Back to Journals » Drug Design, Development and Therapy » Volume 12
Pharmacokinetic interaction between fimasartan and atorvastatin in healthy male volunteers
Authors Choi Y, Lee S ![]() , Jang IJ
, Jang IJ ![]() , Yu KS
, Yu KS ![]()
Received 10 February 2018
Accepted for publication 3 April 2018
Published 24 July 2018 Volume 2018:12 Pages 2301—2309
DOI https://doi.org/10.2147/DDDT.S165171
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Yewon Choi, SeungHwan Lee, In-Jin Jang, Kyung-Sang Yu
Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, Seoul, Republic of Korea
Introduction: Major cardiovascular risk factors, including hypertension and dyslipidemia, are often comorbidities, frequently leading to concurrent prescription of angiotensin receptor blockers and 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins). The study’s objective was to evaluate the effect of coadministration of fimasartan and atorvastatin on their pharmacokinetics (PKs).
Subjects and methods: In a randomized, open-label, three-period, six-sequence, crossover, multiple-dose study, 36 healthy subjects received 120 mg fimasartan, 40 mg atorvastatin, or both (based on their assigned sequence) once daily for 7 days in each period, with a 7-day washout between periods. Blood samples for the PK analysis of fimasartan, atorvastatin, and the 2-hydroxy atorvastatin metabolite were collected up to 48 h after the last dose.
Results: The coadministration of fimasartan and atorvastatin was well tolerated and led to an increase in the peak concentration and area under the concentration–time curve at steady state of fimasartan by 2.18-fold (95% confidence interval [CI], 1.79–2.65) and 1.35-fold (95% CI, 1.26–1.43) and those of atorvastatin increased by 1.82-fold (95% CI, 1.51–2.18) and 1.12-fold (95% CI, 1.04–1.22), respectively.
Conclusion: Coadministration increased the systemic exposures of fimasartan and atorvastatin, but the clinical significance of this finding needs to be evaluated with respect to exposure responses and clinical outcomes.
Keywords: angiotensin receptor blocker, statin, HMG-CoA inhibitor, fixed dose combination, FDC
Introduction
Angiotensin receptor blockers (ARBs) are generally well tolerated and considered highly effective in lowering blood pressure (BP) among patients with essential hypertension through their action on the renin–angiotensin system.1,2 As ARBs protect the kidney in diabetes mellitus and reduce mortality and morbidity in patients with heart failure, they are widely prescribed due to their favorable safety profile.1,3 Fimasartan is a recently available type 1-selective ARB, approved in the Republic of Korea in 2010. Fimasartan has been shown to be efficacious in reducing BP with good tolerability. In ambulatory BP-monitoring studies in hypertensive patients, once-daily fimasartan maintained a reduced BP profile over dosing intervals and was well tolerated in patients with mild-to-moderate essential hypertension.4 In a large-scale, open-label observational study in a large population of patients with arterial hypertension, the safety, efficacy, and patient compliance of fimasartan were found to be excellent.5 Fimasartan is rapidly absorbed, reaching first peak plasma concentration at 0.5–3.0 h, with a second minor peak in most subjects, and an absolute bioavailability of 18.6%. The terminal elimination half-life of fimasartan is 9–16 h; more than 85% is excreted unchanged by extrarenal pathways (ie, fecal elimination or biliary excretion), with the remainder metabolized by CYP3A4.6,7 In vitro studies have shown that fimasartan is also a substrate of OATP1B1, OATP2B1, and OATP1B3 and is transported by MDR1, MRP2, or BCRP.8–10 No significant drug interactions were observed when used in combination with hydrochlorothiazide, amlodipine, warfarin, or digoxin.11
Statins, also known as HMG-CoA reductase inhibitors, are used in patients with hypercholesterolemia and mixed dyslipidemia for reducing total-cholesterol, low-density lipoprotein cholesterol, very-low-density lipoprotein cholesterol, apolipoprotein B, and triglycerides, and for increasing high-density lipoprotein cholesterol (HDL-C). Statins lower the risk of cardiovascular disease in at-risk patients. Statins are usually prescribed in combination with other drugs, such as antihypertensive or antidiabetic drugs, to treat comorbidities and reduce overall cardiovascular risk. Atorvastatin is rapidly absorbed after oral administration, and its bioavailability is 14%, with an extensive first-pass effect.12 Atorvastatin is metabolized by CYP3A4 into the active metabolites, 2-hydroxy atorvastatin and 4-hydroxy atorvastatin, and is subject to cellular membrane transport by OATP and P-glycoprotein. Alterations of atorvastatin pharmacokinetics (PKs) with inhibitors of these systems (eg, itraconazole, cyclosporine, and grapefruit) have been demonstrated.12,13 Atorvastatin is also known as a weak inhibitor of CYP3A4, OATP, and P-glycoprotein, affecting the PKs of other drugs.14,15
Hypertension and dyslipidemia are major cardiovascular risk factors with high prevalence, which often exist as comorbidities.1 In clinics, antihypertensives and antihyperlipidemics are frequently prescribed to patients in combination. Potential drug–drug interactions (DDIs) should be considered important in ARB–statin combination therapy, because increased statin levels may be associated with statin-induced myopathy or rhabdomyolysis and increased ARB levels may cause hypotension and related symptoms.16–18 Therefore, the assessment of DDIs between these two agents is important to determine appropriate treatment regimens to maximize therapeutic efficacy and to minimize patient risks.
Because fimasartan and atorvastatin are frequently coadministered to patients, the assessment of DDIs of the two drugs was needed. The effect of fimasartan on the PKs of atorvastatin was investigated previously;19 however, the effect of atorvastatin on the PKs of fimasartan has been unknown. For these reasons, this study was designed to evaluate the effect of coadministration of fimasartan and atorvastatin on the PKs of each drug at steady state after multiple administrations.
Subjects and methods
Study population
Healthy Korean male subjects between the ages of 19 and 50, with body mass indexes (BMIs) of 19.0–28.0 kg·−2 were eligible for this study. Subjects who had significant hypersensitivity to fimasartan or atorvastatin, any significant disease or medical history, an HDL-C level of <35 mg·dL−1, systolic blood pressure (SBP) >140 or <100 mmHg, and diastolic blood pressure (DBP) >90 or <65 mmHg were excluded. Subjects were prohibited from taking any medication without prior consent of the investigator, consuming beverages containing caffeine or alcohol, and smoking from 24 h prior to the first dose until the end of the study.
Study design
This study was designed as a randomized, open-label, three-period, six-sequence, crossover, multiple-dose study. Subjects were randomly assigned into six sequence groups and received 120 mg of fimasartan, 40 mg of atorvastatin, or both concomitantly, once daily for 7 days according to their treatment sequence (Figure 1). Blood samples for PK analysis were collected at 0 (pre-dose), 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, and 48 h after the last dose of each period. After the last dose of the first and second period, subjects had a 7-day washout before starting the following period. The doses of fimasartan (120 mg) and atorvastatin (40 mg) in this study were determined based on common doses used in clinics, and were considered appropriate in terms of safety and therapeutic efficacy. Since the two drugs are prescribed for long-term use for chronic disease, the PKs and safety evaluation were conducted at steady state. The duration of drug administration needed to reach steady state was determined based on previous PK studies of fimasartan and atorvastatin.6,13
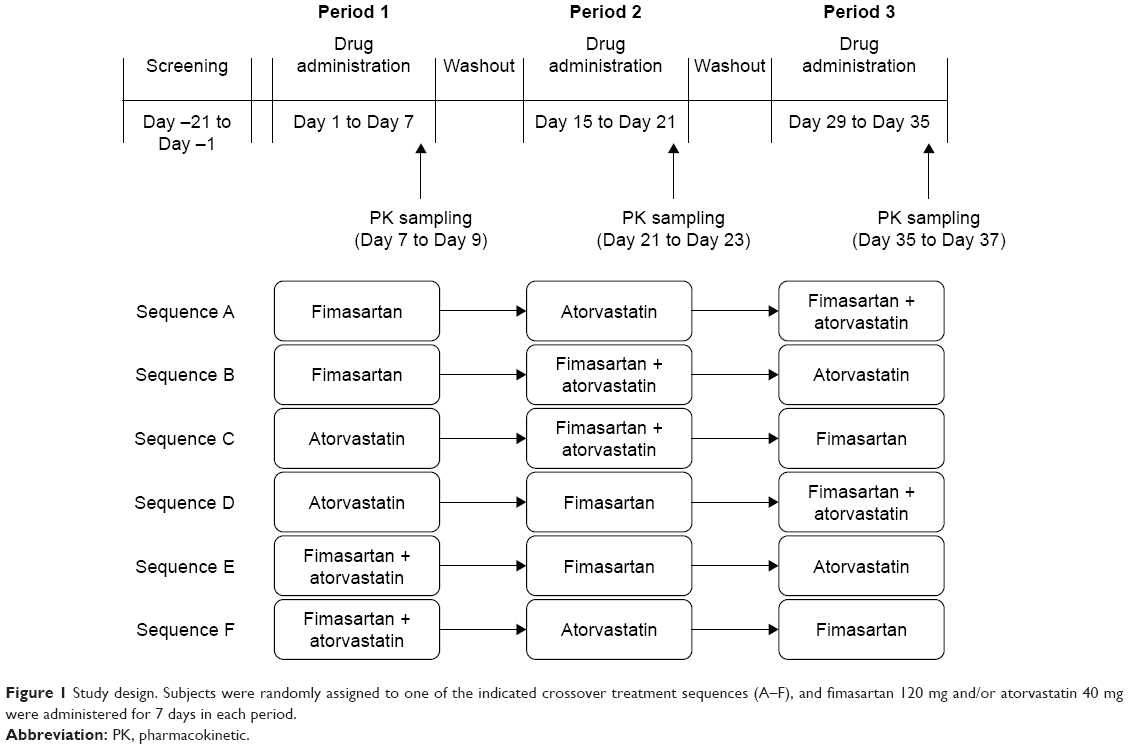 | Figure 1 Study design. Subjects were randomly assigned to one of the indicated crossover treatment sequences (A–F), and fimasartan 120 mg and/or atorvastatin 40 mg were administered for 7 days in each period. |
This study protocol (ClinicalTrials.gov registry identifier: NCT02397590) was approved by the Institutional Review Board of Seoul National University Hospital (IRB no 06-2015-0730). All subjects provided written informed consent prior to enrollment.
Quantification of plasma levels of fimasartan, atorvastatin, and 2-hydroxy atorvastatin
The plasma concentration of fimasartan was measured using a validated high-performance liquid chromatography (HPLC; Agilent 1260 series; Agilent Technologies, Santa Clara, CA, USA) coupled with tandem mass spectrometry (MS/MS; API 4000 Qtrap, AB SCIEX; SCIEX, Framingham, MA, USA). The sample preparation for plasma involved a simple protein precipitation with acetonitrile. Fifty microliters of plasma was thoroughly mixed with 450 μL of internal standard (IS; BR-A563 2.5 ng·mL−1 in 100% acetonitrile) and vortexed for 1 min. This mixture was injected onto the LC–MS/MS. The plasma samples were separated under gradient conditions using a mobile phase that consisted of 0.05% formic acid and 100% acetonitrile, and the samples were run at a flow rate of 0.35 mL·min−1. Fimasartan and IS were separated on an Poroshell 120 C18 column (3.0 × 50 mm, 2.7 μm; Agilent Technologies) and detected in a positive turbospray ionization mode. The plasma concentrations were extrapolated from the standard curve by comparing the ratio of the peak area of compound with that of IS. The method was validated over the range of 0.5–500 ng·mL−1. Precisions and accuracies of back-calculated concentrations were <4.662% and 95.87%–103.5%, respectively.
Plasma concentrations of atorvastatin and 2-hydroxy atorvastatin were also determined using an HPLC (Agilent 1260 series) coupled with MS/MS (Agilent 6490 Quadrupole). The sample preparation for plasma involved a simple protein precipitation with methanol. A mixture of 50 μL of deproteinized plasma and 200 μL of IS (atorvastatin-d5, 0.5 ng·mL−1 in methanol containing 0.1% formic acid) were vortexed for 1 min and separated under gradient conditions using a mobile phase that consisted of 5 mM ammonium formate and 100% acetonitrile, at a flow rate of 0.3 mL·min−1. Atorvastatin, 2-hydroxy atorvastatin, and IS were separated on an Xbridge C18 column (2.1 × 50 mm, 3.5 μm; Waters Corporation, Milford, MA, USA) and detected in the positive electrospray ionization mode with multiple reaction monitoring. The plasma concentrations were extrapolated from the standard curve by comparing the ratio of the peak area of compound with that of IS. The method was validated over the range of 0.1–150 ng·mL−1, and precisions and accuracies of back-calculated concentrations were <2.746% and 93.67%–103.8%, respectively.
PK and statistical analysis
Individual PK parameters of fimasartan, atorvastatin, and 2-hydroxy atorvastatin were analyzed using a non-compartmental analysis method with WinNonlin (Version 6.4; Certara, Princeton, NJ, USA). The area under the concentration–time curve for dose interval at steady state (AUCτ,ss), maximum plasma concentration at steady state (Cmax,ss), time to reach Cmax at steady state (Tmax,ss), terminal half-life (t1/2), and other PK parameters were calculated.
Statistical analysis was performed using SAS 9.4 (SAS Institute Inc., Cary, NC, USA). Descriptive statistics for data were calculated. Log-transformed Cmax,ss and AUCτ,ss of each drug and its metabolite, if applicable, are presented as the ratio of the geometric means of coadministration of fimasartan and atorvastatin to fimasartan or atorvastatin alone. The log-transformed parameters were analyzed and compared by treatment using a mixed-effect model analysis of variance with the period, sequence, and treatment as fixed effects.
Safety assessment
Adverse events (AEs) were monitored throughout the study by the investigators’ questionnaire. The course, severity, outcomes, and relationships to the investigational drugs were evaluated by the investigator. Laboratory tests, 12-lead electrocardiograms (ECGs), and physical examinations were also assessed during the study. Vital signs, including SBP and DBP, were measured in the sitting position at 0 h (pre-dose) on Days 1 and 3 in each period, and at 0 (pre-dose), 2, 4, 12, 24, and 48 h after the last dose in each period.
Results
Subjects
A total of 38 subjects, aged 31.0 ± 6.5 years, weighing 67.9 ± 6.9 kg with a BMI of 22.67 ± 2.02 kg·m−2 were randomized into six groups. Each group had its own sequence of drug administration, and demographic characteristics were similar across the groups (P = 0.4932, 0.6108, and 0.5700 for age, weight, and BMI, respectively). Among the 38 subjects, two subjects withdrew consent prior to the first dose, and 36 subjects were administered the investigational drugs at least once and were included in the safety analysis. One subject withdrew consent after the second period so that 35 subjects completed the drug administrations and PK samplings. The PK analysis was conducted in these 35 subjects who completed the study.
Pharmacokinetics
After 7-day multiple administrations of 120 mg to reach steady state, fimasartan was rapidly absorbed and the plasma concentration reached a maximum at 0.5 h at median. Secondary peaks were observed within 4–8 h in most subjects. The Cmax,ss and AUCτ,ss of fimasartan increased by 2.18- and 1.35-fold, respectively, when coadministered with atorvastatin, compared to those of fimasartan alone. The Tmax,ss and t1/2 of fimasartan were not significantly changed by coadministration with atorvastatin (Figure 2, Table 1).
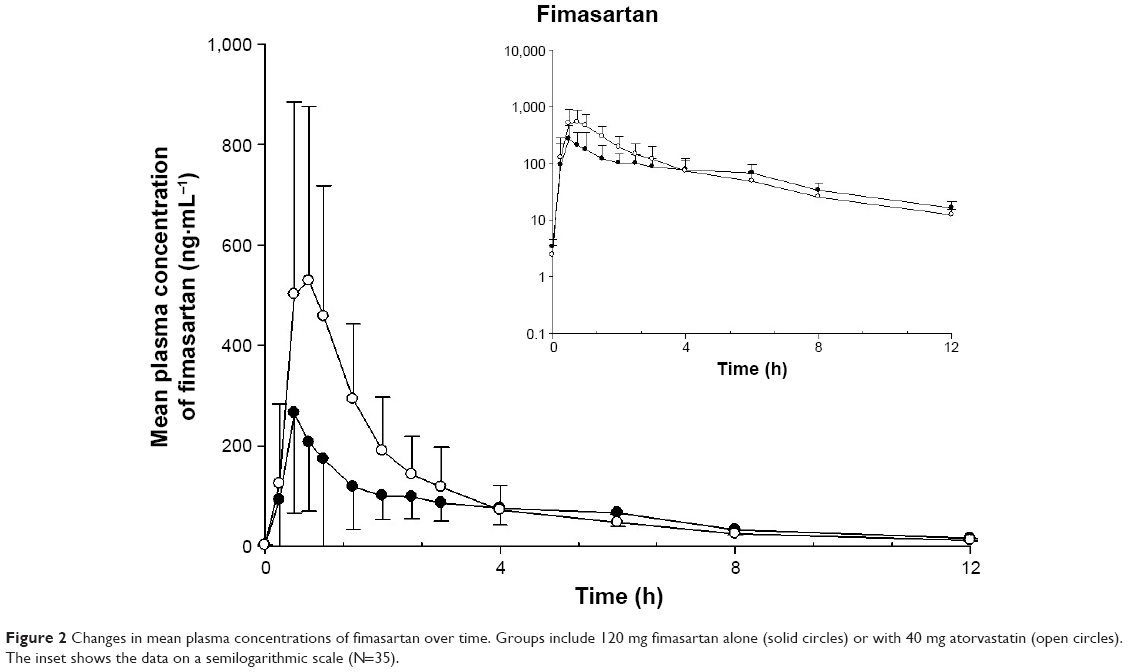 | Figure 2 Changes in mean plasma concentrations of fimasartan over time. Groups include 120 mg fimasartan alone (solid circles) or with 40 mg atorvastatin (open circles). The inset shows the data on a semilogarithmic scale (N=35). |
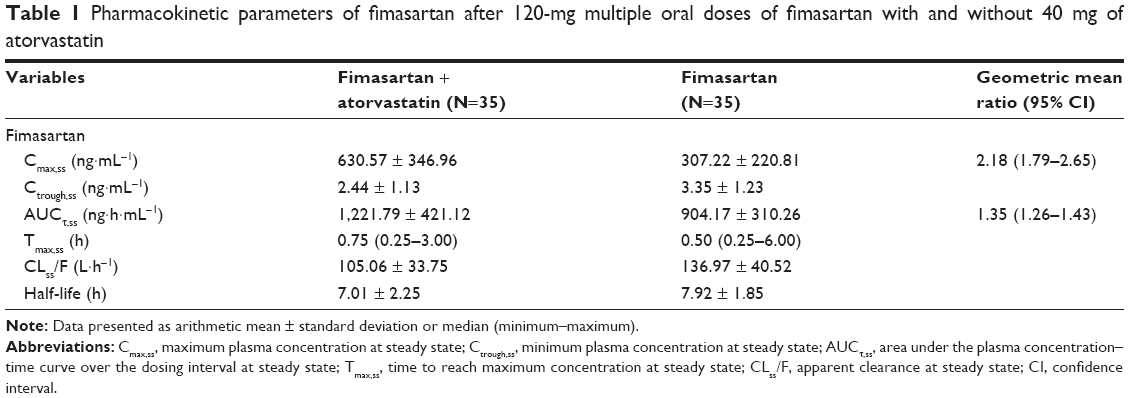 | Table 1 Pharmacokinetic parameters of fimasartan after 120-mg multiple oral doses of fimasartan with and without 40 mg of atorvastatin |
Atorvastatin and its 2-hydroxy metabolite reached their maximum concentrations at 1.0 and 2.0 h at median, respectively. The Cmax,ss and AUCτ,ss of atorvastatin increased by 1.82- and 1.12-fold, respectively, when the two drugs were coadministered, compared to those of atorvastatin alone. For 2-hydroxy atorvastatin, the Cmax,ss and AUCτ,ss increased by 2.68- and 1.35-fold, respectively, after coadministration. Fimasartan coadministration did not affect the Tmax,ss and t1/2 of atorvastatin and 2-hydroxy atorvastatin in terms of clinical significance. The ratio of the AUCτ,ss of 2-hydroxy atorvastatin to the AUCτ,ss of atorvastatin increased 1.20-fold after the coadministration of fimasartan (Figure 3, Table 2).
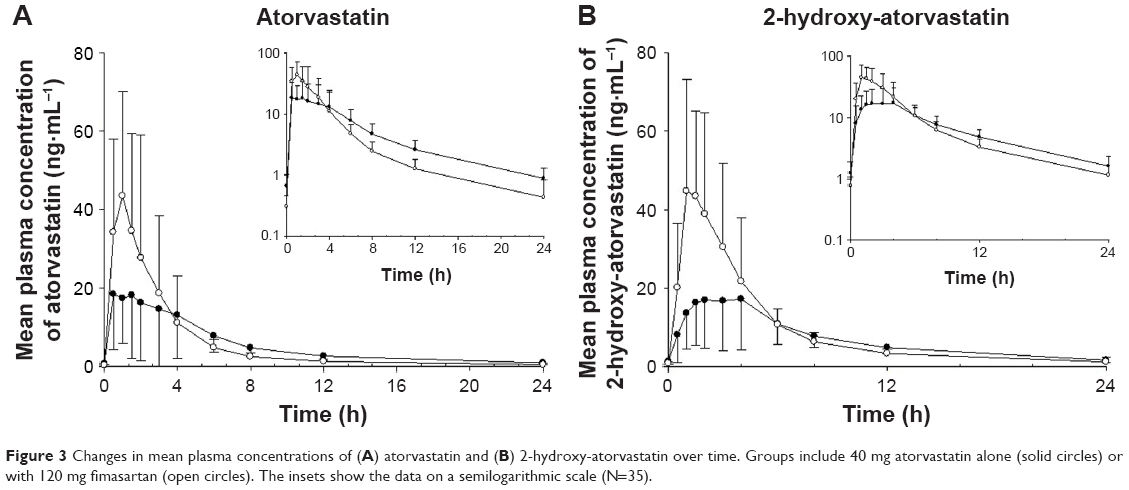 | Figure 3 Changes in mean plasma concentrations of (A) atorvastatin and (B) 2-hydroxy-atorvastatin over time. Groups include 40 mg atorvastatin alone (solid circles) or with 120 mg fimasartan (open circles). The insets show the data on a semilogarithmic scale (N=35). |
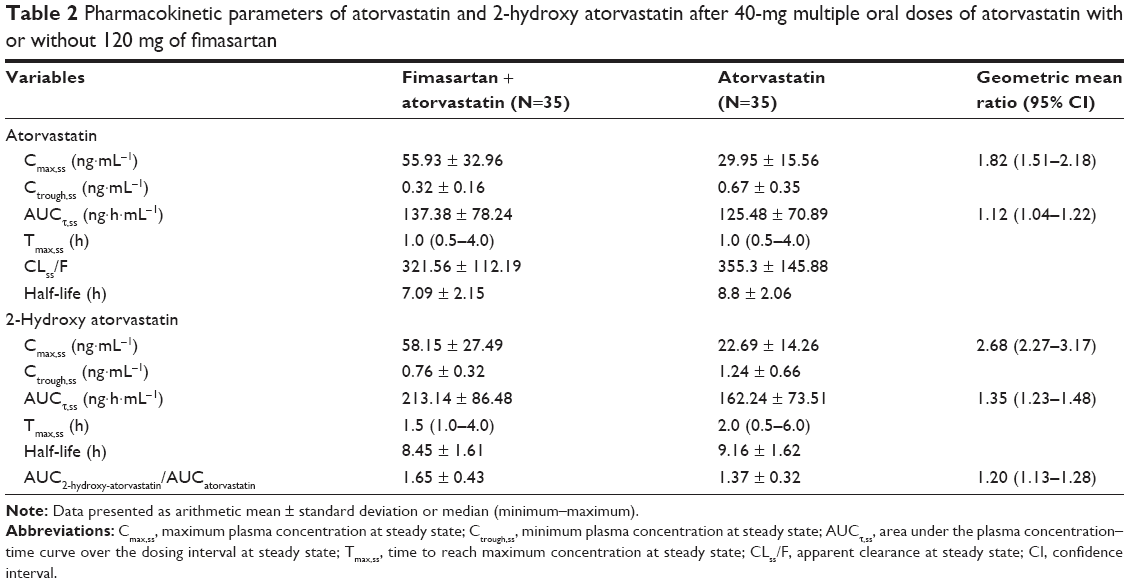 | Table 2 Pharmacokinetic parameters of atorvastatin and 2-hydroxy atorvastatin after 40-mg multiple oral doses of atorvastatin with or without 120 mg of fimasartan |
Safety and tolerability
Seven-day multiple coadministrations of fimasartan and atorvastatin were well tolerated, as well as single-drug administration of either fimasartan or atorvastatin in healthy subjects. A total of 15 AEs were considered to be related to the investigational drugs. Eight events, including dizziness, orthostatic hypotension, and blurred vision, occurred in four subjects with fimasartan monotreatment, whereas five events, including dizziness, were reported in three subjects with cotreatment. Two events, including chest discomfort and headache, occurred with atorvastatin treatment (Table 3). No serious AE was reported, and no participant discontinued the study due to an AE. All of the events were considered mild, and the subjects recovered without sequelae. There was no clinically significant change in vital signs, ECGs, clinical laboratory tests, or physical examinations during the study.
 | Table 3 ADRs by treatment and SOC |
Discussion
This study was designed to evaluate the PK DDI between fimasartan and atorvastatin. Several studies have been conducted to evaluate drug interactions of other statins and ARB agents. Kang et al conducted a drug interaction study between fimasartan and rosuvastatin in healthy subjects. In that study, the Cmax,ss and AUCτ,ss of fimasartan were slightly increased by 1.109- and 1.159-fold, respectively, and the Cmax,ss and AUCτ,ss of rosuvastatin changed by 1.090- and 0.870-fold, respectively.20 Son et al investigated drug interactions between telmisartan and rosuvastatin, and observed 1.1783- and 2.0128-fold increases in the AUCτ,ss and Cmax,ss, respectively, for rosuvastatin, and 1.1717- and 1.3501-fold increases in the AUCτ,ss and Cmax,ss, respectively, for telmisartan.21 In other studies, drug interactions between statin agents (ie, rosuvastatin, atorvastatin, or simvastatin) and ARBs (ie, olmesartan, valsartan, and losartan) were investigated, but no significant drug interactions were reported.22–25 Regarding fimasartan and atorvastatin, the effect of fimasartan on the PKs of atorvastatin was assessed in a previous study, which indicated that fimasartan increased the exposure of atorvastatin by 1.42- to 2.45-fold.19 However, the effect of atorvastatin on fimasartan exposure has not been investigated. To the best of our knowledge, the current study is the first to evaluate the effect of atorvastatin on the PKs of fimasartan. In addition, the effect of fimasartan on atorvastatin PK was confirmed in the current study.
Fimasartan is known to be a substrate of CYP3A4 and UGT1A3 enzymes and to be transported by OAT1, OATP1B1, OATP1B3, and BCRP (Figure 4). Fimasartan also inhibits the activity of the OATP1B1 transporter.6,8,19,26 Atorvastatin is a substrate and an inhibitor of CYP3A4, a substrate of UGT1A1 and UGT1A3 enzymes, and also a substrate of P-glycoprotein, OATP1B1, OATP1B3, and BCRP transporters.12,27–31
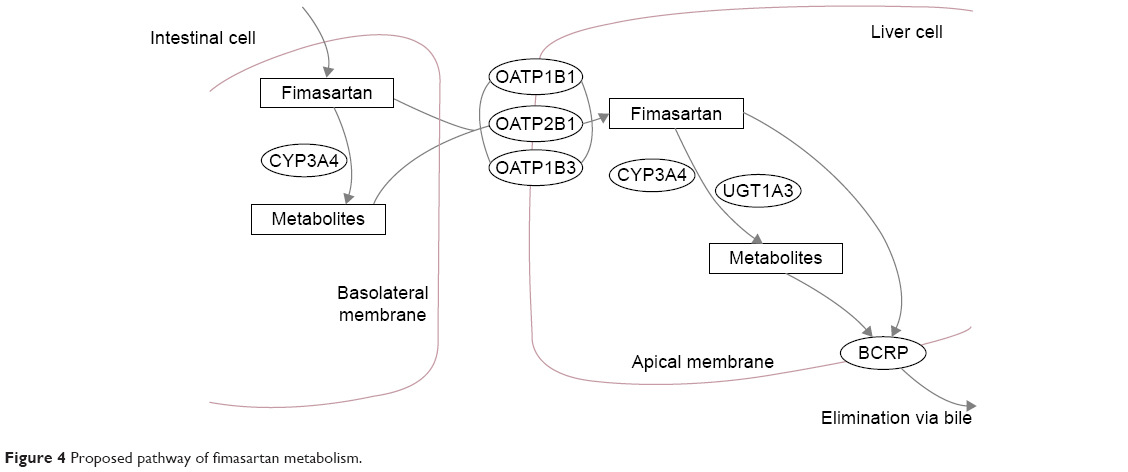 | Figure 4 Proposed pathway of fimasartan metabolism. |
When atorvastatin is administered with fimasartan, it is expected that the activity of CYP3A4 would be weakly inhibited and the first-pass effect of fimasartan would be reduced, thus increasing systemic exposure to fimasartan. As the inhibitory effect of atorvastatin is weak (defined by the US Food and Drug Administration guidelines as increasing the AUC of sensitive index substrates from >1.25- to <2-fold),32 the extent of drug interaction with fimasartan was also expected to be weak. We were able to confirm this in our study, whereby the AUCτ,ss of fimasartan increased by only 1.35-fold after the coadministration with atorvastatin.
Fimasartan and atorvastatin are both substrates of OATP1B1 and OATP1B3 transporters, and the activities of OATP1B1 and OATP1B3 are supposedly inhibited by competition during the coadministration of the two compounds. Thus, the hepatic uptake of fimasartan should be decreased, and its systemic exposure after the coadministration with atorvastatin should be increased due to the competitive inhibition of the transporters, especially at high concentrations near the Tmax. Because of this and the fact that the Tmax of both drugs are similar, we believe that the Cmax,ss of fimasartan increased to a greater extent than AUCτ,ss did (2.18- and 1.35-fold, respectively).
Atorvastatin is metabolized to 2-hydroxy atorvastatin by CYP3A4. In a previous in vitro study using human hepatic microsomal CYP isoenzymes, the IC50 value for the inhibition of CYP3A4 by fimasartan was found to be 283–494 μM.33 The average Cmax,ss of fimasartan following co-treatment in our study was 630.57 ng·mL−1 (~1.3 μM). Because the IC50 values were at least 218-fold greater than the Cmax,ss values, it is less likely that the activity of CYP3A4 is inhibited by fimasartan with typical dosages.
Instead of occurring via CYP inhibition, the increased Cmax,ss of atorvastatin and its metabolite may be explained by the competitive inhibition of OATP1B1 and OATP1B3, as mentioned above. In the concentration–time profile of atorvastatin, it was observed that the rate of concentration decrease after Cmax,ss was more rapid with a steeper slope after coadministration than after the administration with atorvastatin alone (Figure 2). The rapid fall of atorvastatin concentration can be explained by the short Tmax of fimasartan (0.75 h, median). Fimasartan may inhibit the transportation of atorvastatin into hepatocytes in a concentration-dependent manner. However, fimasartan is eliminated rapidly after the Tmax. As the fimasartan concentration declines, the competitive inhibition of OATP1B1 and OATP1B3 would be attenuated, and atorvastatin would be transported into hepatocytes rapidly, quickly decreasing atorvastatin concentrations after the increased Cmax,ss.
Nonetheless, the aforementioned drug interaction may be less meaningful in clinical settings. Considering the wide therapeutic range and highly variable PK parameters of fimasartan (CV% of 71.88 at maximum), the extent of increase in exposures due to drug interactions is less than the interindividual variability in the PK parameters when the drugs are administered alone. In addition, the relationship between exposure and response (ie, BP-lowering effect) was not significant when the AUCτ,ss ranged from 500 to 2,500 ng·h·mL−1. In addition, the incidences of fimasartan-related AEs were similar between fimasartan treatment and fimasartan–atorvastatin co-treatment, although the exposure of fimasartan differed by 1.35- to 2.18-fold between the two cohorts. Thus, the increases in exposures to both drugs seem to be less significant in terms of tolerability in our study, although safety and tolerability should be confirmed in relevant patient populations in future studies. Assessments of clinical endpoints would be helpful for the evaluation of the clinical significance of drug interactions between fimasartan and atorvastatin.
In conclusion, the systemic exposures of fimasartan and atorvastatin were increased by the coadministration of the drugs. The concurrent use of fimasartan and atorvastatin for 7 days was well tolerated in healthy male subjects. Clinical significance needs to be evaluated further in the context of exposure response and through future studies with clinical outcomes.
Acknowledgments
The authors deeply appreciate all of the support of the staff at the Clinical Trial Center, Seoul National University Hospital, during this study. This work was supported by Boryung Pharmaceutical, Co, Ltd, Seoul, Republic of Korea.
Disclosure
The authors report no conflicts of interest in this work.
References
Aronow WS, Fleg JL, Pepine CJ, et al. ACCF/AHA 2011 expert consensus document on hypertension in the elderly: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation. 2011;123(21):2434–2506. | ||
Matchar DB, McCrory DC, Orlando LA, et al. Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers for treating essential hypertension. Ann Intern Med. 2008;148(1):16–29. | ||
Redon J, Fabia MJ. Efficacy in angiotensin receptor blockade: a comparative review of data with olmesartan. J Renin Angiotensin Aldosterone Syst. 2009;10(3):147–156. | ||
Lee H, Kim KS, Chae SC, Jeong MH, Kim DS, Oh BH. Ambulatory blood pressure response to once-daily fimasartan: an 8-week, multicenter, randomized, double-blind, active-comparator, parallel-group study in Korean patients with mild to moderate essential hypertension. Clin Ther. 2013;35(9):1337–1349. | ||
Park JB, Sung KC, Kang SM, Cho EJ. Safety and efficacy of fimasartan in patients with arterial hypertension (Safe-KanArb study): an open-label observational study. Am J Cardiovasc Drugs. 2013;13(1):47–56. | ||
Ghim JL, Paik SH, Hasanuzzaman M, et al. Absolute bioavailability and pharmacokinetics of the angiotensin II receptor antagonist fimasartan in healthy subjects. J Clin Pharmacol. 2016;56(5):576–580. | ||
Chi YH, Lee H, Paik SH, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of fimasartan following single and repeated oral administration in the fasted and fed states in healthy subjects. Am J Cardiovasc Drugs. 2011;11(5):335–346. | ||
Jeong E-S, Kim Y-W, Kim H-J, et al. Glucuronidation of fimasartan, a new angiotensin receptor antagonist, is mainly mediated by UGT1A3. Xenobiotica. 2015;45(1):10–18. | ||
Yi S, Kim T-E, Yoon SH, et al. Pharmacokinetic interaction of fimasartan, a new angiotensin II receptor antagonist, with amlodipine in healthy volunteers. J Cardiovasc Pharmacol. 2011;57(6):682–689. | ||
Lee H, Yang H-M, Lee H-Y, et al. Efficacy and tolerability of once-daily oral fimasartan 20 to 240 mg/d in Korean patients with hypertension: findings from two Phase II, randomized, double-blind, placebo-controlled studies. Clin Ther. 2012;34(6):1273–1289. | ||
Lee HY, Oh BH. Fimasartan: a new angiotensin receptor blocker. Drugs. 2016;76(10):1015–1022. | ||
Lennernas H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet. 2003;42(13):1141–1160. | ||
Lipotor [package insert]. New York, NY: Pfizer Global Pharmaceuticals; 2009 [cited January 4, 2018]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/020702s056lbl.pdf. Accessed January 4, 2018. | ||
Mc Donnell CG, Harte S, O’Driscoll J, O’Loughlin C, Van Pelt FN, Shorten GD. The effects of concurrent atorvastatin therapy on the pharmacokinetics of intravenous midazolam. Anaesthesia. 2003;58(9):899–904. | ||
Boyd RA, Stern RH, Stewart BH, et al. Atorvastatin coadministration may increase digoxin concentrations by inhibition of intestinal P-glycoprotein-mediated secretion. J Clin Pharmacol. 2000;40(1):91–98. | ||
Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther. 2006;80(6):565–581. | ||
Omar MA, Wilson JP, Cox TS. Rhabdomyolysis and HMG-CoA reductase inhibitors. Ann Pharmacother. 2001;35(9):1096–1107. | ||
Chatzizisis YS, Koskinas KC, Misirli G, Vaklavas C, Hatzitolios A, Giannoglou GD. Risk factors and drug interactions predisposing to statin-induced myopathy: implications for risk assessment, prevention and treatment. Drug Saf. 2010;33(3):171–187. | ||
Shin KH, Kim TE, Kim SE, et al. The effect of the newly developed angiotensin receptor II antagonist fimasartan on the pharmacokinetics of atorvastatin in relation to OATP1B1 in healthy male volunteers. J Cardiovasc Pharmacol. 2011;58(5):492–499. | ||
Kang WY, Kim EH, Seong SJ, et al. Pharmacokinetic drug interaction study using fimasartan and rosuvastatin in healthy volunteers. Int J Clin Pharmacol Ther. 2016;54(12):992–1003. | ||
Son M, Kim Y, Lee D, et al. Pharmacokinetic interaction between rosuvastatin and telmisartan in healthy Korean male volunteers: a randomized, open-label, two-period, crossover, multiple-dose study. Clin Ther. 2014;36(8):1147–1158. | ||
Ahmed T, Kollipara S, Gautam A, et al. Bioavailability and interaction potential of atorvastatin and losartan on co-administration in healthy human subjects. J Bioequiv Bioavailab. 2009;01(01):018–027. | ||
Roh H, Son H, Lee D, Chang H, Yun C, Park K. Pharmacokinetic interaction between rosuvastatin and olmesartan: a randomized, open-label, 3-period, multiple-dose crossover study in healthy Korean male subjects. Clin Ther. 2014;36(8):1159–1170. | ||
Jung JA, Lee SY, Kim JR, et al. A pharmacokinetic and pharmacodynamic drug interaction between rosuvastatin and valsartan in healthy subjects. Drug Des Devel Ther. 2015;9:745–752. | ||
Sunkara G, Reynolds CV, Pommier F, Humbert H, Yeh C, Prasad P. Evaluation of a pharmacokinetic interaction between valsartan and simvastatin in healthy subjects. Curr Med Res Opin. 2007;23(3):631–640. | ||
Kim JW, Yi S, Kim T-E, et al. Increased systemic exposure of fimasartan, an angiotensin ii receptor antagonist, by ketoconazole and rifampicin. J Clin Pharmacol. 2013;53(1):75–81. | ||
Lau YY, Huang Y, Frassetto L, Benet LZ. Effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther. 2007;81(2):194–204. | ||
Shitara Y, Sugiyama Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol Ther. 2006;112(1):71–105. | ||
Jacobsen W, Kuhn B, Soldner A, et al. Lactonization is the critical first step in the disposition of the 3-hydroxy-3-methylglutaryl-coa reductase inhibitor atorvastatin. Drug Metab Dispos. 2000;28(11):1369–1378. | ||
Fukazawa I, Uchida N, Uchida E, Yasuhara H. Effects of grapefruit juice on pharmacokinetics of atorvastatin and pravastatin in Japanese. Br J Clin Pharmacol. 2004;57(4):448–455. | ||
Backman JT, Luurila H, Neuvonen M, Neuvonen PJ. Rifampin markedly decreases and gemfibrozil increases the plasma concentrations of atorvastatin and its metabolites. Clin Pharmacol Ther. 2005;78(2):154–167. | ||
Patel G, King A, Dutta S, et al. Evaluation of the effects of the weak CYP3A inhibitors atorvastatin and ethinyl estradiol/norgestimate on lomitapide pharmacokinetics in healthy subjects. J Clin Pharmacol. 2016;56(1):47–55. | ||
Kanarb® [investigator’s brochure, Version 7.0]. Seoul: Boryung Pharmaceutical Co., Ltd; 2014. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.