")
Back to Journals » International Journal of Nanomedicine » Volume 11
Pharmacokinetics and safety of DTS-108, a human oligopeptide bound to SN-38 with an esterase-sensitive cross-linker in patients with advanced malignancies: a Phase I study
Authors Coriat R, Faivre SJ, Mir O, Dreyer C, Ropert S, Bouattour M , Desjardins R, Goldwasser F, Raymond E
Received 10 April 2016
Accepted for publication 1 June 2016
Published 21 November 2016 Volume 2016:11 Pages 6207—6216
DOI https://doi.org/10.2147/IJN.S110274
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Romain Coriat,1 Sandrine J Faivre,2 Olivier Mir,3 Chantal Dreyer,2 Stanislas Ropert,3 Mohammed Bouattour,2 Robert Desjardins,4 François Goldwasser,3 Eric Raymond5
1Gastroenterology and Digestive Oncology Unit, Cochin Teaching Hospital, Université Paris Descartes Sorbonne Paris Cité, Paris, 2Department of Medical Oncology, Beaujon Teaching Hospital, Université Paris Diderot, Paris 7, Clichy, 3Department of Medical Oncology, Cochin Teaching Hospital, Université Paris Descartes Sorbonne Paris Cité, Paris, France; 4Drais Pharmaceuticals, Bridgewater, NJ, USA; 5Groupe Hospitalier Paris Saint-Joseph, Paris, France
Background: DTS-108 is a hydrosoluble prodrug, where the SN-38 moiety is covalently linked to a 20-amino acid vector peptide by a specific esterase-sensitive cross-linker, releasing 7-ethyl-10-hydroxycampthotecin (SN-38) by esterase bond cleavage.
Methods: The pharmacokinetics of DTS-108, adverse events graded according to NCI-CTCv3.1, dose-limiting toxicities at cycle 1, the maximum tolerated dose (MTD), and the recommended Phase II dose (RP2D) of intravenous DTS-108 (1–2 hours) every 2 weeks were evaluated in a first-in-human Phase I study in patients with advanced/metastatic carcinomas, according to an accelerated dose escalation design. SN-38 and SN-38 glucuronide (SN-38G) levels were evaluated with fluorescence high-performance liquid chromatography (HPLC) test, then liquid chromatography–tandem mass spectrometry (LC/MS/MS) methods.
Results: Forty-two patients received DTS-108 across 14 dosing cohorts (range 3–416 mg/m2). At 416 mg/m2, three out of six patients had grade 4 neutropenia thereby defining the MTD and the RP2D at 313 mg/m2. Fluorescence HPLC was inaccurate to quantify DTS-108 and its metabolites (SN-38 and SN-38G). New processes and analytical LC/MS/MS methods for testing SN-38 were implemented. At a dose of 313 mg/m2, mean DTS-108, SN-38, and SN-38G area under the plasma concentration–time curve to infinity (coefficients of variation %) were 439,293 (24%), 1,992 (34%), and 4,538 (46%) h·ng/mL. Stable disease (according to Response Evaluation Criteria in Solid Tumors) was observed in nine patients.
Conclusion: Assessing SN-38 concentration using fluorescence HPLC is questionable since this method failed to monitor dose escalation of DTS-108, a new topoisomerase I inhibitor, due to ex vivo degradation. LC/MS/MS methods were consistent in evaluating SN-38 exposures allowing drug monitoring. The maximum tolerated dose of DTS-108 was 416 mg/m2. The RP2D for intravenous DTS-108 was 313 mg/m2 every 2 weeks in patients with advanced/metastatic solid tumors.
Keywords: Phase I, topoisomerase I inhibitor, irinotecan, fluorescence HPLC
Introduction
7-Ethyl-10-hydroxycampthotecin (SN-38) is a very potent topoisomerase I inhibitor that exhibits antiproliferative and proapoptotic effects in a large number of human carcinoma models. The poor solubility1,2 and the toxicity profile of SN-38 manifesting as diarrhea and bone marrow toxicity3–5 rendered this molecule unsuitable for human delivery. Irinotecan (CPT-11, Campto®, Camptosar®, Pfizer, Inc., New York, NY, USA) appeared as a paradigm for prodrug delivery of SN-38, exhibiting a broad spectrum of antitumor activity6,7 but retaining diarrhea and neutropenia as dose-limiting toxicities (DLTs).8 Irinotecan has intrinsically very poor antitumor effects but can be converted into SN-38 by carboxylesterases that are predominantly present in the liver.9 SN-38 generated from irinotecan is eventually glycuro-conjugated for metabolic degradation by UGT1A1.10,11 The metabolic biotransformation of irinotecan into SN-38 was shown to be variable among individuals and poorly efficient, with only 2%–8% of the administered dose of irinotecan being converted into SN-38 in humans.12 Another limitation has been the occurrence of delayed, severe, and potentially life-threatening diarrhea that is thought to be due to SN-38 accumulation in the bowel. Severe diarrhea was observed in up to 40% of treated patients in early clinical trials,13–15 and this toxicity has been a dose-limiting factor to explore high dose of irinotecan.16,17 Subsequently, attempts were made to lower biliary excretion of SN-38, limit glycuro-conjugation, and increase the area under the plasma concentration–time curve (AUC) of SN-38, thereby increasing activity and reducing systemic toxicity.18–20
Over the last 15 years, while topoisomerase I inhibitors have been recognized as major anticancer drugs for several malignancies, it has become apparent that significant clinical advantages could be derived from the direct administration of SN-38. Novel drug delivery technologies of SN-38 were engineered to bypass hepatic bioactivation and catabolism, while intending to reach high plasma and tissue exposure. Novel drug delivery techniques also sought to limit gastrointestinal toxicity and interpatient pharmacokinetic (PK) variability.21 Based on this rationale, a novel water-soluble conjugate (DTS-108) was developed by linking SN-38 to a highly charged 20-amino acid oligopeptide of human origin (Figure 1).22 The SN-38 oligopeptide cross-linker can be cleaved chemically by esterases that are primarily found in blood, in contrast to irinotecan, which is cleaved primarily by hepatic carboxylesterases releasing SN-38 in the liver where it is subsequently glucuronidated and secreted in bile as SN-38G. This technology sought to enhance tumor delivery properties by taking advantage of the PK of the highly soluble oligopeptide in the plasma and reducing SN-38 toxicity by lowering the systemic exposure of SN-38 especially in the bone marrow and gut.21 In preclinical models, DTS-108 was able to deliver significantly higher plasma levels of SN-38 with lower toxicity compared to irinotecan, resulting in a broader therapeutic potential. Based on these encouraging preclinical results, we conducted a Phase I study based on PK and tolerability assessments with the aim of ascertaining the DLTs, the maximal tolerated dose (MTD), and the recommended Phase II dose (RP2D) of DTS-108 in patients with advanced malignancies.
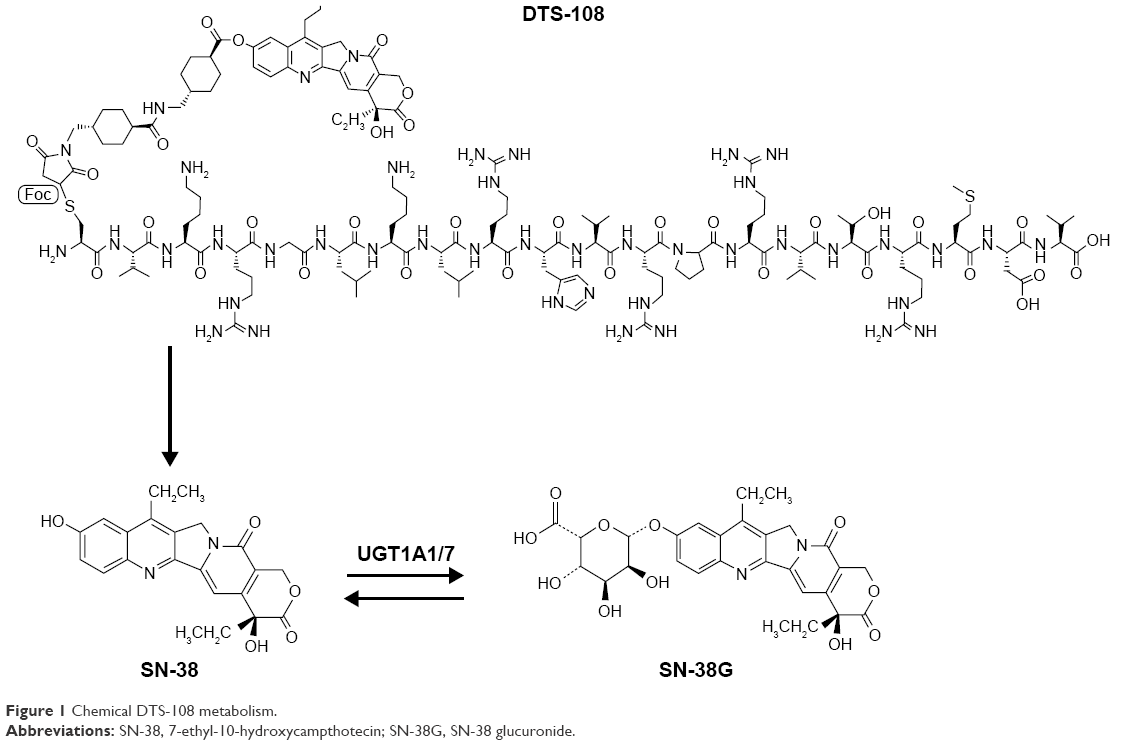 | Figure 1 Chemical DTS-108 metabolism. |
Patients and methods
Patient selection
This first-in-human Phase I clinical trial was designed as a dose-escalation study of DTS-108 given as a single agent on a bi-weekly schedule until progression, unacceptable toxicity, and/or refusal in patients with advanced malignancies. The study was conducted in accordance with International Conference on Harmonization good clinical practice standards with approval by a French ethical committee (Affsaps). All patients provided written informed consent according to the National and International guidelines.
Patients eligible for this study met the following criteria: histologically confirmed diagnosis of solid tumor refractory to standard therapy or for which no standard therapy existed; age >18 years; life expectancy ≥3 months; Eastern Cooperative Oncology Group performance status <2; no chemotherapy, hormonal therapy, immunotherapy, or radiotherapy within 4 weeks before treatment with DTS-108 and no immunosuppressive agents within 3 weeks preceding study entry; and adequate renal function with creatinine clearance ≥60 mL/min/1.73 m2. Based on known toxicities of campthotecin derivatives, patients who entered into this trial were also required to have no chronic severe diarrhea at baseline and no history of inflammatory bowel disease.
Pretreatment and follow-up examinations
Toxicity was evaluated over bi-weekly clinical and laboratory examinations and graded using the National Cancer Institute Common Toxicity Criteria, version 3.1 (http://ctep.cancer.gov). Tumors were measured by computed tomography scans 4 weeks before starting DTS-108 and then every 6 weeks (three cycles) and at the end of the treatment or at the withdrawal visit. Tumor response was evaluated according to Response Evaluation Criteria in Solid Tumors, v1.023 in four categories: complete response, partial response, stable disease, and disease progression.
Drug profile and administration
The synthesis of DTS-108 was conducted with the 20-amino acid DPV1047 Vectocell peptide (CVKRGLKLRHVRPRVTRMDV), which was synthesized as a trifluoroacetate salt by NeoMPS Inc. (San Diego, CA, USA) as previously published.21 DTS-108 is a soluble prodrug of the topoisomerase inhibitor SN-38, the active metabolite of irinotecan. In DTS-108, the SN-38 moiety is covalently linked to a 20-amino acid peptide through a specific cross-linker, which allows the release of SN-38 through esterase bond cleavage. DTS-108 was designed to achieve therapeutic levels of SN-38 while reducing gastrointestinal exposure to SN-38 to reduce the incidence of severe diarrhea, one of the major DLTs associated with irinotecan administration.18–21 DTS-108 was administered by intravenous infusion over 2 hours every 2 weeks (1 day of dosing followed by 13 days of rest).
Dose-escalation procedure
This Phase I, open-label, one-arm study was conducted to evaluate the DTS-108 safety and tolerability in escalating dose cohorts of patients with advanced or metastatic solid tumors. The primary objectives of the study were to determine the DLTs, identify the MTD, and the RP2D of DTS-108.
Dose escalation occurred in two phases – accelerated and standard – following a modification of the accelerated dose titration design described by Simon et al24 and discussed in Eisenhauer et al.25 During the accelerated titration phase (dose levels 1–4), dose levels increased by 100% once safety was established at a given dose level; during the standard titration phase (dose levels 5–16), dose levels increased by 33% once safety was established. In the absence of safety issues, one patient was enrolled at each dose level during the accelerated titration phase (dose levels 1 [3 mg/m2] to 4 [24 mg/m2]), and three patients were enrolled at each dose level during the standard titration phase (dose levels 5 [32 mg/m2] to 16 [735 mg/m2]). During the standard titration phase (dose levels 5–16; three patients per dose level), escalation to the next higher dose level proceeded once all three patients completed one cycle of treatment without experiencing a DLT. The MTD was defined as the dose level at which no more than one of the six patients experienced a DLT during cycle 1, while two or more patients at the next higher dose level experienced a DLT during cycle 1.
PK methods
Testing of bioanalytical samples was performed at two laboratories (Avogadro’s Lab Supply Inc., Shamong, NJ, USA and MicroConstants Inc., San Diego, CA, USA). DTS-108, SN-38, and SN-38G have a molecular weight of 3,499 kDa, 392 kDa, and 369 kDa, respectively. DTS-108 is highly soluble in water as SN-38G while SN-38 is insoluble. DTS-108, SN-38, and SN-38G exist in lactone (pH<7) and carboxylate (pH>7) forms. Relative fluorescence response is 1.78 in lactone and 1.00 in carboxylate. A separation phase was conducted with high-performance liquid chromatography (HPLC). A detection using fluorescence was performed (excitation 370 nm, emission 540 nm) to identify SN-38 and SN-38G concentration. Initial plasma sample analysis was performed at the Avogadro’s laboratory using a HPLC fluorescence method to simultaneously quantify all three components (parent molecule DTS-108 and metabolites SN-38 and SN-38 glucuronide [SN-38G]). Based on stability experiments, the amount of SN-38 formed ex vivo during sample collection and processing was controlled with the procedures developed by MicroConstants Inc. When we noticed that the amount of SN-38 that formed immediately ex vivo in drawn sample was leading to overestimation of concentrations in the plasma, we subsequently revised the sampling process to allow immediate freezing and revised the HPLC method used. The original HPLC fluorescence method was modified, validated, and used to analyze DTS-108 and SN-38G in the plasma. The calibration curve for DTS-108 was obtained by fitting the peak height ratios of DTS-108/internal standard and the standard concentrations to a log-transformed linear equation using SoftMax Pro (Molecular Devices LLC, Sunnyvale, CA, USA). Initially, the DTS-108 peak height ratios were fit to the same regression as SN-38G. Methods were applicable for measuring concentrations of SN-38, SN-38G, and DTS-108 ranging from 1 to 500 ng/mL, 1 to 500 ng/mL, and 200 to 100,000 ng/mL, respectively.
Acidified human plasma method for HPLC with mass spectrometric detection
Human plasma samples containing SN-38, SN-38-d3 as the internal standard, tri-potassium ethylenediaminetetraacetic acid as the anticoagulant, and phosphoric acid as the preservative were diluted with water and extracted using a mixture of hexane and methyl tertiary-butyl ether. The peak heights of SN-38 and the internal standard were acquired using MassLynx v 4.1 (Waters Corporation, Milford, MA, USA).
Results
Patient characteristics
Forty-two patients were enrolled in the dose-escalation Phase I study (Table 1). Median age at enrolment was 59.2 years (range 31–66 years). The majority of patients had Eastern Cooperative Oncology Group performance status 0–1, multiple-site metastatic diseases, and experienced relapse after prior chemotherapy with a median of two prior chemotherapy regimens, with nearly all including platinum compounds.
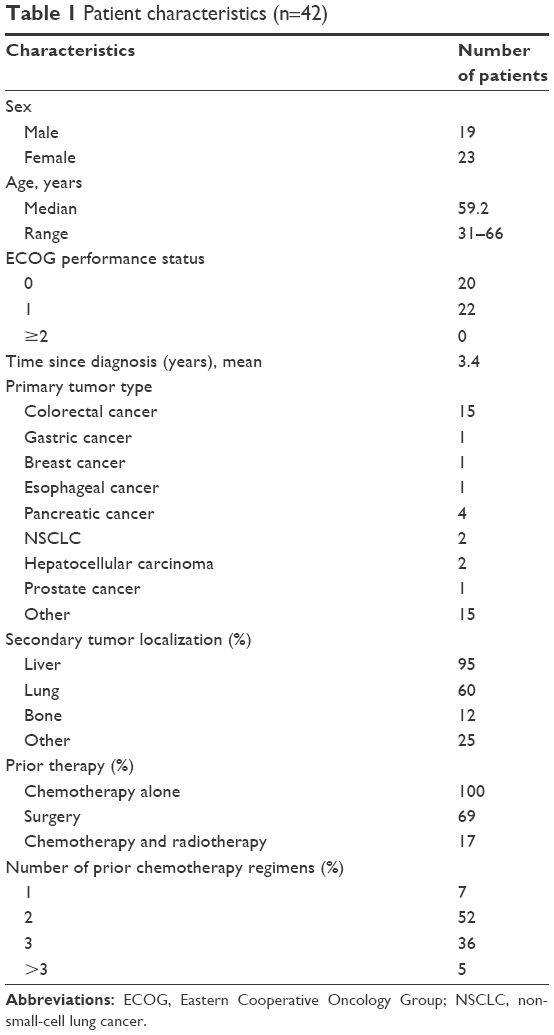 | Table 1 Patient characteristics (n=42) |
Dose-limiting toxicities
Neutropenia was the DLT of DTS-108. The incidence of treatment-emergent neutropenia by severity and dose cohort is summarized in Table 2. Decreased neutrophil counts were observed at a dose of 56 mg/m2 with increasing frequency at the dose of 416 mg/m2. Grade 3 neutropenia was observed in one of four patients at 235 mg/m2, four of six patients at 313 mg/m2, and four of six patients at 416 mg/m2. One of the patients who received DTS-108 at 313 mg/m2 and three at 416 mg/m2 did not recover to grade 1 at day 15 following the first infusion and required dose reduction. Neutropenia was therefore considered dose limiting at the dose of 416 mg/m2, and the MTD was determined as a dose of 313 mg/m2.
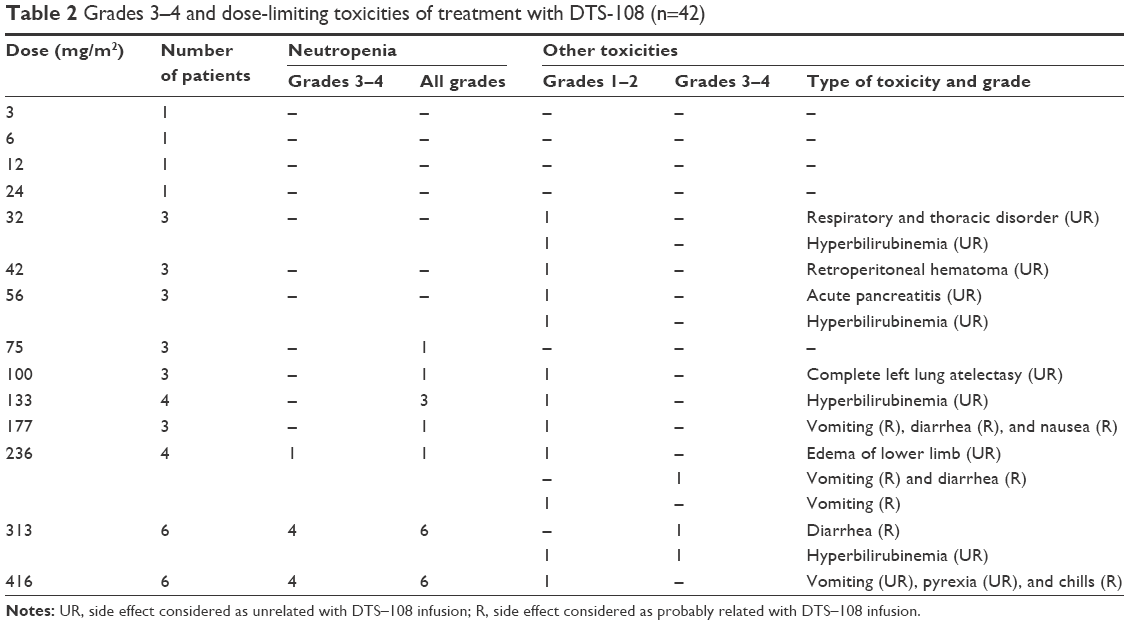 | Table 2 Grades 3–4 and dose-limiting toxicities of treatment with DTS-108 (n=42) |
Toxicity
Adverse events related to DTS-108 infusion were mainly nausea (42.9%), asthenia (42.9%), diarrhea (40.5%), vomiting (33.3%), anemia (31.0%), rash (21.4%), and anorexia (21.4%). Other adverse events were experienced in less than 20% of cases and are summarized in Table 3. As expected in the population of patients with advanced and refractory metastatic solid tumors, many of whom had liver metastases, there were numerous abnormalities in serum chemistry, especially at baseline with moderate elevations of aspartate transaminase, alanine transaminase, gamma-glutamyl transpeptidase, alkaline phosphatase, and lactate dehydrogenase. The majority of patients enrolled in this study were ultimately withdrawn due to disease progression. Three patients treated with DTS-108 doses of 32 mg/m2, 56 mg/m2, and 133 mg/m2, respectively, had elevated bilirubin levels attributed to their progressive disease. There was no evidence of nephrotoxicity associated with the administration of DTS-108. Histamine class I infusion-related reactions (itching urticaria) were observed with increasing frequency and severity at doses >75 mg/m2 and required antihistamine premedication. At doses of 313 mg/m2 and 416 mg/m2, skin reactions to infusion were seen in two of six and six of six patients, respectively.
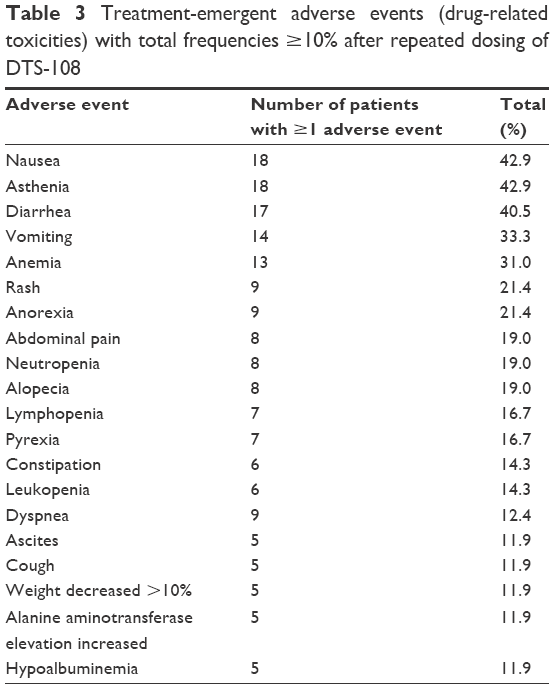 | Table 3 Treatment-emergent adverse events (drug-related toxicities) with total frequencies ≥10% after repeated dosing of DTS-108 |
DTS-108 was evaluated for potential cardiovascular effects and there were no clinically significant drug-related changes in the QRS morphology or interval measurements. There were no instances of a postdose QTc >500 ms. There was one instance of a change from baseline QTc >60 ms in one patient receiving 416 mg/m2 with a predose QTc of 391 ms and a QTc of 484 ms (+24%) at the end of dosing in cycle 3. There were four and six instances of a postdose QTc >450 ms and 60 ms > QTc >30 ms, respectively. These sporadic events bore no relationship to dose, and there was no evidence of a significant effect of DTS-108 on cardiac repolarization as reflected by the analysis of the QTc intervals or change from the baseline QTc intervals in this study.
Pharmacokinetic analysis
Patient samples from 3 mg/m2 to 42 mg/m2 were tested using the Avogadro method. During patient sample testing at Avogadro, incurred sample reanalyses failed to meet the acceptance criteria (two of three of samples within 20% of original value). The investigation on sample stability concluded that DTS-108 degrades forming SN-38 during the blood collection process, frozen plasma storage, and sample extraction process. The ex vivo degradation of DTS-108 to SN-38 did not impact the SN-38G values since this metabolite can only be generated in vivo. The potential implications for the calculation of PK parameters of DTS-108, SN-38, and SN-38G would result in lower concentrations of DTS-108, elevated concentrations of SN-38, but no change in the concentrations of SN-38G since the glucuronidation pathway is only mediated by a UGT-catalyzed metabolic process.
PK parameters for DTS-108, SN-38, and SN-38G are presented in Table S1. Despite the degradation of DTS-108, the variability in the exposure parameters (Cmax and AUCinf) was relatively low with coefficients of variation for Cmax ranging from 13% to 39% and for AUCinf from 9% to 40%. The results indicate that Cmax and AUCinf both appear to increase linearly with an increase in dose (Figure 2), except with the SN-38 Cmax.
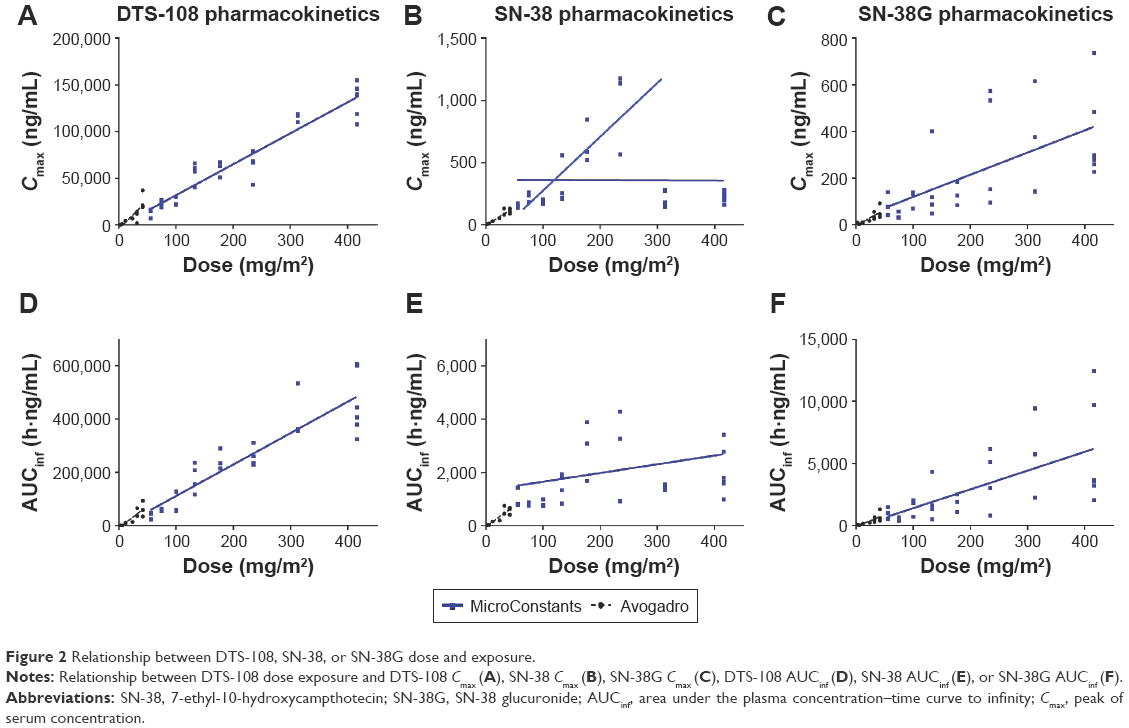 | Figure 2 Relationship between DTS-108, SN-38, or SN-38G dose and exposure. |
The ratios of the AUCinf for SN-38 to DTS-108 are the most stable for the MicroConstants’ data and an indication of the relative exposure, which ranges from 0.25% to 0.53%. As shown in Figure 2, the SN-38G results show a relatively high variability (typically >50%) at all dose levels and a trend toward an increase in exposure with an increase in dose. As expected, the Avogadro and MicroConstants analyses provided similar results, since the plasma stability issue does not affect SN-38G concentrations. The ratios of the AUCs for SN-38G and SN-38, which represent an indication of the relative conversion of SN-38 to SN-38G, are relatively constant over the dose range. On average, the range of the ratios is 0.78–2.63. Due to the elevated values of SN-38, the most reliable ratio data were obtained with the MicroConstants analysis, and these individual values ranged from 1.45 to 4.43.
The accuracy of the method was determined by comparing the mean measured concentrations with the theoretical concentrations of the compound in the quality control samples. For the DTS-108 quality control samples, the deviation of the mean from theoretical values did not exceed ±1.88%. For the SN-38 quality control samples, the deviation of the mean from theoretical values did not exceed ±5.00%. For the SN-38G quality control samples, the deviation of the mean from theoretical values did not exceed ±3.67%.
Antitumor activity
Forty-two patients received DTS-108 treatment and all were assessable for response. Tumor control (complete response + partial response + stable disease) occurred in six patients (Table 4). The median progression-free survival in the study was 11 months (95% confidence interval [CI], 3–17 months, and 95% CI). No patient experienced an objective response.
 | Table 4 Treatment-confirmed tumor response related to dose exposure |
Discussion
In this Phase I trial of DTS-108, a novel water-soluble conjugate of SN-38, the DLT was ascertained by the occurrence of grade 3 neutropenia in cycle 1 at a dose of 416 mg/m2. Limitations in dosing were further defined by dosing delays for subsequent cycles at 416 mg/m2, dose reductions to 313 mg/m2 in three of six patients, infusion reactions inadequately controlled by premedication causing dose interruptions, and prolongation of the infusion duration from 1 to 2 hours in all six patients receiving the 416 mg/m2 dose. The MTD was found to be the 313 mg/m2 dose level based on the observation that five of the six patients treated at this dose level had no dose-limiting neutropenia, and the overall improvement in the management of infusion reactions.
Vomiting and diarrhea were observed in several patients in this study including two cases of grade 3 vomiting and two cases of grade 3 diarrhea (Tables 2 and 3). Cases of grade 3 diarrhea occurred in two patients with extenuating circumstances consisting of a partial bowel obstruction due to tumor in one and a history of a total colectomy in the other. Diarrhea was less problematic with the administration of DTS-108 than historically reported at the MTD of irinotecan. When DTS-108 is administered intravenously, the cleavage of the ester bond and release of SN-38 occurred in the blood by the action of plasma esterases. This distinguishes it from irinotecan, which is cleaved primarily by hepatic carboxylesterases releasing SN-38 in the liver where it is subsequently glucuronidated and secreted in the bile as SN-38G. The SN-38G is then cleaved by bacterial glucuronidases, allowing the release of SN-38 in the bowel thereby producing diarrhea. The location of the cleavage may explain the low incidence of severe diarrhea in our study.
While objective tumor responses were not observed in this study, stable disease status was maintained for six cycles or longer in seven patients. The dose selected for subsequent evaluation in RP2D with less extensively pretreated disease is 313 mg/m2, to be administered as a 2-hour infusion, every 14 days.
Regarding the analytical part of the PK study, fluorescence HPLC was initially used to quantify DTS-108 and its metabolites (SN-38 and SN-38G). Sample stability analysis of this method showed that SN-38 values were inaccurate because DTS-108 degraded, forming ex vivo SN-38 during the blood collection, plasma storage, and/or sample extraction. New processes and analytical liquid chromatography–tandem mass spectrometry methods for testing SN-38 were implemented allowing a better evaluation of plasma levels of DTS-108 and its metabolites (SN-38 and SN-38G). SN-38 is 95% bound to plasma proteins.26 The maximum concentrations of SN-38 are reached ~1 hour after the beginning of a short intravenous irinotecan infusion.26 It has been previously identified that SN-38 plasma decay follows closely that of the parent compound with an apparent terminal half-life ranging from 6 to 30 hours.26
The total body clearance of irinotecan was estimated to be 31.6 L/h.1 The volume of distribution at steady state of irinotecan was 263 L, suggesting extensive distribution into the peripheral compartments.1 Irinotecan is cleaved primarily by hepatic carboxylesterases releasing SN-38 in the liver, where it is subsequently glucuronidated and secreted in the bile as SN-38G. DTS-108 is cleaved chemically by esterases that are found in the blood allowing a higher diffusion and taking advantage of the PK of the highly soluble oligopeptide in the plasma and reduces the SN-38 toxicity by lowering the systemic exposure of SN-38, especially in the bone marrow and gut.21 Our study reinforced the preclinical models and identified the reduced incidence of digestive toxicity while it identified skin toxicity. In animal models, DTS-108 has been identified to induce immediate effects resembling the symptoms of an infusion reaction. In dogs, these effects have always been observed with the same moderate severity and always resolved spontaneously within a few hours of infusion, over a relatively broad dose range (4.5–53 mg free base/kg). The most likely hypothesis to explain this effect was an anaphylactoid reaction to DTS-108 that would result from its ability to directly induce histamine release in vivo. This was confirmed by the demonstration of histamine release induced by DTS-108 in vitro in dog and human basophils and by measurement of the plasma histamine levels in dogs associated with the intravenous administration of DTS-108. In our study, we therefore allowed for treatment of infusion reactions, antihistamines, and/or corticosteroids. The second hypothesis of the skin toxicity would be a true allergic reaction. In line with this hypothesis, the first infusion reaction occurred in Cohort 8 during the administration of 75 mg/m2 DTS-108 in cycle 8. The manifestations resolved spontaneously after the infusion and the clinical symptoms played for a true allergic reaction.
Similar infusion reactions occurred in all three patients treated in Cohort 11 at a dose of 177 mg/m2, and in the beginning of Cohort 12 (235 mg/m2) all patients were pretreated with antihistamines. The infusion reactions were controlled in all but two of six patients treated in Cohort 13 at a dose of 313 mg/m2. However, all six patients enrolled in Cohort 14 at a dose of 416 mg/m2 experienced infusion reactions requiring interruption of the infusion in most cases and retreatment with antihistamines. The infusion reactions persisted despite a protocol amendment to allow infusion of the drug over a period of 2 hours rather than the originally specified 1-hour infusion. Infusion reactions did not cause discontinuation of any patient in Cohort 14, though three patients had dosage reductions to 313 mg/m2 for subsequent infusions due to the development of neutropenia.
At a dose of 313 mg/m2, DTS-108 allowed to reach a SN-38 AUC of 1,992 h·ng/mL, four times higher than that observed with irinotecan given at a dose of 350 mg/m2.27
To date, no such analytical limitation has been reported with other conjugated formulations of SN-38.28–31 However, our findings in this trial illustrate the potential caveats of early pharmacological evaluation of investigational new drugs, stressing the need to systematically question the relevance of analytical data.
Regarding the toxicity profile of DTS-108, neutropenia was dose limiting, whereas diarrhea was mild and easily manageable. This may be due to the differential expression of esterases in the bone marrow and digestive tract.32–34 Indeed, high local esterase activity could lead to an increased release of SN-38 from its prodrug DTS-108, thereby contributing to toxicity.
Conclusion
In conclusion, DTS-108 given as a 2-hour infusion at a dose of 313 mg/m2 is tolerated with only moderate digestive toxicity and without clinically significant hematologic toxicity. The PKs of DTS-108 showed a short t1/2 (<2 hours) and a significant exposure to SN-38 at the 313 mg/m2 dose level. Therefore, based on its preliminary antitumor activity, safety profile, and PK profile, we recommend Phase II testing of DTS-108 given as 2-hour infusions once every 2 weeks, at a dose level of 313 mg/m2.
Acknowledgments
We gratefully acknowledge the nursing staff of Departments of Medical Oncology, Hôpital Beaujon and Hôpital Cochin, for their assistance. Drais Pharmaceuticals sponsored this clinical trial.
Data have been presented at the American Society for Clinical Oncology meeting 2012 in Chicago (abstract no 2557) and at the European Society for Clinical Oncology meeting 2012 in Vienna (European Union Drug Regulating Authorities Clinical Trials registration number: 2008-004452-74).
Disclosure
RD is the director at Drais Pharmaceuticals and owns stock options in Drais Pharmaceuticals. The other authors report no conflicts of interest in this work.
References
Mathijssen RH, van Alphen RJ, Verweij J, et al. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin Cancer Res. 2001;7(8):2182–2194. | ||
Garcia-Carbonero R, Supko JG. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin Cancer Res. 2002;8(3):641–661. | ||
Gupta E, Lestingi TM, Mick R, Ramirez J, Vokes EE, Ratain MJ. Metabolic fate of irinotecan in humans: correlation of glucuronidation with diarrhea. Cancer Res. 1994;54(14):3723–3725. | ||
Araki E, Ishikawa M, Iigo M, Koide T, Itabashi M, Hoshi A. Relationship between development of diarrhea and the concentration of SN-38, an active metabolite of CPT-11, in the intestine and the blood plasma of athymic mice following intraperitoneal administration of CPT-11. Jpn J Cancer Res. 1993;84(6):697–702. | ||
Kudoh S, Fukuoka M, Masuda N, et al. Relationship between the pharmacokinetics of irinotecan and diarrhea during combination chemotherapy with cisplatin. Jpn J Cancer Res. 1995;86(4):406–413. | ||
Jaxel C, Kohn KW, Wani MC, Wall ME, Pommier Y. Structure-activity study of the actions of camptothecin derivatives on mammalian topoisomerase I: evidence for a specific receptor site and a relation to antitumor activity. Cancer Res. 1989;49(6):1465–1469. | ||
Bissery MC, Vrignaud P, Lavelle F, Chabot GG. Experimental antitumor activity and pharmacokinetics of the camptothecin analog irinotecan (CPT-11) in mice. Anticancer Drugs. 1996;7(4):437–460. | ||
Vanhoefer U, Harstrick A, Achterrath W, Cao S, Seeber S, Rustum YM. Irinotecan in the treatment of colorectal cancer: clinical overview. J Clin Oncol. 2001;19(5):1501–1518. | ||
Slatter JG, Su P, Sams JP, Schaaf LJ, Wienkers LC. Bioactivation of the anticancer agent CPT-11 to SN-38 by human hepatic microsomal carboxylesterases and the in vitro assessment of potential drug interactions. Drug Metab Dispos. 1997;25(10):1157–1164. | ||
Iyer L, Hall D, Das S, et al. Phenotype-genotype correlation of in vitro SN-38 (active metabolite of irinotecan) and bilirubin glucuronidation in human liver tissue with UGT1A1 promoter polymorphism. Clin Pharmacol Ther. 1999;65(5):576–582. | ||
Iyer L, King CD, Whitington PF, et al. Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J Clin Invest. 1998;101(4):847–854. | ||
Senter PD, Beam KS, Mixan B, Wahl AF. Identification and activities of human carboxylesterases for the activation of CPT-11, a clinically approved anticancer drug. Bioconjug Chem. 2001;12(6):1074–1080. | ||
Hartmann JT, Lipp HP. Camptothecin and podophyllotoxin derivatives: inhibitors of topoisomerase I and II – mechanisms of action, pharmacokinetics and toxicity profile. Drug Saf. 2006;29(3):209–230. | ||
Alimonti A, Gelibter A, Pavese I, et al. New approaches to prevent intestinal toxicity of irinotecan-based regimens. Cancer Treat Rev. 2004;30(6):555–562. | ||
Xie R, Mathijssen RH, Sparreboom A, Verweij J, Karlsson MO. Clinical pharmacokinetics of irinotecan and its metabolites in relation with diarrhea. Clin Pharmacol Ther. 2002;72(3):265–275. | ||
Shimada Y, Yoshino M, Wakui A, et al. Phase II study of CPT-11, a new camptothecin derivative, in metastatic colorectal cancer. CPT-11 Gastrointestinal Cancer Study Group. J Clin Oncol. 1993;11(5):909–913. | ||
Fukuoka M, Niitani H, Suzuki A, et al. A phase II study of CPT-11, a new derivative of camptothecin, for previously untreated non-small-cell lung cancer. J Clin Oncol. 1992;10(1):16–20. | ||
Goncalves E, de Costa L, Abigerges D, Armand JP. A new enkephalinase inhibitor as an alternative to loperamide in the prevention of diarrhea induced by CPT-11. J Clin Oncol. 1995;13(8):2144–2146. | ||
Stringer AM, Gibson RJ, Logan RM, et al. Irinotecan-induced mucositis is associated with changes in intestinal mucins. Cancer Chemother Pharmacol. 2009;64(1):123–132. | ||
Takasuna K, Hagiwara T, Watanabe K, et al. Optimal antidiarrhea treatment for antitumor agent irinotecan hydrochloride (CPT-11)-induced delayed diarrhea. Cancer Chemother Pharmacol. 2006;58(4):494–503. | ||
Meyer-Losic F, Nicolazzi C, Quinonero J, et al. DTS-108, a novel peptidic prodrug of SN38: in vivo efficacy and toxicokinetic studies. Clin Cancer Res. 2008;14(7):2145–2153. | ||
De Coupade C, Fittipaldi A, Chagnas V, et al. Novel human-derived cell-penetrating peptides for specific subcellular delivery of therapeutic biomolecules. Biochem J. 2005;390(Pt 2):407–418. | ||
Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–216. | ||
Simon R, Freidlin B, Rubinstein L, Arbuck SG, Collins J, Christian MC. Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst. 1997;89(15):1138–1147. | ||
Eisenhauer EA, O’Dwyer PJ, Christian M, Humphrey JS. Phase I clinical trial design in cancer drug development. J Clin Oncol. 2000;18(3):684–692. | ||
Chabot GG. Clinical pharmacokinetics of irinotecan. Clin Pharmacokinet. 1997;33(4):245–259. | ||
Armand JP, Ducreux M, Mahjoubi M, et al. CPT-11 (irinotecan) in the treatment of colorectal cancer. Eur J Cancer. 1995;31A(7–8):1283–1287. | ||
Ramesh M, Ahlawat P, Srinivas NR. Irinotecan and its active metabolite, SN-38: review of bioanalytical methods and recent update from clinical pharmacology perspectives. Biomed Chromatogr. 2010;24(1):104–123. | ||
Yao Y, Su X, Xie Y, et al. Synthesis, characterization, and antitumor evaluation of the albumin-SN38 conjugate. Anticancer Drugs. 2013;24(3):270–277. | ||
Veltkamp SA, Witteveen EO, Capriati A, et al. Clinical and pharmacologic study of the novel prodrug delimotecan (MEN 4901/T-0128) in patients with solid tumors. Clin Cancer Res. 2008;14(22):7535–7544. | ||
Patnaik A, Papadopoulos KP, Tolcher AW, et al. Phase I dose-escalation study of EZN-2208 (PEG-SN38), a novel conjugate of poly(ethylene) glycol and SN38, administered weekly in patients with advanced cancer. Cancer Chemother Pharmacol. 2013;71(6):1499–1506. | ||
Guichard S, Terret C, Hennebelle I, et al. CPT-11 converting carboxylesterase and topoisomerase activities in tumour and normal colon and liver tissues. Br J Cancer. 1999;80(3–4):364–370. | ||
Khanna R, Morton CL, Danks MK, Potter PM. Proficient metabolism of irinotecan by a human intestinal carboxylesterase. Cancer Res. 2000;60(17):4725–4728. | ||
Ahmed F, Vyas V, Cornfield A, et al. In vitro activation of irinotecan to SN-38 by human liver and intestine. Anticancer Res. 1999;19(3A):2067–2071. |
Supplementary material
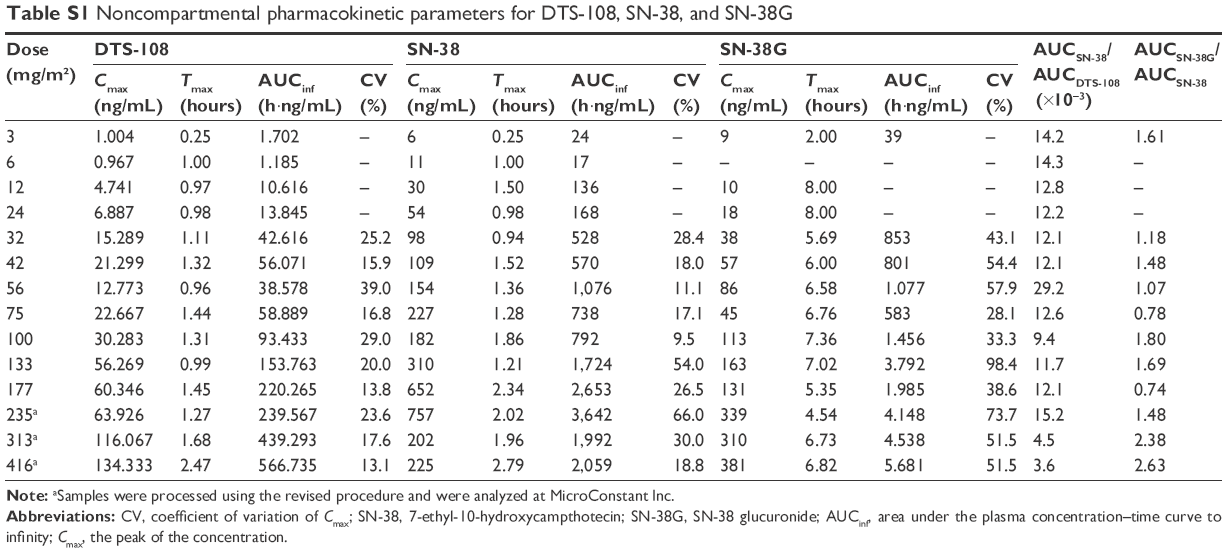 | Table S1 Noncompartmental pharmacokinetic parameters for DTS-108, SN-38, and SN-38G |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.