Back to Journals » Drug Design, Development and Therapy » Volume 11
Pharmacokinetics and tolerability of DA-8031, a novel selective serotonin reuptake inhibitor for premature ejaculation in healthy male subjects
Authors Shin D ![]() , Lee S
, Lee S ![]() , Yi S, Yoon SH, Cho JY
, Yi S, Yoon SH, Cho JY ![]() , Bahng MY, Jang IJ
, Bahng MY, Jang IJ ![]() , Yu KS
, Yu KS ![]()
Received 6 November 2016
Accepted for publication 13 January 2017
Published 10 March 2017 Volume 2017:11 Pages 713—723
DOI https://doi.org/10.2147/DDDT.S126861
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Dongseong Shin,1 SeungHwan Lee,2 Sojeong Yi,2 Seo Hyun Yoon,2 Joo-Youn Cho,2 Mi Young Bahng,3 In-Jin Jang,2 Kyung-Sang Yu2
1Clinical Trials Center, Gachon University Gil Medical Center, Incheon, 2Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, 3Department of Product Development, Dong-A ST, Seoul, Korea
Objective: DA-8031 is a selective serotonin reuptake inhibitor under development for the treatment of premature ejaculation. This is the first-in-human study aimed at evaluating the pharmacokinetics and tolerability of DA-8031 and its metabolites (M1, M2, M4, and M5) in the plasma and urine after administration of a single oral dose in healthy male subjects.
Methods: A dose block-randomized, double-blind, placebo-controlled, single ascending dose study was conducted. Subjects received either placebo or a single dose of DA-8031 at 5, 10, 20, 40, 60, 80, or 120 mg. DA-8031 and its four metabolites were analyzed in the plasma and urine for pharmacokinetic evaluation. The effect of genetic polymorphisms of cytochrome-P450 (CYP) enzymes on the pharmacokinetics of DA-8031 was evaluated.
Results: After a single dose, plasma DA-8031 reached the maximum concentration at a median of 2–3 h and was eliminated with terminal elimination half-life of 17.9–28.7 h. The mean renal clearance was 3.7–5.6 L/h. Dose-proportional pharmacokinetics was observed over the dose range of 20–80 mg. Among the metabolites, M4 had the greatest plasma concentration, followed by M5 and M1. Subjects with CYP2D6 intermediate metabolizer had significantly greater dose-normalized Cmax and AUC0–t of DA-8031 as well as smaller metabolic ratios than those subjects with CYP2D6 extensive metabolizer. The most common adverse events were nausea, dizziness, and headache, and no serious adverse events were reported.
Conclusion: In conclusion, the systemic exposure of DA-8031 was increased proportionally to the dose within 20–80 mg. Genetic polymorphisms of CYP2D6 had an effect on the systemic exposure of DA-8031. DA-8031 was well tolerated after single doses of 80 mg or less.
Keywords: DA-8031, pharmacokinetics, selective serotonin reuptake inhibitor, premature ejaculation, first-in-human
Introduction
Premature ejaculation (PE) is the most common male sexual dysfunction.1,2 Approximately 20%–30% of men complained of PE, and 76.9% of males with typical erectile function reported various degrees of PE.3–5 Psychological issues and neurogenic conditions have been suggested as etiological factors of PE.6 Anxiety or conditioning toward rapid ejaculation was attributed to psychological causes, and either hyposensitivity of 5-hydroxytryptamine 2C (5-HT2c) receptors or hypersensitivity of the 5-HT1a receptors was associated with the neurogenic etiology.6,7 Selective serotonin reuptake inhibitors (SSRIs) are capable of increasing the activity of serotonergic cells in the nucleus paragigantocellularis, thereby inhibiting the expulsion phase of ejaculation by modulating bulbospongiosus muscle activity and impairing the emission phase by blocking the rise in seminal vesicular pressure.8,9
Although several SSRIs have been used off-label (especially dapoxetine, which was approved for on-demand treatment of PE), on-demand SSRI therapies have limited effects because of their pharmacokinetic (PK) and pharmacodynamic characteristics.10,11 Therefore, daily SSRIs are expected to be the first choice of treatment in patients with primary PE.10,12 Because daily SSRI medication may cause priapism, loss of bone mineral density, QTc interval prolongation, reduced libido and erectile dysfunction, as well as the short-term adverse effects of excess yawning, nausea, sweating, fatigue, and diarrhea due to the serotonin subreceptor selectivity and interaction with other neurotransmitter receptors, a novel daily SSRI with high tolerability and that is effective against PE is still required.13,14
DA-8031 is a potent SSRI under clinical investigation for the treatment of PE.9 From preclinical studies, DA-8031 was shown to have a high selectivity and specificity for serotonin transporters compared with other monoamine transporters and to increase ejaculation latency time without altering the post-ejaculatory interval.15,16 Furthermore, DA-8031 had a low affinity to other receptors, and thus it could contribute to fewer adverse effects.8 Regarding the pharmacokinetics of DA-8031, <1% of DA-8031 was excreted in urine, feces, or bile in its unchanged chemical structure, and bioavailability was greater in the high-dose groups in the preclinical study (investigator’s brochure, unpublished data on file). DA-8031 is metabolized into five metabolites (M1, M2, M4, M5, and M6) through hepatic metabolism in Figure 1 (investigator’s brochure, unpublished data on file).
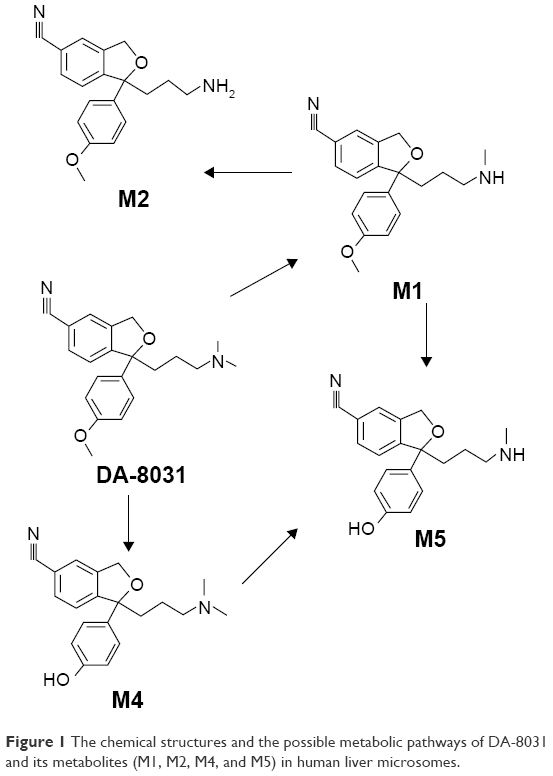 | Figure 1 The chemical structures and the possible metabolic pathways of DA-8031 and its metabolites (M1, M2, M4, and M5) in human liver microsomes. |
This human-based study aimed to evaluate the PK and tolerability profiles of DA-8031 after a single oral administration in healthy male subjects and is the first study to do so in humans.
Methods
Study design and population
This dose block-randomized, double-blind, placebo-controlled, single ascending dose study (registered at ClinicalTrials.gov [NCT01104948]) was approved by the Institutional Review Board of Seoul National University Hospital and conducted in accordance with the Declaration of Helsinki and Korean Good Clinical Practice. The objective and contents of this study were fully explained, and written informed consent was obtained from all subjects for this study before screening.
Subjects aged 20–45 years were screened based on medical history, physical examination, 12-lead electrocardiogram (ECG), vital signs, and clinical laboratory tests. Subjects who met the predefined criteria were randomly assigned to sequentially escalating dose groups of 5, 10, 20, 40, 60, 80, and 120 mg (eight subjects for DA-8031 and two subjects for placebo in each group). Dose escalation was determined based on the 2-day tolerability results after the final dosing in the previous dose group.
Bioanalytical methods
Blood samples to measure the plasma concentration of DA-8031 and its metabolites (N-demethyl-DA-8031 [M1], N,N-didemethyl-DA-8031 [M2], O-demethyl-DA-8031 [M4], and N,O-didemethyl-DA-8031 [M5]) were collected at pre-dose (0 h) and 1, 2, 3, 4, 5, 6, 8, 12, 24, 36, 48, 72, 96, and 120 h after dosing. At each time point, 6 mL of blood was obtained in heparin tubes and separated by 3,000 rpm centrifugation for 10 min at 4°C. Three aliquots of supernatant were stored at −20°C until further analysis.
Urine concentration and volume were measured during the following time intervals – predose (0 h), 0–4, 4–8, 8–12, 12–24, 24–48, 48–72, 72–96, and 96–120 h. Three 5 mL urine samples were obtained in polypropylene tubes and stored at −20°C. Blood and urine samples were analyzed by the Seoul National University Hospital Biomedical Research Institute, Seoul, Korea.
Plasma concentrations of DA-8031 and its metabolites (M1, M2, M4, and M5) were analyzed separately using a high-performance liquid chromatography (HPLC) (Agilent 1260 infinity; Agilent Technologies, Santa Clara, CA, USA)–tandem mass spectrometry (MS/MS) (API 4000 QTRAP, Applied Biosystems, Foster City, CA, USA) system in the positive ionization mode. Chromatographic separation was performed at 25°C using an Atlantis dC18 column (100×2.1 mm, 3 μm, Waters, Milford, MA, USA) for DA-8031, M4, and M5, and a GeminiNX C18 column (50×2.0 mm, 5 μm, Phenomenex, Torrance, CA, USA) for M1 and M2 was operated under reversed-phase conditions with mobile phases A (10 mM ammonium acetate in water) and B (acetonitrile) – for DA-8031, phase A:phase B=10:90 (v/v); for M1 and M2, phase A:phase B=20:80 (v/v); and for M4 and M5, phase A:phase B=50:50 (v/v). The standard curve ranges of DA-8031 and its metabolites were 0.5–200.0 ng/mL and 0.5–25.0 ng/mL, respectively. The coefficient of variation of the inter-assay accuracy and precision did not exceed 15.0%.
Urine concentrations of DA-8031 were quantified using HPLC (Agilent 1100 series, Agilent Technologies) with MS/MS (API 3000, Applied Biosystems) in the positive ionization mode. For M4 and M5, HPLC (Agilent 1200 series, Agilent Technologies) with MS/MS (API 3200, Applied Biosystems) was used. DA-8031 was separated on an Atlantis dC18 reversed-phase column (100×2.1 mm, 3 μm, Waters, Dublin, Ireland) at 25°C with mobile phases A (10 mM ammonium acetate in water) and B (acetonitrile) – phase A:phase B=30:70 (v/v). M4 and M5 were separated on an Atlantis dC18 reversed-phase column (100×2.1 mm, 5 μm, Waters, Dublin, Ireland) at 20°C with mobile phases A (10 mM ammonium acetate in water) and B (acetonitrile) – phase A:phase B=30:70 (v/v). The standard curve ranges of DA-8031 and its metabolites were 5.0–2,000.0 ng/mL and 2.0–500.0 ng/mL, respectively. The coefficient of variation of the inter-assay accuracy and precision did not exceed 15.0%.
PK analysis
The PK parameters of DA-8031 and its metabolites were estimated by a non-compartmental method using WinNonlin® 6.3 (Pharsight Co, Mountain View, CA, USA). The maximum observed plasma concentration (Cmax) after a single oral administration was directly obtained from the plasma concentration–time profiles. The area under the plasma concentration–time curve (AUC) was calculated using the linear-up/log-down trapezoidal method. The elimination rate constant λz was estimated by regression of the log-linear decrease of the plasma concentration–time profile, and the terminal elimination half-life was calculated as ln(2)/λz. The metabolic ratios (MRs) were determined by the equation AUCmetabolite/AUCDA-8031. Renal clearance (CLR) was estimated as the cumulative amount of unchanged drug excreted in urine (Ae) divided by the AUC.
Safety and tolerability
Twelve-lead ECGs were recorded using MAC 5000 and MAC 5500 systems (GE Healthcare, Milwaukee, WI, USA) before dosing (0 h) and at 2, 4, 8, 24, and 72 h after dosing. ECG recordings were analyzed using the automated Marquette 12-lead ECG Analysis Program. The ventricular rate, PR interval, QT interval, and QRS duration were observed directly from automated ECG interval readings, and the RR interval and heart rate-corrected QT intervals (QTc) were calculated. The QTc were obtained using Bazett’s formula (QTcB = QT/[RR interval]1/2) and Fridericia’s formula (QTcF = QT/[RR interval]1/3). ECGs were independently reviewed by two cardiologists. Continuous ECG monitoring was performed from 0 to 6 h. To assess the cardiovascular effect, a QTc interval longer than 450 ms was considered as QTc prolongation.17 Adverse event (AE) monitoring, vital signs, orthostatic blood pressure, 12-lead ECG/continuous ECG monitoring, computerized impedance cardiography, and clinical laboratory tests were assessed to evaluate the tolerability.
Genotyping
DNA was extracted from whole blood using the Gentra Puregene DNA Isolation Kit (Qiagen, Hilden, Germany). A total of 1,936 single nucleotide polymorphisms of 225 PK-related genes were analyzed by DMET™ Plus (Affymetrix, Santa Clara, CA, USA) at DNA Link. Co. Ltd. (Seoul, Korea). In this study, five cytochrome-P450 (CYP) enzymes (CYP2C19, CYP2C8, CYP2D6, CYP3A4, and CYP3A5) were selected based on nonclinical study results to evaluate the PK variability according to the genetic polymorphism. For the CYP2C19 genotype, subjects were classified into four groups: ultra-rapid metabolizer (UM) (*17/*17, *1/*17), extensive metabolizer (EM) (*1/*1), intermediate metabolizer (IM) (*1/*2, *1/*3), and poor metabolizer (PM) (*2/*2, *3/*3, *2/*3). For the CYP2D6 genotype, *1/*1, *2/*2, *1/*2, *1/*10, *2/*10, *1/*41, and *2/*41 were classified as EM, and *10/*10, *10/*41, and *2/*14 as IM. The CYP3A4 genotype was classified into the following two groups: EM (*1/*1) and IM (*1/*16). The CYP3A5 genotype was categorized into three groups: EM (*1/*1), IM (*1/*3), and PM (*3/*3).
Statistical analysis
SPSS 22.0 (SPSS Korea, Seoul, Korea) was used for statistical analysis. For dose proportionality evaluation, dose-normalized Cmax (Cmax/D) and dose-normalized AUC0–t (AUC0–t/D) were compared using analysis of variance (ANOVA), and log-transformed Cmax and AUC0–t values were tested by linear regression using a power model.
The PK parameters such as Cmax/D and AUC0–t/D were based on the log-normal distribution, and the MRs of M1, M4, and M5 were were based on the normal distribution. The Cmax/D, AUC0–t/D, and MRs of M1, M4, and M5 were compared according to the genotype of the selected genes. Associations between CYP enzyme genotypes and the PK parameters of systemic exposure were evaluated using a linear model with dose administered as the independent variable.
The maximum QTcB (QTcBmax) and QTcF (QTcFmax) intervals of each dosage group were compared with the placebo group using one-way ANOVA followed by post hoc analyses with Dunnett’s t-test, and the maximum changes in QTc intervals from baseline were compared with the placebo group using Welch ANOVA with Dunnett’s T3 post hoc comparison test. The homogeneity of variance was confirmed by Levene’s test. To assess the correlations between the QTc interval and systemic exposures of DA-8031 and its metabolites, partial correlation coefficients adjusted to each dosage group were used. Statistical significance was defined as P<0.05.
Results
Demographic characteristics
A total of 72 healthy Korean male subjects were enrolled with two subjects dropping out. In the 70 subjects, the age, weight, height, and body mass index were 26.1±4.4 years, 68.0±5.2 kg, 174.3±5.0 cm, and 22.4±1.4 kg/m2 (mean ± standard deviation), respectively, and these demographic characteristics of the subjects were not significantly different among the dosage groups (P=0.513, P=0.587, P=0.615, and P=0.139, respectively).
PK characteristics
The mean plasma concentration–time profiles for DA-8031 after a single oral dose are described in Figure 2. Two subjects in the 120 mg dose group were excluded from PK evaluation due to vomiting within 2 h after drug administration. Following a single dose, DA-8031 reached peak plasma concentration at median time of 2–3 h. The mean terminal elimination half-life and apparent clearance of DA-8031 were 17.9–28.7 h and 23.7–73.0 L/h, respectively (Table 1). Regarding urinary excretion, the fraction of unchanged drug excreted in urine (fe) of DA-8031 was 0.09–0.18, and the mean renal clearance (CLR) was 3.7–5.6 L/h. The fe and CLR were not significantly different over the entire dosage range (P=0.156 and P=0.269, respectively).
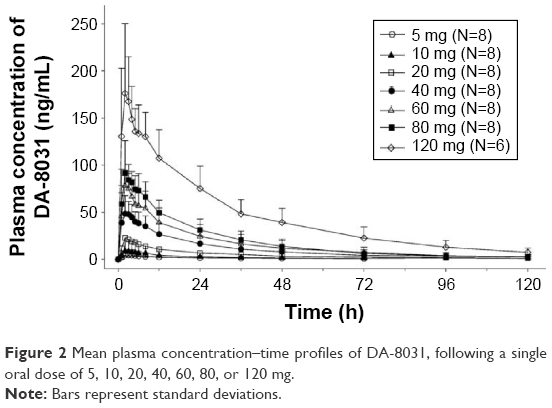 | Figure 2 Mean plasma concentration–time profiles of DA-8031, following a single oral dose of 5, 10, 20, 40, 60, 80, or 120 mg. |
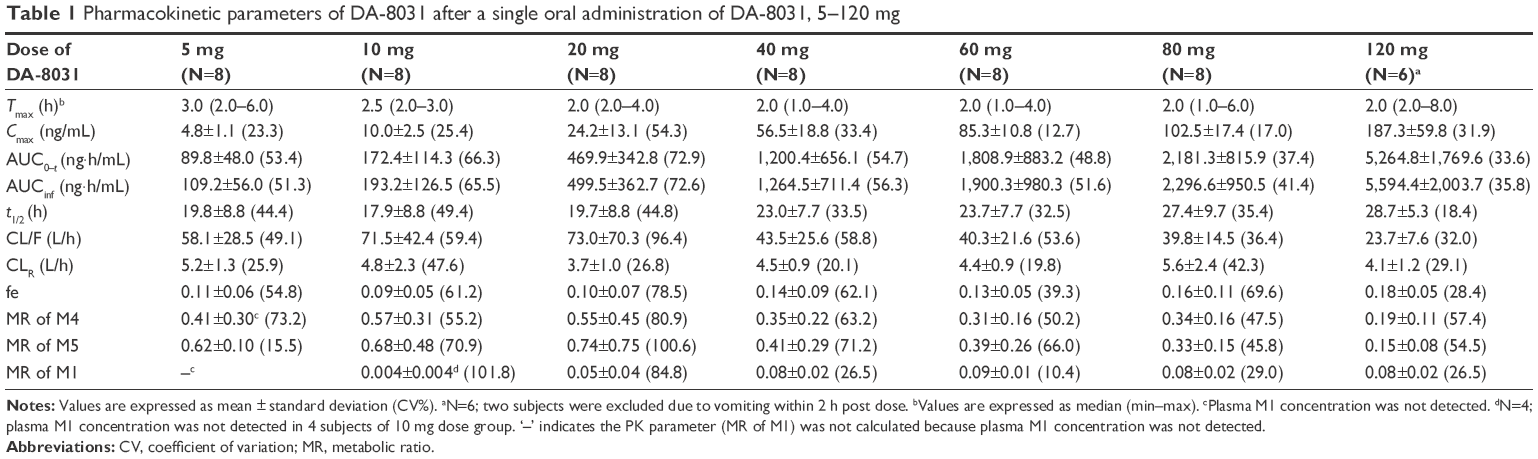 | Table 1 Pharmacokinetic parameters of DA-8031 after a single oral administration of DA-8031, 5–120 mg |
Regarding dose proportionality, the 95% confidence intervals (CIs) of the slope of Cmax and AUC0–t in the power model were 1.07–1.23 and 1.14–1.43, respectively, in 5–120 mg dose range. Significant differences of the dose-normalized PK parameters were observed among all dose ranges (P=0.043 for Cmax/D and P=0.015 for AUC0–t/D). However, in the 20–80 mg dose group, the estimated slopes (95% CI) of Cmax and AUC0–t were 1.15 (0.92–1.38) and 1.30 (0.87–1.70), respectively. In the ANOVA analysis, Cmax/D and AUC0–t/D were not significantly different within the dose range of 20–80 mg (P=0.710 and P=0.788, respectively). On the basis of these results, dose-proportional PK characteristics were observed over the dose range of 20–80 mg.
For the pharmacokinetics of the DA-8031 metabolites, the Cmax of M4 was highest followed by M5 and M1. The AUC0–t of M5 was greater than that of M4 at a dose <80 mg; however, M4 was greater than M5 inversely at the 120 mg dose. M4 and M1 were produced faster than M5, and M4 was eliminated more rapidly than M5 and M1 (Figure S1). The plasma M2 concentration was too low compared with the other metabolites to estimate any PK characteristics. The MRs of M4, M5, and M1 were 0.19–0.57, 0.15–0.74, and 0.004–0.09, respectively (Table 1). The MRs of M4 and M1 were not different among the dose groups (P=0.124 and P=0.067, respectively); however, a significant difference was observed in M5 (P=0.022). In the dose range of 20–80 mg, the MRs of M4 and M5 were not different (P=0.290 and P=0.623), but the MR of M1 was different among the doses (P=0.049). Of the administered oral doses of DA-8031, 10.7%–16.5% and 5.1%–9.9% were excreted in the urine as the metabolites M4 and M5, respectively.
Effect of genetic polymorphism on pharmacokinetics
There were 19 IM subjects and 35 EM subjects according to the CYP2D6 genotype. The dose-normalized Cmax and AUC0–t values as well as the MRs of M1, M4, and M5 were significantly different according to CYP2D6 genotype (Table 2). The mean dose-normalized Cmax and AUC0–t values were significantly higher, and the MRs of M4 and M5 were lower in the IM CYP2D6 subjects compared to EM subjects. CYP2C19, CYP3A4, and CYP3A5 did not exert a significant effect on pharmacokinetics. CYP2C8 was excluded from the analysis, because all subjects had the same genotype (wild-type).
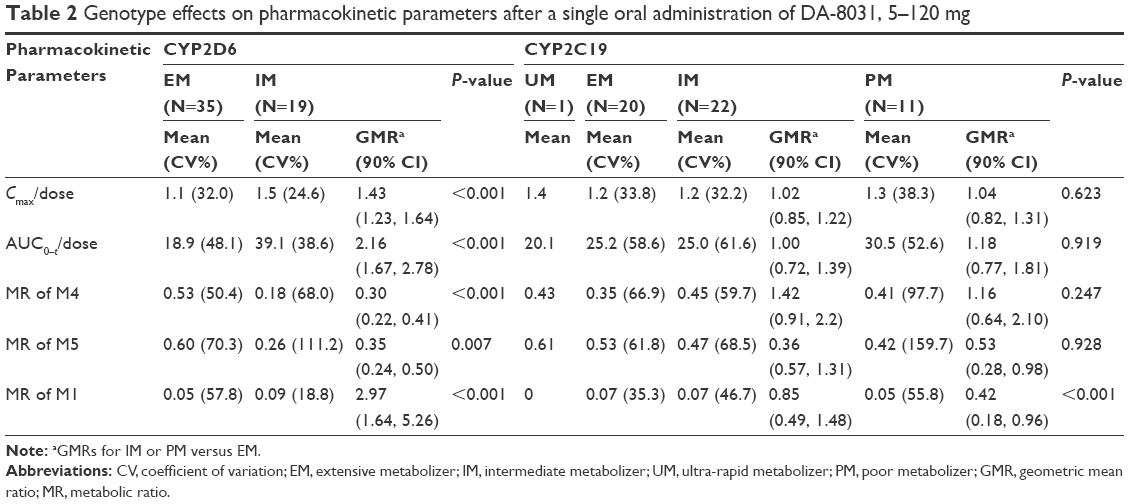 | Table 2 Genotype effects on pharmacokinetic parameters after a single oral administration of DA-8031, 5–120 mg |
Safety and tolerability
The 63 drug-related AEs occurred in 27 subjects (54 in 21 DA-8031 subjects and 9 in 6 placebo subjects). All AEs were of mild or moderate severity, and the moderate AEs were 1 case of eye pain and 4 cases of nausea in the highest (120 mg) dosage group. The most frequent drug-related AE associated with DA-8031 was nausea (11 cases) followed by decreased orthostatic blood pressure (9 cases). The frequency and intensity of AEs considered to be DA-8031 related increased according to the administered dose. None of the subjects either developed any serious AEs or discontinued the study due to an AE.
Regarding vital signs and orthostatic blood pressure, a decrease of systolic blood pressure (>20 mmHg) or a decrease of diastolic blood pressure (>10 mmHg) was observed in 15 of 71 subjects. The frequency was not significantly different among the placebo and multiple DA-8031 dose groups, and no accompanying symptoms were reported.
Prolonged QTcB and QTcF intervals longer than 450 ms were observed in the groups receiving a 60 mg dose or higher. The frequency of QTc prolongation was highest in the 120 mg group (Table 3), and the mean QTcBmax and QTcFmax were longer than 450 ms in the 120 mg dose group (Table 3 and Figure 3). The maximum changes in both the QTcB and QTcF from baseline (predose) were significantly different between the 120 mg dose of DA-8031 and placebo (QTcB interval: P<0.001; QTcF interval: P<0.001) (Table 3). The 90% CI of the difference between DA-8031 and placebo was 21–55 ms for the maximum change in QTcB and 16–60 ms for the maximum change in QTcF. These differences were significant according to the dose groups, but genetic variation of CYP2C19 and CYP2D6 had little effect on QTc changes between DA-8031 and placebo.
 | Table 3 Effect of DA-8031 on the QTcB and QTcF intervals after a single oral administration of placebo or DA-8031, 5–120 mg |
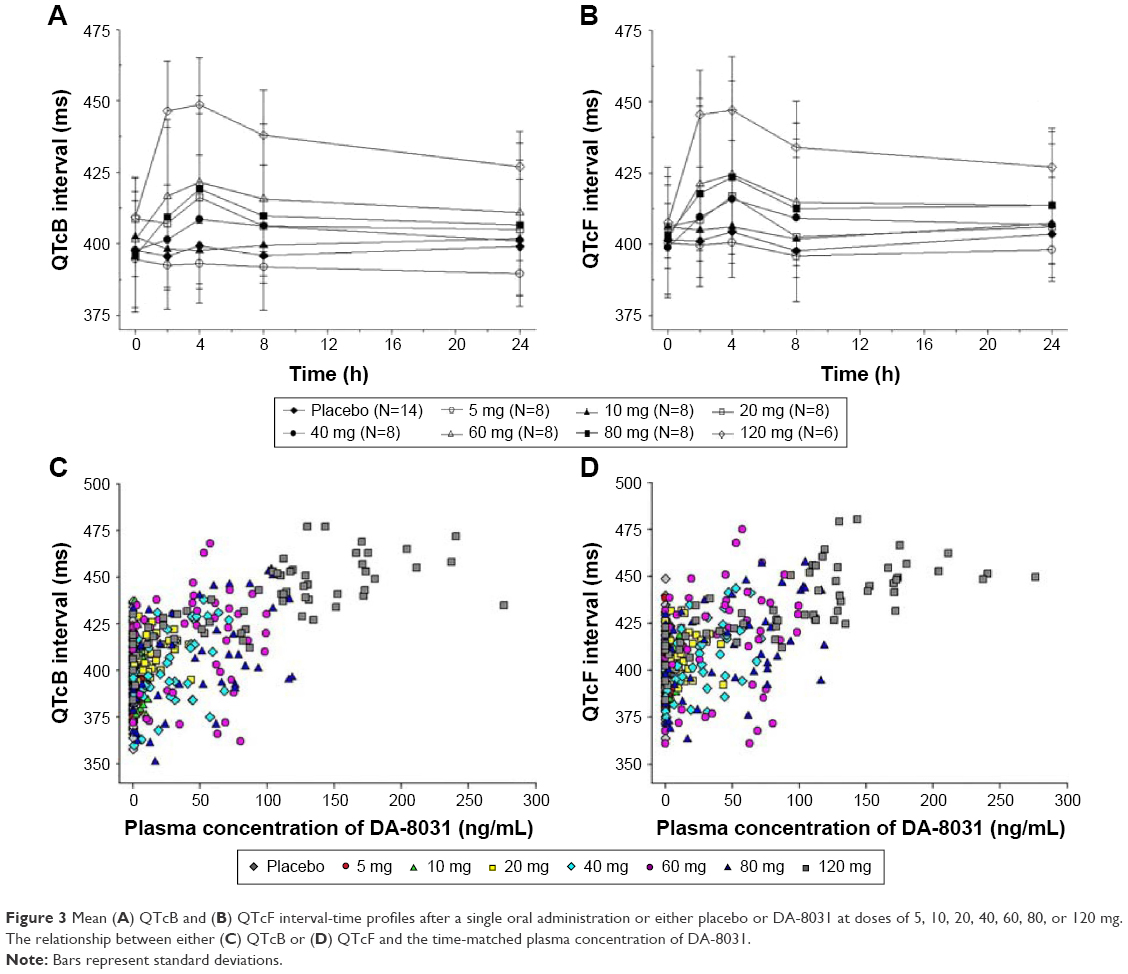 | Figure 3 Mean (A) QTcB and (B) QTcF interval-time profiles after a single oral administration or either placebo or DA-8031 at doses of 5, 10, 20, 40, 60, 80, or 120 mg. The relationship between either (C) QTcB or (D) QTcF and the time-matched plasma concentration of DA-8031. |
Considering the possibility of the QTc interval overestimating and inaccurately detecting a terminal T-wave based on the automated 12-lead ECG Analysis Program, most of the case of prolonged QTc over 450 ms in the dose range of 60–80 mg were clinically insignificant after review of the ECG findings by two independent cardiologists. As a result, QTc prolongation was thought to be a concerning issue only when 120 mg DA-8031 was administered.
Discussion
In this first-in-human clinical study, the pharmacokinetics and safety profiles of single ascending dose of DA-8031 up to 120 mg were provided. DA-8031 was rapidly absorbed at a median time of 2–3 h and eliminated with a mean terminal t1/2 of 17.9–28.7 h, both of which are suitable for a once-daily regimen.
After single oral administration of DA-8031, the Cmax and AUC0–t for plasma DA-8031 levels increased in dose-proportional manner within the range of 20–80 mg. When a 120 mg dose of DA-8031 was administered, the Cmax and AUC0–t increased supra-proportionally, and the CL/F was reduced compared to lower doses. Since CLR was similar over the entire range of doses, the increased exposure of 120 mg of DA-8031 was presumably due to the reduced hepatic clearance by the saturation of enzyme capacity.18
Genetic polymorphisms in CYP2D6 and CYP2C19 were involved in the metabolism of DA-8031 and can exert influence on the individual systemic exposure of SSRIs.19 Furthermore, genetic polymorphisms also could affect the risk profile for side effects and the overall efficacy of the medication via PK variability of the parent drug and its active metabolites.19 In the studies with human cDNA-expressed CYPs, DA-8031 was primarily metabolized by CYP enzymes. After oral administration, DA-8031 was directly metabolized to M4 and M1 followed by M5, which is formed from M4 and M1 (investigator’s brochure, unpublished data on file). CYP2C8, 2C19, 2D6, and 3A4 played a role in the production of M4 and M1, and CYP2D6 was involved in the formation of M5 (investigator’s brochure, unpublished data on file). In addition, the formation of M1 and M4 was mainly inhibited under the presence of the CYP2C8 antibody and the CYP2D6 antibody, respectively (investigator’s brochure, unpublished data on file). In the present study, the metabolic parameters were significantly different according to the CYP2C19 and CYP2D6 genotypes. The differences in the exposure of the parent drug DA-8031 and the MRs for all metabolites (M1, M4, and M5) were observed in the two CYP2D6 genotypes (EM and IM). The subjects with CYP2D6 EM had lower Cmax and AUC0–t values, higher MRs, and a shorter t1/2 than the subjects with CYP2D6 IM. On the other hand, the CYP2C19 genotype had a limited effect on the metabolism of M1, which is a minor metabolite. These results suggest that the genetic variability of CYP2D6 could have an effect on the PK variability of DA-8031, leading to possible variations in the frequencies in AE occurrences and which would ultimately have an effect on optimized treatment based on different systemic exposure to DA-8031.19,20
Regarding safety and tolerability, DA-8031 was well tolerated up to 80 mg; the most commonly reported AEs were nausea, dizziness, headache, or prolonged QT based on an ECG. These findings were consistent with previous reports that AEs during SSRI therapy were dose dependent and that profiles of AEs such as nausea, diarrhea, and dizziness were caused by serotonergic effects.20,21 With respect to QTc prolongation, although SSRIs are not generally regarded as QTc-prolonging agents, occasional cases of QTc prolongation with SSRI and even fatal cardiotoxicities such as torsade de pointes have been reported.22 In a preclinical study, a high dose of DA-8031 induced hERG current inhibition and QT prolongation. The QTc interval, which was longer than 450 ms, was associated with the development of a dangerous arrhythmia.23 In our results, QTc intervals longer than 450 ms were prominently observed in groups receiving a 120 mg dose. The QTc interval after administration of DA-8031 was prolonged proportionally to the systemic exposure of DA-8031 rather than its metabolites (Figure 3), but influence according to the CYP2C19 and CYP2D6 genotypes on the QTc prolongation were not significant. Because QTc prolongation could be influenced by lipophilicity or bulkiness of the molecular structure due to varying degrees of potassium channel-blocking activity, the polar metabolites might have a reduced effect on the QTc interval.24 Prolonged QTc associated with SSRI is known to be relatively small and clinically insignificant in patients with no other risk factors such as old age, female gender, organic heart disease, hypokalemia, and a history of long QT or drug-induced arrhythmias. Moreover, the pharmacologically active dose for DA-8031 was expected to be 20–40 mg in humans (investigator’s brochure, unpublished data on file). Based on these results, the influence of DA-8031 on QTc prolongation would be limited in expected active dose ranges.25
Conclusion
We investigated the pharmacokinetics and safety of DA-8031 as a new SSRI for the treatment of PE in humans. After a single oral dose, DA-8031 was rapidly absorbed and eliminated by first-order kinetics. PK linearity was suggested within the dose range of 20–80 mg. CYP2D6 was presumed to be involved in the metabolism of DA-8031, and polymorphisms of CYP2D6 had an effect on the systemic exposure and the MR of DA-8031. Single oral administration of DA-8031 was well tolerated up to 80 mg. DA-8031 doses of 120 mg may cause QTc prolongation, which should be considered in the design of further studies evaluating the safety and efficacy of DA-8031 in PE patients.
Acknowledgments
This study was sponsored by Dong-A ST, Seoul, Korea. The investigation was conducted at the Clinical Trials Center, Seoul National University Hospital.
Disclosure
Mi Young Bahng is an employee of Dong-A ST. The other authors report no conflicts of interest in this work.
References
Althof SE, McMahon CG, Waldinger MD, et al. An update of the International Society of Sexual Medicine’s guidelines for the diagnosis and treatment of premature ejaculation (PE). J Sex Med. 2014;11(6):1392–1422. | ||
Association AP. The diagnostic and statistical manual of mental disorders. 5th edn. Washington, DC: American Psychiatric Association; 2013. | ||
Porst H, Montorsi F, Rosen RC, Gaynor L, Grupe S, Alexander J. The Premature Ejaculation Prevalence and Attitudes (PEPA) survey: prevalence, comorbidities, and professional help-seeking. Eur Urol. 2007;51(3):816–823; discussion 824. | ||
Shaeer O. The global online sexuality survey (GOSS): The United States of America in 2011 Chapter III – premature ejaculation among English-speaking male Internet users. J Sex Med. 2013;10(7):1882–1888. | ||
Shaeer O, Shaeer K. The Global Online Sexuality Survey (GOSS): ejaculatory function, penile anatomy, and contraceptive usage among Arabic-speaking Internet users in the Middle East. J Sex Med. 2012;9(2):425–433. | ||
Donatucci CF. Etiology of ejaculation and pathophysiology of premature ejaculation. J Sex Med. 2006;3(Suppl 4):303–308. | ||
Waldinger MD. The neurobiological approach to premature ejaculation. J Urol. 2002;168(6):2359–2367. | ||
Jeon HJ, Kim HS, Lee CH, et al. Candidate molecule for premature ejaculation, DA-8031: in vivo and in vitro characterization of DA-8031. Urology. 2011;77(4):1006 e1017–1021. | ||
Kang KK, Ahn GJ, Sung JH, Kim SH, Kim H, Lee S. Ejaculatory responses are inhibited by a new chemical entity, DA-8031, in preclinical rodent models of ejaculation. Urology. 2013;81(4):920 e913–928. | ||
Hatzimouratidis K, Amar E, Eardley I, et al. Guidelines on male sexual dysfunction: erectile dysfunction and premature ejaculation. Eur Urol. 2010;57(5):804–814. | ||
Waldinger MD, Schweitzer DH, Olivier B. On-demand SSRI treatment of premature ejaculation: Pharmacodynamic limitations for relevant ejaculation delay and consequent solutions. J Sex Med. 2005;2(1):121–131. | ||
Serefoglu EC, Saitz TR. New insights on premature ejaculation: a review of definition, classification, prevalence and treatment. Asian J Androl. 2012;14(6):822–829. | ||
Xin ZC, Zhu YC, Yuan YM, et al. Current therapeutic strategies for premature ejaculation and future perspectives. Asian J Androl. 2011;13(4):550–557. | ||
Ferguson JM. SSRI antidepressant medications: adverse effects and tolerability. Prim Care Companion J Clin Psychiatry. 2001;3(1):22–27. | ||
Kang KK, Sung JH, Kim SH, Lee S. Effect of DA-8031, a novel oral compound for premature ejaculation, on male rat sexual behavior. Int J Urol. 2014;21(3):325–329. | ||
Park HS, Jung IS, Lim NH, et al. Proof of mechanism study of a novel serotonin transporter blocker, DA-8031, using [11C]DASB positron emission tomography and in vivo microdialysis. Urology. 2014;84(1):245 e241–e247. | ||
Rautaharju PM, Surawicz B, Gettes LS, et al. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol. 2009;53(11):982–991. | ||
Shand DG, Hammill SC, Aanonsen L, Pritchett EL. Reduced verapamil clearance during long-term oral administration. Clin Pharmacol Ther. 1981;30(5):701–706. | ||
Probst-Schendzielorz K, Viviani R, Stingl JC. Effect of cytochrome P450 polymorphism on the action and metabolism of selective serotonin reuptake inhibitors. Expert Opin Drug Metab Toxicol. 2015;11(8):1219–1232. | ||
Preskorn SH. Clinically relevant pharmacology of selective serotonin reuptake inhibitors. An overview with emphasis on pharmacokinetics and effects on oxidative drug metabolism. Clin Pharmacokinet. 1997;32(Suppl 1):1–21. | ||
Pryor JL, Althof SE, Steidle C, et al. Efficacy and tolerability of dapoxetine in treatment of premature ejaculation: an integrated analysis of two double-blind, randomised controlled trials. Lancet. 2006;368(9539):929–937. | ||
Haddad PM, Anderson IM. Antipsychotic-related QTc prolongation, torsade de pointes and sudden death. Drugs. 2002;62(11):1649–1671. | ||
Metzger E, Friedman R. Prolongation of the corrected QT and torsades de pointes cardiac arrhythmia associated with intravenous haloperidol in the medically ill. J Clin Psychopharmacol. 1993;13(2):128–132. | ||
De Ponti F, Poluzzi E, Montanaro N. QT-interval prolongation by non-cardiac drugs: lessons to be learned from recent experience. Eur J Clin Pharmacol. 2000;56(1):1–18. | ||
Beach SR, Kostis WJ, Celano CM, et al. Meta-analysis of selective serotonin reuptake inhibitor-associated QTc prolongation. J Clin Psychiatry. 2014;75(5):e441–e449. |
Supplementary material
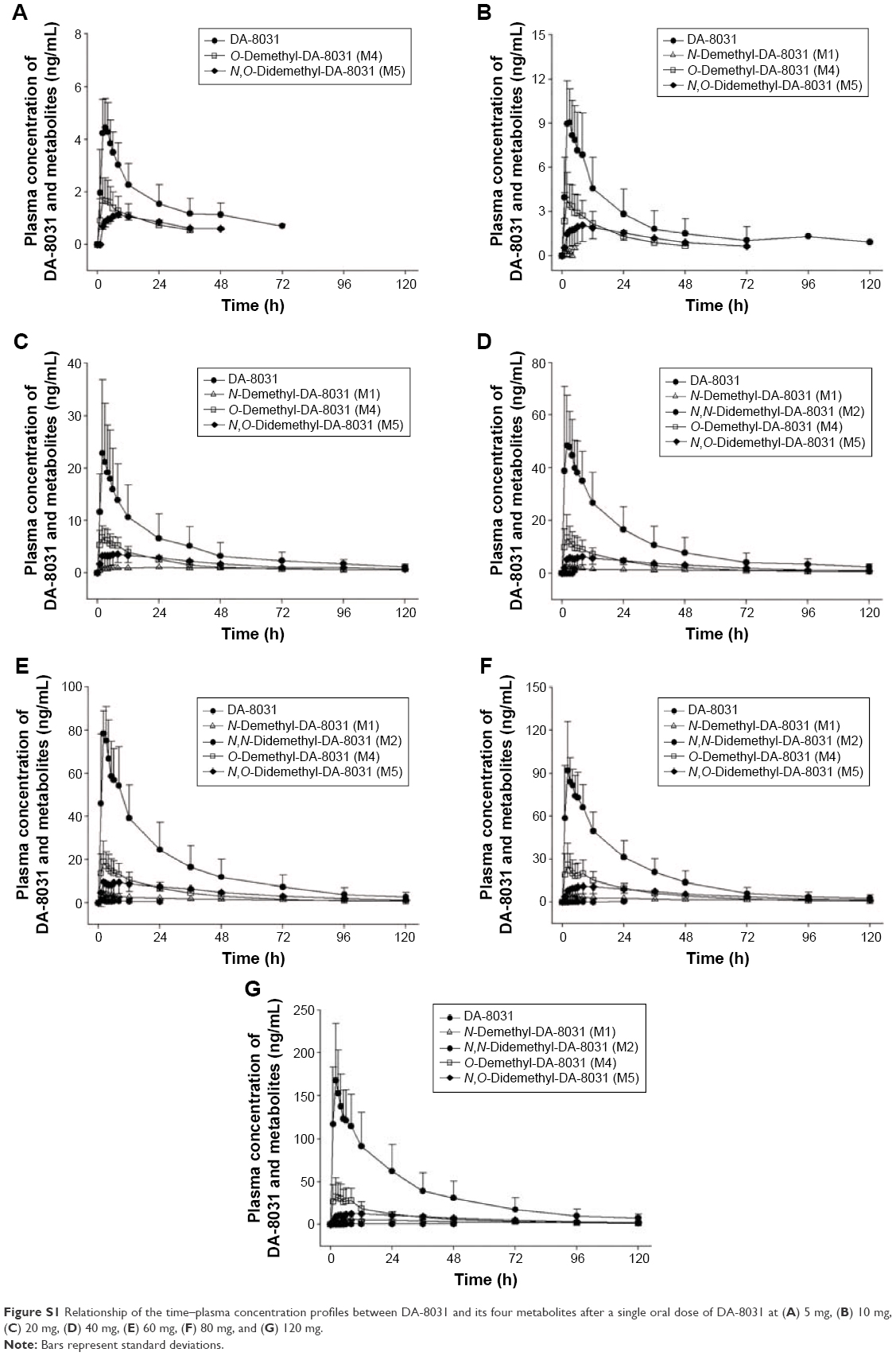 | Figure S1 Relationship of the time–plasma concentration profiles between DA-8031 and its four metabolites after a single oral dose of DA-8031 at (A) 5 mg, (B) 10 mg, (C) 20 mg, (D) 40 mg, (E) 60 mg, (F) 80 mg, and (G) 120 mg. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.