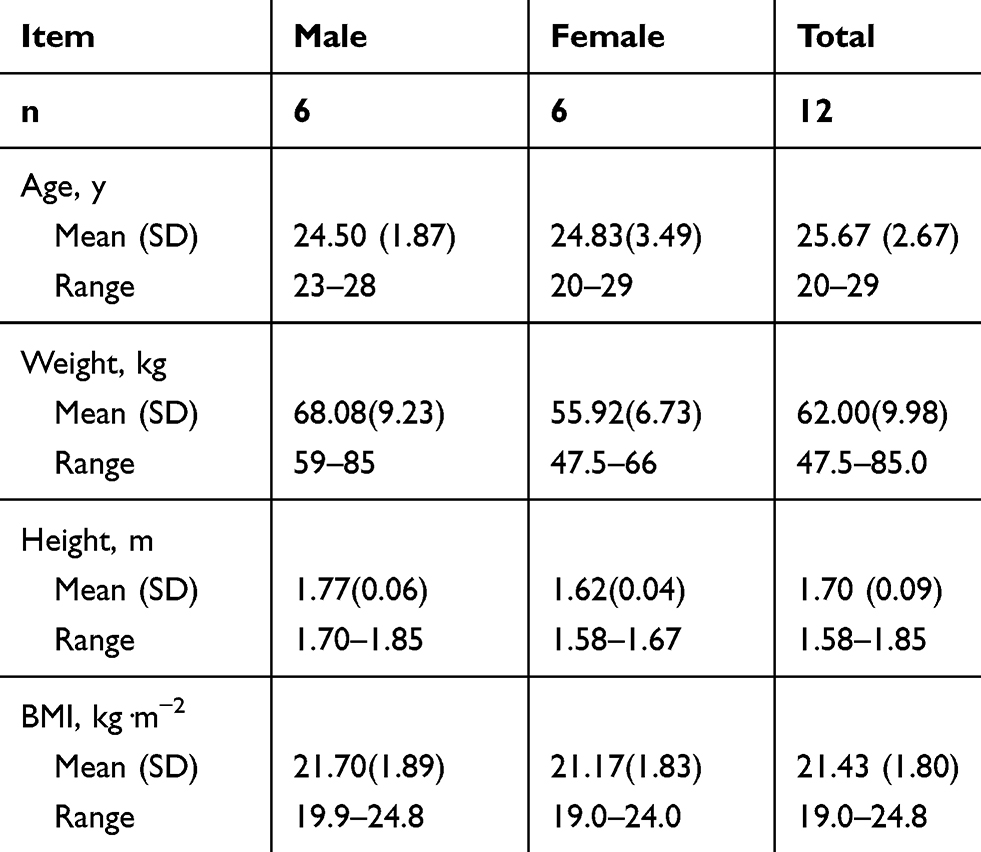 |
Table 1 Demographic Data for the Subjects Receiving Cefotetan Disodium |
Safety
All subjects were included in the safety analysis. There were no deaths or SAEs. In the single-dose phase, the most common adverse event (AE) was phlebitis at the site of injection (2/12 subjects [16.7%]). In the multiple-dose phase, the most common AEs were as follows: phlebitis at the site of injection (5/12 subjects [41.7%]), epistaxis (2/12 subjects [16.7%]), and arm weakness (2/12 subjects [16.7%]).
All AEs were mild and transient, which the subjects could tolerate and required no intervention. None of the subjects withdrew from the study or were suspended by the investigators because of AEs. Vital signs, physical examinations, ECGs, and clinical laboratory assessments remained within normal limits for all subjects. There were no clinically meaningful differences between single and multiple groups.
Pharmacokinetics
Single-Dose Phase
The mean plasma concentration-time profile of cefotetan disodium following administration of single intravenous doses 0.5–2.0 g is illustrated in Figure 1. Pharmacokinetic parameters are presented in Table 2. The mean Cmax values for the 0.5, 1.0 and 2.0 g group were 69.49 μg·mL−1, 132.03 μg·mL−1 and 237.75 μg·mL−1, respectively. The mean AUClast values were 278.29 μg·h·mL−1, 543.25 μg·h·mL−1 and 1003.8 μg·h·mL−1, respectively. The mean AUC∞values were 284.42 μg·h·mL−1, 551.38 μg·h·mL−1 and 1020.18 μg·h·mL−1, respectively. The mean t1/2 values were 4.21 h, 4.39 hand 4.27 h, respectively, which was independent of the dose. The mean CL values were 1.81 L·h−1, 1.86 L·h−1 and 2.02 L·h−1, respectively. There were no significant differences in CL among groups. The mean Vd values were 10.80 L, 11.78 L and 12.25 L, respectively.
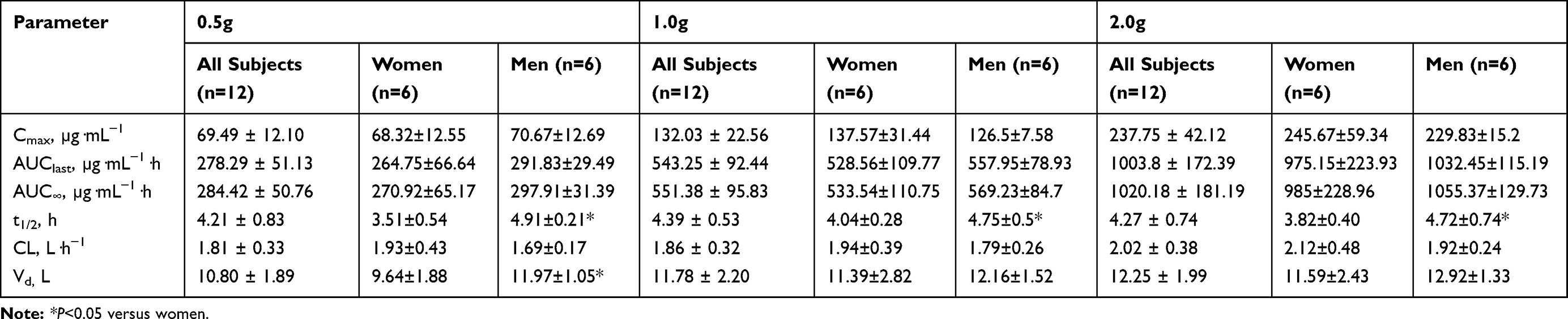 |
Table 2 Pharmacokinetic Parameters of Cefotetan Disodium After Administration of Single Intravenous Doses. Values are Expressed as Mean±SD |
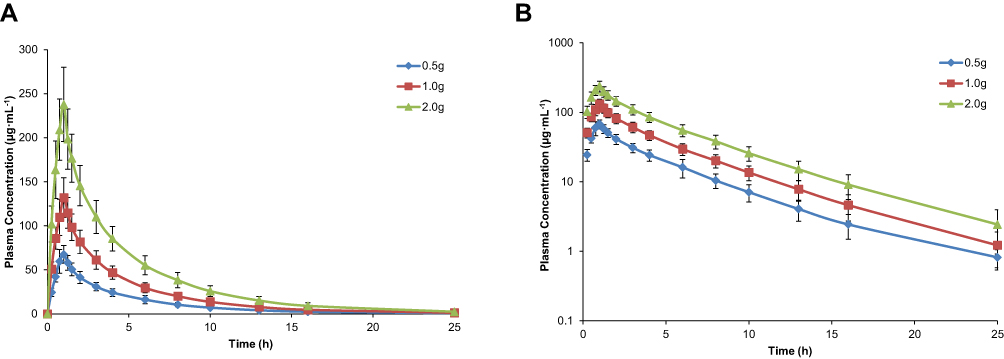 |
Figure 1 Mean (SD) plasma concentration-time profile of cefotetan after administration of single intravenous doses (0.5 g, 1.0 g, 2.0 g) (n=12). Linear scale (A); Semi-log scale (B). Error bars represent standard deviation (SD). |
For the relationship between non-compartmental parameters (AUC∞, AUClast, and Cmax) and doses, analysis results indicated that the estimated proportionality coefficients (β) for AUC∞, AUClast, and Cmax were 0.949 (95% CI: 0.868–1.094), 0.950 (95% CI: 0.865–1.088), and 0.949 (95% CI: 0.900–1.136), respectively.
Multiple-Dose Phase
The mean plasma concentration–time profiles of cefotetan following administration of multiple intravenous doses of cefotetan disodium 1.0 g BID are presented in Figure 2 and the pharmacokinetic parameters are shown in Table 3.
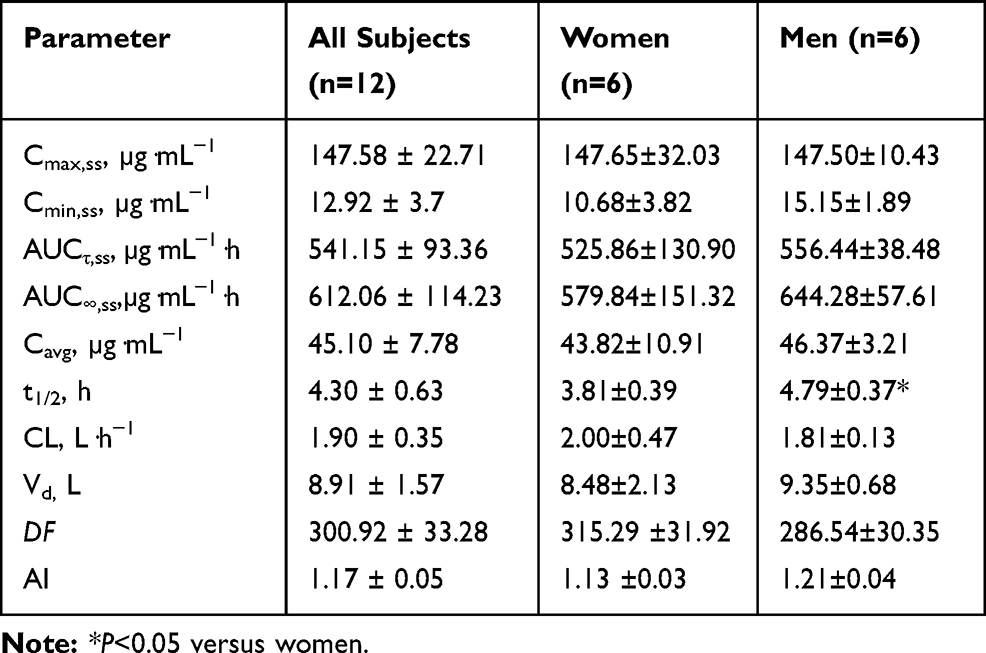 |
Table 3 Pharmacokinetic Parameters of Cefotetan Disodium After Administration of Multiple Intravenous Doses of 1.0 g BID. Values are Expressed as Mean±SD |
 |
Figure 2 Mean (SD) plasma concentration-time profile of cefotetan after administration of multiple intravenous doses (1.0 g) (n=12). Linear scale (A); Semi-log scale (B). Error bars represent standard deviation (SD). |
After administration of multiple intravenous doses of cefotetan disodium 1.0 g BID, based on plasma concentration of drug in pre-3rd, −4th, −5th, −6th and −7th dose samples, the steady state was achieved after administration of cefotetan disodium for 5 consecutive doses. The mean values of Cmax,ss, Cmin,ss, AUCτ,ss, AUC∞,ss, Cavg, t1/2, CL, and Vd, were 147.58 µg·mL−1, 12.92 µg·mL−1, 541.15 µg·mL−1·h, 612.06 µg·mL−1·h, 45.10 µg·mL−1, 4.30 h, 1.90 L·h−1 and 8.91 L, respectively. The accumulation ratio was found to be in the range of 1.07 and 1.27, indicating that there was no accumulation after multiple intravenous doses of cefotetan disodium.
Except for Vd, there were no significant differences in other pharmacokinetic parameters between single and multiple doses.
Gender Effect
With 2 exceptions (Tables 2 and 3), all in the single- and multiple-dose phase, there were no significant differences in the pharmacokinetic parameters of cefotetan disodium between female and male subjects. In the group that received the 0.5 g single dose, Vd was significantly greater in men than in women (11.97 L vs 9.61 L; P < 0.05). Another one of the exceptions occurred in both single and multiple doses: t1/2 was significantly longer in men than in women (4.91 h vs 3.51 h (0.5 g, single dose), 4.75 h vs 4.04 h (1.0 g, single dose), 4.72 h vs 3.82 h (2.0 g, single dose), 4.79 h vs 3.81 h (1.0 g, multiple dose); P < 0.05).
Discussion
Cefotetan disodium is a second-generation cephalosporin derivative with high-efficiency antimicrobial activities against ESBLs-induced infection, as well as the mixed infection which is induced by aerobic and anaerobic bacteria, and it has long t1/2. Cefotetan disodium for injection has been used in the clinic for many years since it was developed and listed. It is worth choosing in the research and development of cefotetan disodium for injection. At present, there is no such product in China. The objective of this study was to investigate the tolerability and pharmacokinetic characteristics of Class 3.1 new drug (according to the “Drug Registration Regulations”), and to meet National Medical Products Administration (NMPA) requirements for marketing of the new generic formulation.
In single-dose study, Cmax and AUC showed a good linear relationship with the dosage after a single dose of 0.5–2.0 g, and this result is consistent with the literature.3,10,13 The mean t1/2 values 4.29 h (2.90 −6.15 h) were consistent with the reported data (3–4 h) of Caucasians and Japanese.3,12–15 Similarly, the mean values of CL 1.90 L·h−1 (1.40–2.73 L·h−1) and Vd 11.61 L (7.25–15.81 L) obtained in the present study is consistent with that reported in literature (1.8–2.9 L·h−1 for CL and 8–13 L for Vd).3,12–15 Plasma t1/2, CL, and Vd were independent of the dose, suggesting that there is no saturation in drug metabolism and elimination within this dose range. The results above also can be concluded that there is no existence of ethnic differences in cefotetan disodium pharmacokinetics.
In multiple-dose study, there are no significant differences in mean trough concentrations between the 5th, 6th and 7th intravenous administration. It means that steady state was reached after 5 consecutive times of intravenous administration. There are no significant differences in the main pharmacokinetic parameters, including Cmax, AUC, t1/2, and CL in the 1.0 g dose group between single- and multiple-dose group (P>0.05). The Vd of the multiple-dose group was 8.91±1.57 L, which is less than that of the single-dose group (11.78±2.19 L) (P<0.05), but the value is also within the range of 8–13 L reported in the literature.3,12–15 Moreover, the accumulation index (AI) in vivo is 1.17±0.05. All these results suggest that there is no significant accumulation of the drug in vivo after repeated administration in this dosage group.
Examination of the effects of gender on the pharmacokinetics of cefotetan disodium found no significant differences in AUC, Cmax, and CL between women and men in either single or multiple-dose study. The exceptions were Vd in the group that received the 0.5-g single dose, and t1/2 in both single and multiple-dose group. Hence, no dose adjustment appears to be necessary based on sex.
Cefotetan disodium showed good safety. AE mostly occurred in multiple administration stages, but no SAE occurred. The main AEs were related to intravenous injection, such as phlebitis, epistaxis, and arm weakness. The degree of these AEs was mild and transient, and the subjects could tolerate it and required no intervention.
This study was done in healthy subjects. Therefore, the pharmacokinetic parameters would not apply for infected patients, since the metabolism in infected users might differ from that of naïve persons. Further studies in infected subjects and population pharmacokinetics should be carried out.
Data Sharing Statement
Individual deidentified participant data is not going to be shared. And all available data have been showed in the article. No other study-related document will be made available.
Acknowledgment
This study was funded by Zhejiang Jianfeng Pharmaceutical Co., Ltd. (Jinhua, China, the manufacturer of Cefotetan disodium) and the National Natural Science Foundation of China (No. 81702862).
Disclosure
All authors report no conflicts of interest in this work.
References
1. Jones RN. Cefotetan: a review of the microbiologic properties and antimicrobial spectrum. Am J Surg. 1988;155(5A):16–23.
2. Ward A, Richards DM. Cefotetan A review of its antibacterial activity, pharmacokinetic properties and therapeutic use. Drugs. 1985;30(5):382–426. doi:10.2165/00003495-198530050-00002
3. Martin C, Thomachot L, Albanese J. Clinical pharmacokinetics of cefotetan. Clin Pharmacokinet. 1994;26(4):248–258. doi:10.2165/00003088-199426040-00002
4. China Food and Drug Administration. Drug registration regulations; 2007. Available from: http://www.nmpa.gov.cn/WS04/CL2174/300629.html.
5. China Food and Drug Administration. Guideline for good clinical practice; 2003. Available from: http://www.nmpa.gov.cn/WS04/CL2077/300595.html.
6. China Food and Drug Administration. Guidelines for clinical pharmacokinetics of chemicals; 2007. Available from: http://www.cde.org.cn/zdyz.do?method=largePage&id=2070.
7. European Medicines Agency. ICH Topic E 6 (R1): guideline for Good Clinical Practice. Note for guidance on good clinical practice. London, UK: CPMP; 2002. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-e6-r1-guideline-good-clinical-practice_en.pdf.
8. World Medical Association. Declaration of Helsinki.
9. Su MX, Liu MH, Di B, et al. Pharmacokinetic differences between the epimers of cefotetan disodium after single intravenous injection in healthy Chinese volunteers. Eur J Drug Metab Pharmacokinet. 2011;36(4):223–228. doi:10.1007/s13318-011-0064-7
10. Kees F, Grobecker H, Naber KG. High-performance liquid chromatographic analysis of cefotetan epimers in human plasma and urine. J Chromatogr. 1984;305(2):363–371. doi:10.1016/S0378-4347(00)83350-2
11. Shi SJ, Liu YN, Li ZF, Zheng H, Lv YN, Chen H. Pharmacokinetics and tolerability of intravenous cefotetan disodium for injection in healthy chinese volunteers: a randomized, open-label, single- and multiple-dose study. Clin Ther. 2010;32(10):1832–1841. doi:10.1016/j.clinthera.2010.09.015
12. Guibert J, Kitzis MD, Yvelin C, Acar JF. Pharmacokinetics of single intravenous and intramuscular doses of cefotetan in normal human volunteers. J Antimicrob Chemother. 1983;11(SupplA):201–206. doi:10.1093/jac/11.suppl_A.201
13. Yates RA, Adam HK, Donnelly RJ, Houghton HL, Charlesworth EA, Laws EA. Pharmacokinetics and tolerance of single intravenous doses of cefotetan disodium in male Caucasian volunteers. J Antimicrob Chemother. 1983;11(Suppl A):185–191. doi:10.1093/jac/11.suppl_A.185
14. Nakagawa K, Koyama M, Tachibana A, Komiya M, Kikuchi Y, Yano K. Pharmacokinetics of Cefotetan (YM09330) in Humans. Antimicrob Agents Chemother. 1982;22(6):935–941. doi:10.1128/AAC.22.6.935
15. Carver PL, Nightingale CH, Quintiliani R. Pharmacokinetics and pharmacodynamics of total and unbound cefoxitin and cefotetan in healthy volunteers. J Antimicrob Chemother. 1989;23(1):99–106. doi:10.1093/jac/23.1.99
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.