Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Pharmacokinetic drug interactions with clopidogrel: updated review and risk management in combination therapy
Authors Wang Z, Chen M, Zhu L, Yu L, Zeng S, Xiang M, Zhou Q ![]()
Received 6 January 2015
Accepted for publication 5 February 2015
Published 19 March 2015 Volume 2015:11 Pages 449—467
DOI https://doi.org/10.2147/TCRM.S80437
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor De Yun Wang
Zhi-Yu Wang,1 Meng Chen,1 Ling-Ling Zhu,2 Lu-Shan Yu,3 Su Zeng,3 Mei-Xiang Xiang,4 Quan Zhou1
1Department of Pharmacy, 2VIP Care Ward, Division of Nursing, the Second Affiliated Hospital, School of Medicine, 3Department of Pharmaceutical Analysis and Drug Metabolism, College of Pharmaceutical Sciences, 4Department of Cardiology, the Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang Province, People’s Republic of China
Background: Coprescribing of clopidogrel and other drugs is common. Available reviews have addressed the drug–drug interactions (DDIs) when clopidogrel is as an object drug, or focused on combination use of clopidogrel and a special class of drugs. Clinicians may still be ignorant of those DDIs when clopidogrel is a precipitant drug, the factors determining the degree of DDIs, and corresponding risk management.
Methods: A literature search was performed using PubMed, MEDLINE, Web of Science, and the Cochrane Library to analyze the pharmacokinetic DDIs of clopidogrel and new P2Y12 receptor inhibitors.
Results: Clopidogrel affects the pharmacokinetics of cerivastatin, repaglinide, ferulic acid, sibutramine, efavirenz, and omeprazole. Low efficacy of clopidogrel is anticipated in the presence of omeprazole, esomeprazole, morphine, grapefruit juice, scutellarin, fluoxetine, azole antifungals, calcium channel blockers, sulfonylureas, and ritonavir. Augmented antiplatelet effects are anticipated when clopidogrel is coprescribed with aspirin, curcumin, cyclosporin,
St John’s wort, rifampicin, and angiotensin-converting enzyme inhibitors. The factors determining the degree of DDIs with clopidogrel include genetic status (eg, cytochrome P540 [CYP]2B6*6, CYP2C19 polymorphism, CYP3A5*3, CYP3A4*1G, and CYP1A2-163C>A), species differences, and dose strength. The DDI risk does not exhibit a class effect, eg, the effects of clopidogrel on cerivastatin versus other statins, the effects of proton pump inhibitors on clopidogrel (omeprazole, esomeprazole versus pantoprazole, rabeprazole), the effects of rifampicin on clopidogrel versus ticagrelor and prasugrel, and the effects of calcium channel blockers on clopidogrel (amlodipine versus P-glycoprotein-inhibiting calcium channel blockers). The mechanism of the DDIs with clopidogrel involves modulating CYP enzymes (eg, CYP2B6, CYP2C8, CYP2C19, and CYP3A4), paraoxonase-1, hepatic carboxylesterase 1, P-glycoprotein, and organic anion transporter family member 1B1.
Conclusion: Effective and safe clopidogrel combination therapy can be achieved by increasing the awareness of potential changes in efficacy and toxicity, rationally selecting alternatives, tailoring drug therapy based on genotype, checking the appropriateness of physician orders, and performing therapeutic monitoring.
Keywords: clopidogrel, drug–drug interactions, drug metabolism, drug transporter, genotype, pharmacokinetics, polypharmacy, pharmacogenetics, P2Y12 receptor inhibitors, risk management
Introduction
The high prevalence of polypharmacy among older adults with cardiovascular disease and other comorbidities is not a problem in itself, but there is a risk of drug interactions in the event of a lack of coordination among care providers.1,2 To guarantee safety in medication use, Joint Commission International requires that medication prescriptions or orders must be reviewed for appropriateness prior to dispensing. Identifying real or potential drug–drug interactions (DDIs) and drug–food interactions is one of the key elements included in the process of appropriateness review.3 For each drug interaction, the object drug is defined as the medication for which the pharmacokinetics and/or pharmacodynamics may be modified by the drug interaction process. The precipitant drug is defined as the medication responsible for affecting the pharmacokinetic and/or pharmacodynamic properties of the object drug.1,2
Clopidogrel is an oral antiplatelet agent used to inhibit blood clots in coronary artery disease, peripheral vascular disease, and cerebrovascular disease, and to prevent myocardial infarction. Plavix® (clopidogrel), marketed jointly by Sanofi-Aventis and Bristol-Myers Squibb, has been the second best-selling drug in the world, with $9.4 billion in global sales in 2010.4 The drug works by irreversibly inhibiting a receptor called P2Y12, an adenosine diphosphate chemoreceptor on platelet cell membranes. Although there are other pathways leading to thrombotic events, and inhibition of P2Y12 is not the sole therapeutic measure in order to limit the thrombotic risk,5 the clinical use of clopidogrel is an effective strategy recommended by the American Heart Association and the American College of Cardiology for inhibiting platelet activity in patients with acute coronary syndrome and for preventing thrombotic events in those undergoing percutaneous coronary intervention (PCI) with stenting.4,6
The DDI profiles of clopidogrel have been reviewed by Egan et al, Shah et al, Siller-Matula et al and Scott et al.7–10 However, these authors only addressed the circumstances under which DDIs occurred when clopidogrel was the object drug or focused on combination therapy with clopidogrel and special classes of medications, such as antiretrovirals,7 proton pump inhibitors (PPIs),8,9 and strong inhibitors or inducers of cytochrome P450 (CYP)3A/CYP2C19.10 Moreover, new interesting findings in clopidogrel-associated DDI studies in recent years have not been reviewed in depth, so clinicians may be still ignorant of those DDIs when clopidogrel is the precipitant drug, the factors determining the degree of DDIs, and the corresponding risk management. In this paper, we investigate this issue in depth and present an updated review, with the intention of improving the reader’s knowledge of the DDIs associated with clopidogrel.
Methods
Search strategy
Potentially relevant literature until December 31, 2014 was identified by performing searches in the following databases: PubMed, MEDLINE, Web of Sciences (additional filter: proceedings paper, meeting abstract, and correction were excluded), and the Cochrane Library. Considering that the DDIs of the new P2Y12 receptor inhibitors (prasugrel, ticagrelor, cangrelor) may be relevant to the issue of clopidogrel, we used the search string “(clopidogrel or prasugrel or ticagrelor or cangrelor) and drug interaction and pharmacokinetics” and an English language filter.
Selection criteria
Two reviewers (ZYW and MXX) independently searched the literature and screened the relevant studies. The numbers of papers identified in the four databases were 211, 205, 145, and 44, respectively. After excluding duplicated literature, 220 papers underwent further assessment. Both in vitro and in vivo studies were considered if they met the following inclusion criteria: studies describing DDIs of P2Y12 receptor inhibitors with statistically significant changes in pharmacokinetic or pharmacodynamic parameters; studies directly comparing the degree of DDIs associated with different P2Y12 receptor inhibitors; and studies excluding the possibility of a DDI between a P2Y12 receptor inhibitor (“A”) and a specific medication (“B”), the therapeutic or structural analog (“C”) of which, however, could exhibit a significant DDI with “A”. Documents such as comments, letters without an experimental study, reviews, meta-analyses, and editorials were excluded despite being retrieved using the search terms. Each reviewer was blinded to the other reviewer during the process of data extraction. In the event of disagreement between the two reviewers, a third reviewer (QZ) was consulted. Fifty-eight original papers on the DDI issue were finally included based on the inclusion/exclusion criteria (Figure 1). Valuable information was summarized by data interpretation.
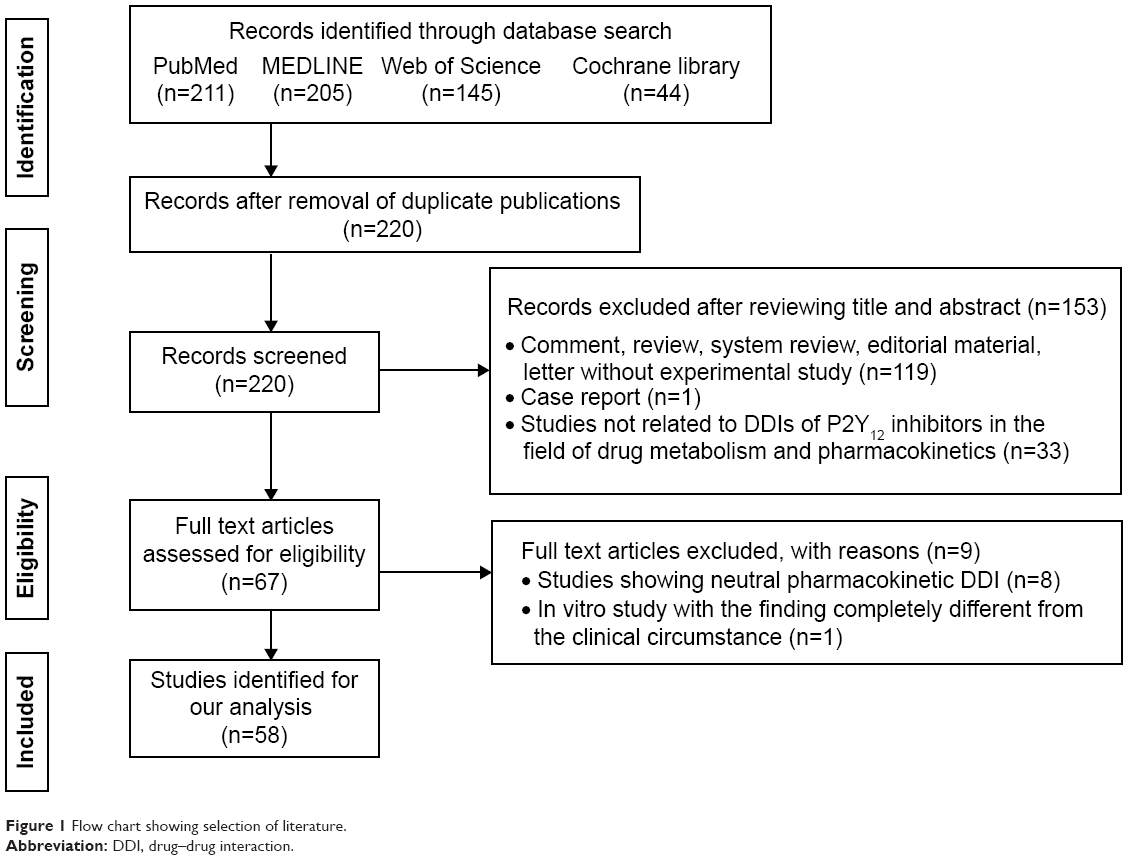 | Figure 1 Flow chart showing selection of literature. |
Results and discussion
Metabolic profiles of P2Y12 inhibitors
The metabolic profiles of P2Y12 inhibitors are summarized in Figure 2. Clopidogrel is a prodrug that is absorbed in the intestine and activated in the liver. P-glycoprotein (P-gp) encoded by ABCB1 is involved in the intestinal absorption of clopidogrel.11 Approximately 85% of clopidogrel is hydrolyzed by esterases into an inactive compound, leaving only 15% available for hepatic metabolism. The conversion of clopidogrel to its active metabolite requires two sequential oxidative steps. The first step leads to formation of 2-oxo-clopidogrel, followed by conversion to the active metabolite. CYP enzymes like CYP1A2, CYP2B6, CYP2C9, CYP2C19, and CYP3A4/5 are involved in the metabolism of clopidogrel. CYP2C19 contributes substantially to both oxidative steps and CYP3A4 contributes substantially to the second oxidative step. Genetic polymorphisms of CYP2C19 are associated with impaired metabolism of clopidogrel.12
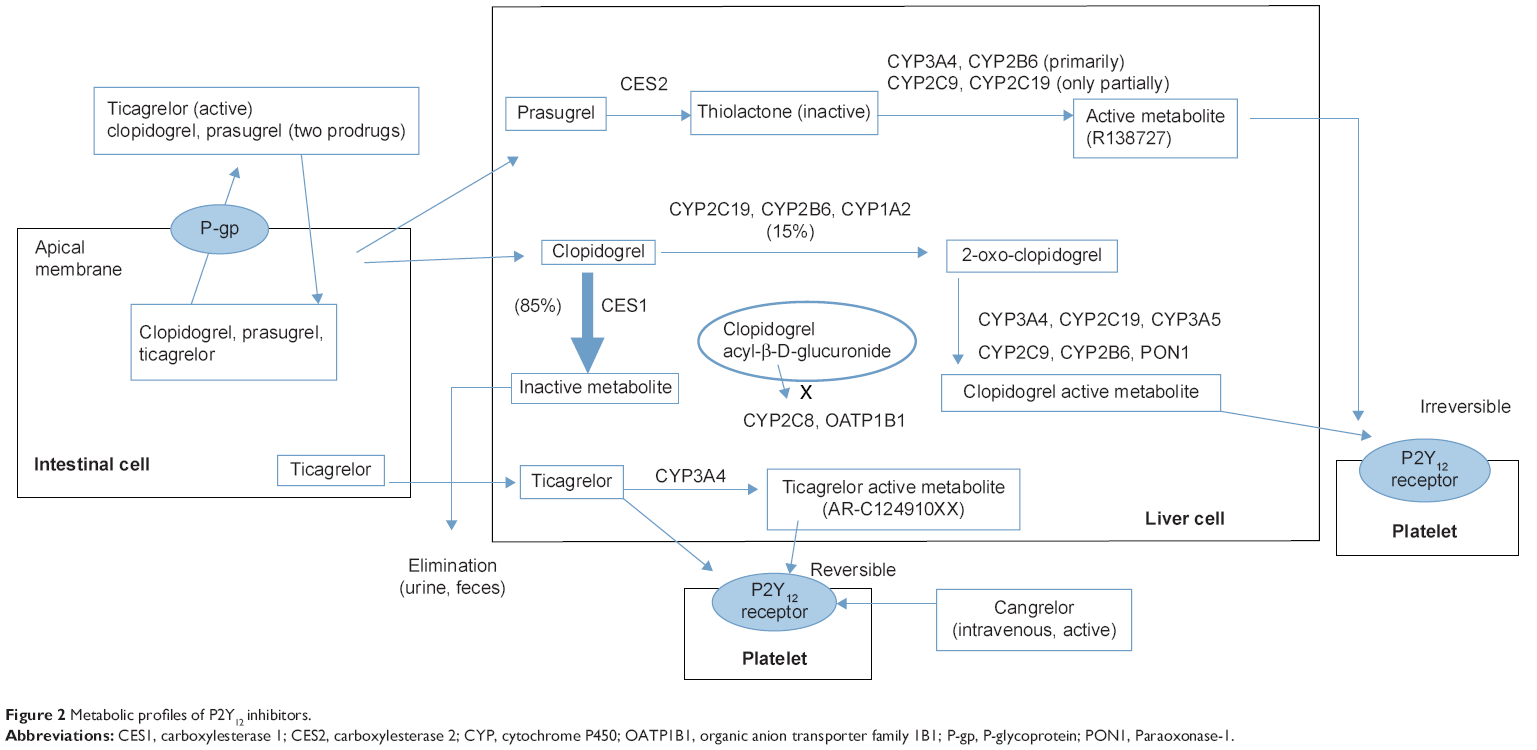 | Figure 2 Metabolic profiles of P2Y12 inhibitors. |
Prasugrel is also a prodrug that is metabolized in a two-step process, activation of which is initiated by plasma esterases, followed by a single CYP-dependent step that primarily involves CYP3A and CYP2B6, and only partially CYP2C9 and CYP2C19. Unlike clopidogrel, prasugrel does not have an analogous inactivation pathway that consumes the majority of the absorbed dose. Prasugrel is subject to efflux via P-gp, although ABCB1 genotypes are not significantly associated with clinical outcomes. Ticagrelor undergoes extensive CYP3A4-mediated metabolism to produce an active metabolite; both the parent drug and the active metabolite can reversibly inhibit the P2Y12 receptor. Absorption of ticagrelor may be affected by ABCB1 polymorphism. Cangrelor is an intravenous, direct-acting, reversible P2Y12 receptor antagonist that does not require metabolic conversion/activation.13
Clopidogrel comedicated as a precipitant drug
Clopidogrel and statins
DDI and risk description
A screening study of DDIs in cerivastatin users revealed an adverse effect of clopidogrel, ie, use of clopidogrel was strongly associated with rhabdomyolysis (odds ratio 29.6; 95% confidence interval 6.1–143). Retrieval of the US Food and Drug Administration Adverse Event Reporting System showed that clopidogrel was used more commonly in patients with rhabdomyolysis receiving cerivastatin (17%) than in those receiving atorvastatin (0%).14 Cerivastatin is primarily metabolized by CYP2C8. Clopidogrel acyl-β-D-glucuronide is a potent time-dependent inhibitor of CYP2C8 in vitro. A physiologically based pharmacokinetic model indicates that inactivation of CYP2C8 by clopidogrel acyl-β-D-glucuronide leads to uninterrupted 60%–85% inhibition of CYP2C8 during daily clopidogrel therapy. Computational modeling further reveals the docking of clopidogrel acyl-β-D-glucuronide at the CYP2C8 active site with its thiophene moiety close to heme. Both epidemiological and in vitro findings suggest that clopidogrel may cause clinically important, dose-dependent DDIs with other medications metabolized by CYP2C8.14,15 Cerivastatin is also a substrate for solute carrier organic anion transporter family member 1B1 (OATP1B1), and clopidogrel and its two metabolites have inhibitory effects on OATP1B1-mediated uptake of cerivastatin (The half maximal inhibitory concentration [IC50] values: clopidogrel, 3.95 μmol/L; 2-oxo clopidogrel, 8.18 μmol/L; clopidogrel acyl glucuronide, 10.9 μmol/L), which also accounts for cerivastatin-induced rhabdomyolysis.16
A population-based cohort study in patients with coronary stent implantation revealed that CYP3A4-metabolizing statin use versus non-use was not associated with an increased rate of major adverse cardiovascular events in patients using clopidogrel after coronary stent implantation.17 The literature also indicates no adverse effects of clopidogrel on the pharmacokinetics of atorvastatin and fluvastatin.18,19 Pinheiro et al evaluated the pharmacokinetic interactions between clopidogrel and rosuvastatin and suggested a beneficial synergism between the two drugs in terms of cardiovascular protection in patients with coronary heart disease.20 Rosuvastatin had a neutral effect on platelet inhibition by clopidogrel. Interestingly, the effect of clopidogrel on elevating plasma levels of rosuvastatin occurred exclusively after the 300 mg loading dose, but not with the 75 mg dose.
DDIs between ticagrelor and atorvastatin or simvastatin were investigated. For atorvastatin, healthy volunteers (n=24) received ticagrelor (loading dose 270 mg; 90 mg twice daily, 7 days) or placebo plus atorvastatin calcium (80 mg; day 5). For simvastatin, volunteers (n=24) received simvastatin 80 mg or ticagrelor (loading dose 270 mg; 180 mg twice daily, 7 days) plus simvastatin (80 mg; day 5). The effects of ticagrelor on the pharmacokinetics of atorvastatin were modest and unlikely to be clinically relevant. However, the maximal plasma concentration (Cmax) and area under the concentration-time curve (AUC) of simvastatin were increased by 81% and 56%, respectively, with ticagrelor. Some individuals experienced two to three-fold increases in simvastatin exposure.21
Implications and risk management
Collectively, the available evidence indicates that clopidogrel is not an object drug susceptible to statins, but a precipitant drug that has clinically relevant and significant DDIs with cerivastatin instead of other statins. Although cerivastatin has been withdrawn from the world market, the DDIs between clopidogrel and cerivastatin provide a big lesson to both clinicians and researchers in the drug metabolism and pharmacokinetics field. Patients receiving ticagrelor should avoid simvastatin doses higher than 40 mg because an increased risk of muscle injury has been observed in patients taking simvastatin at the highest approved dose (80 mg) compared with patients taking lower doses of simvastatin.
Clopidogrel and insulinotropic agents
DDI and risk description
Repaglinide, nateglinide, and mitiglinide are three insulinotropic agents that mediate the release of insulin from beta-cells. Repaglinide is primarily metabolized by CYP2C8; moreover, it is also a substrate of OATP1B1. In healthy subjects, the AUC of repaglinide was increased 5.1-fold by a 300 mg loading dose of clopidogrel and 3.9-fold by sustained treatment with 75mg clopidogrel daily.15 DDI studies of clopidogrel and other insulinotropic agents are not available. Nateglinide is predominantly metabolized by CYP2C9 (70%) and CYP3A4 (30%), on which clopidogrel has little inhibitory effect.22 The metabolism of mitiglinide is different to that of repaglinide and nateglinide. Glucuronidation represents a main elimination pathway for mitiglinide; UDP-glucuronosyltransferases 1A3 and 2B7 are important catalytic enzymes in the carboxyl-glucuronidation of mitiglinide in the human liver.23
Implications and risk management
Clopidogrel should not be comedicated with repaglinide, to avoid occurrence of potential hypoglycemic events. Ticagrelor has been reported to show little inhibition of CYP2C8,24 and so may be an alternative to clopidogrel for patients on repaglinide therapy. Theoretically, nateglinide and mitiglinide may be alternatives to repaglinide when patients are concomitantly using clopidogrel. However, this needs to be confirmed.
Clopidogrel and ferulic acid
Ferulic acid (4-hydroxy-3-methoxycinnamic acid), a compound isolated from herbs with the action of “activating the blood circulation to dissipate blood stasis”, has large potential for development as a useful drug in the treatment of cardiovascular disease. Clopidogrel is often combined with ferulic acid-containing herbs, eg, Danggui (Angelicae sinensis) and Chuangxiong (Rhizoma chuanxiong) to treat cardiovascular disease in clinical practice. Coadministration of clopidogrel (7 mg/kg, oral) resulted in a 79.7% increase in the AUC of ferulic acid (10 mg/kg, oral) in rats. The Cmax of ferulic acid was significantly increased by 74.3% in the presence of clopidogrel (P<0.01). Moreover, the time taken to reach Cmax (Tmax) of ferulic acid in the presence of clopidogrel was 3.76 times slower than when administered alone.25 The notable increases in AUC, Cmax, and Tmax of ferulic acid caused by coadministration indicate that clopidogrel improves the bioavailability of ferulic acid possibly by metabolic inhibition, decreased systemic elimination, or local interactions of ferulic acid with clopidogrel in the intestine. Close monitoring for potential DDIs may be necessary in patients who are receiving combined therapy with clopidogrel and ferulic acid-containing herbs (eg, Danggui and Chuangxiong).
Clopidogrel and sibutramine
DDI and risk description
Sibutramine is an oral anorexiant. It is metabolized by CYP2B6 and CYP2C19 into two active metabolites, M1 (mono-desmethyl sibutramine) and M2 (di-desmethyl sibutramine). The efficacy and safety of sibutramine are related to the plasma concentrations of its active metabolites. Clopidogrel is a mechanism-based inhibitor of CYP2B6 and CYP2C19.26,27 A study confirmed that the effects of clopidogrel on the disposition of sibutramine, M1, and M2 were related to CYP2B6*6 polymorphism. Administration of clopidogrel (300 mg on the first day and then 75 mg once daily for 6 days) increased the plasma concentrations of sibutramine and M1, significantly increased the half-life and AUC(0-infinity) of sibutramine (242% and 227% of control phase, respectively), and decreased the apparent oral clearance of sibutramine (31% of control phase).28 Moreover, pretreatment with clopidogrel did not cause a statistically significant change in the Cmax of sibutramine in CYP2B6*1/*1 subjects, but it did in subjects with the CYP2B6*6/*6 genotype (11.2±3.70 ng/mL [control] versus 20.3±7.85 ng/mL [clopidogrel], P<0.05). Coadministration of clopidogrel, combined with the CYP2B6 genotype, increased the interindividual variation in the pharmacokinetics of sibutramine, M1, and M2. The AUC of M1 plus M2 in CYP2B6*6/*6 subjects pretreated with clopidogrel was 1.65-fold that in the CYP2B6*1/*1 subjects in the control phase.29
Implications and risk management
M1 and M2 metabolites account predominantly for the inhibition of neurotransmitter reuptake in vivo and the potential cardiovascular adverse events of sibutramine. Clopidogrel may increase the side effects of sibutramine. Careful treatment planning is required when clopidogrel is comedicated with sibutramine, especially in patients with a CYP2B6 functional deficit genotype.
Clopidogrel and digoxin
DDI and risk description
Digoxin is a substrate of renal and intestinal P-gp. The therapeutic range for digoxin is a serum concentration of 0.5–1.0 ng/mL. Peeters et al assessed the safety and pharmacodynamic/pharmacokinetic compatibility of clopidogrel with digoxin in healthy male subjects who ingested digoxin 0.25 mg and clopidogrel 75 mg once daily in steady-state conditions.30 The plasma pharmacokinetics and urinary excretion of digoxin for days 10 and 20 were very similar. The clinical, cardiac, and biological evidence indicates that administration of clopidogrel does not enhance the cardiac effects of digoxin.
The potential for DDI between digoxin and ticagrelor was also examined in healthy volunteers.31 Compared with placebo, coadministration of ticagrelor (400 mg once daily) increased the digoxin steady-state Cmax by 75%, steady-state minimum plasma concentration by 31%, and mean AUC by 28%. The renal clearance of digoxin was unaffected in the presence of ticagrelor. In vitro studies have shown that ticagrelor is a substrate and inhibitor of P-gp.32 Increased exposure to digoxin by ticagrelor could be explained by inhibition of intestinal P-gp by ticagrelor and reduced efflux of digoxin into the intestine.
Implications and risk management
Clopidogrel can be added to digoxin for long-term management of patients with cardiac disease. However, serum concentrations of digoxin should be monitored when initiating or changing ticagrelor therapy.
Clopidogrel and efavirenz
DDI and risk description
The medication regimens for patients with human immunodeficiency virus infection and cardiovascular comorbidities are complex and require careful assessment for potentially serious DDIs. Efavirenz is a non-nucleoside reverse transcriptase inhibitor. It is extensively metabolized, predominantly through CYP2B6-mediated 8-hydroxylation, and the 8-hydroxyefavirenz undergoes secondary metabolism to 8,14-dihydroxyefavirenz (catalyzed exclusively by CYP2B6). A population pharmacokinetic study in healthy male Korean subjects showed that clopidogrel could reduce both formation and elimination clearances of 8-hydroxyefavirenz by 22% and 19%, respectively (P<0.05).33 Jiang et al studied the clopidogrel-efavirenz DDI in 17 healthy Korean subjects pregenotyped for the CYP2B6*6 allele. The AUC and Cmax of efavirenz and the AUC of 8-hydroxyefavirenz were significantly higher in the clopidogrel phase than in the placebo phase (P<0.05). The placebo to clopidogrel phase ratio for Cmax, AUC(0,120h), and AUC(0,48h) of 8,14-dihydroxyefavirenz were 0.37, 0.80, and 0.60, respectively.34 The underlying mechanism may be the potent mechanism-based inhibitory effect of clopidogrel on CYP2B6,26 a major enzyme responsible for metabolizing efavirenz. Jiang et al also observed more significant genetic effects in the clopidogrel phase than in the placebo phase. Statistically significant differences in the AUC of efavirenz among genotypes (*1/*1, *1/*6, *6/*6) were not observed in the placebo phase, but were observed in the clopidogrel phase (P<0.05).33 This finding is consistent with in vitro study results showing that CYP2B6*6 is more susceptible than CYP2B6*1 to metabolic inhibition by clopidogrel.35
Implications and risk management
Clopidogrel may decrease the elimination of efavirenz in patients with human immunodeficiency virus, and therefore potentially increase its adverse effects, especially in patients with the CYP2B6*6 genotype. A combination such as efavirenz-ticagrelor may be a better choice, since ticagrelor has little influence on CYP2B6.
Clopidogrel and CYP2C19 substrates
DDI and risk description
Clopidogrel (300 mg on the first day and then 75 mg once daily for 3 consecutive days) could significantly inhibit CYP2C19-dependent hydroxylation of omeprazole (a valid and sensitive method for evaluating the phenotypic activity of CYP2C19). The AUC(0-infinity) of omeprazole in the presence of clopidogrel increased significantly in CYP2C19 homozygous extensive metabolizers, but not in poor metabolizers.36 Nishiya et al compared mechanism-based inhibition of CYP2C19 in human liver microsomes by clopidogrel and prasugrel. Clopidogrel was a potent mechanism-based inhibitor of CYP2C19, with the maximal rate of enzyme inactivation being 0.0557 per minute and an inhibitor concentration that supports half the maximal rate of inactivation of 14.3 μM. In contrast, prasugrel did not inactivate CYP2C19.27 Ticagrelor exhibits little inhibition of CYP2C19.24
Implications and risk management
The prevalence of extensive metabolizers of CYP2C19 exceeds 50% in the population. Given that many substrate drugs of CYP2C19 are widely prescribed and the period of clopidogrel treatment is comparatively long, their interactions with clopidogrel need to be explored. Coadministration of prasugrel or ticagrelor would not cause a clinically relevant interaction with CYP2C19 substrates.
DDIs potentially attenuating the antiplatelet effects of clopidogrel comedicated as an object drug
PPI and clopidogrel
DDI and risk description
Comparative studies have suggested that the extent to which a PPI reduces exposure to the active metabolite of clopidogrel and attenuates its antithrombotic effect differs between the PPIs.8,9,37–40 Omeprazole and esomeprazole have a greater effect on the CYP2C19-mediated conversion of clopidogrel to its active metabolite and the effect of clopidogrel on platelet reactivity (omeprazole > esomeprazole > lansoprazole > dexlansoprazole).38 Concomitantly administered pantoprazole or rabeprazole does not affect the pharmacokinetics and antiplatelet efficacy of clopidogrel.37,41,42
Ferreiro et al reported that even a 12-hour separation of dosing could not prevent drug interactions between omeprazole and clopidogrel; they also observed an interesting phenomenon whereby pharmacodynamic interaction with platelet reactivity changes only occurred in the maintenance phase of clopidogrel treatment (75 mg once daily), but not in the acute phase following a loading dose (600 mg) of clopidogrel,43 indicating that a high loading dose of clopidogrel may overcome the pharmacokinetic DDI between clopidogrel and omeprazole.
Moceri et al observed that esomeprazole could reduce the effect of clopidogrel (75 mg/day) in patients with stable coronary artery disease, with a 38.6% loss in P2Y12 reaction units (PRU, P<0.001) and an eight-fold increase in the prevalence of low responders to clopidogrel. However, doubling the dose of clopidogrel could restore the loss of antiplatelet effect induced by esomeprazole.44 Collet et al reported that the higher platelet inhibitory effect obtained by doubling the clopidogrel maintenance dose (150 mg) could be neutralized by coadministration of lansoprazole, whereas this DDI was not observed with prasugrel 10 mg.45 Oral ranitidine has no significant DDI with concomitantly administered prasugrel or clopidogrel.46 Famotidine is considered to be a safe anti-acid agent for patients taking clopidogrel due to the minimal inhibition of activation of clopidogrel by CYP2B6, CYP2C19, and CYP3A4.47
Implications and risk management
Clinicians should avoid prescribing omeprazole and esomeprazole for patients taking clopidogrel. The potential of PPIs to attenuate the efficacy of clopidogrel could be minimized by use of pantoprazole, dexlansoprazole, or rabeprazole, rather than esomeprazole or omeprazole. A daily dose of 150 mg of clopidogrel has been suggested in the American College of Cardiology/American Heart Association recommendations for patients at very high risk of stent thrombosis. Therefore, the clopidogrel/esomeprazole interaction can be diminished by increasing the dose of clopidogrel to 150 mg or replacing esomeprazole with ranitidine or famotidine. Compared with clopidogrel, prasugrel may be an alternative P2Y12 inhibitor that can escape the adverse DDIs induced by PPIs.45,48
Morphine and clopidogrel
DDI and risk description
Morphine is the recommended treatment for the pain of myocardial infarction. Coprescribing of morphine and clopidogrel is common. A randomized, double-blind, placebo-controlled, crossover trial revealed that morphine could significantly delay the absorption of clopidogrel, reduce the AUC of the active metabolite of clopidogrel by 34%, and retard its antiplatelet effects in 24 healthy subjects who received a loading dose of 600 mg clopidogrel together with placebo or 5 mg morphine intravenously. Morphine delayed the maximal inhibition of platelet aggregation on average by 2 hours (P<0.001) and abolished the three-fold prolongation in collagen adenosine diphosphate-induced closure times in extensive and rapid metabolizers.49 In the placebo phase, median Cmax and AUC values of the active metabolite of clopidogrel decreased in the following order: rapid > extensive > intermediate > poor metabolizers. However, morphine caused a “poor metabolizer phenotype” in individuals genetically prone to extensively metabolize clopidogrel. Morphine reduced the levels of the active metabolite to concentrations usually observed in intermediate or poor metabolizers who are prone to poor clinical outcome. Delayed activity of prasugrel and ticagrelor in the presence of morphine was also observed in patients with ST-segment elevation myocardial infarction (STEMI).50 The underlying mechanism might be that opiates inhibit gastric emptying and delay absorption of oral drugs.
Implications and risk management
The novel and potentially relevant DDIs between morphine and oral P2Y12 receptor inhibitors indicate that coadministration of morphine should be avoided, if possible. More caution should be exercised regarding administration of morphine in patients with STEMI. Cangrelor, an intravenous direct-acting P2Y12 receptor inhibitor, might theoretically be a ideal choice in patients with STEMI receiving morphine.
Scutellarin and clopidogrel
DDI and risk description
Erigeron breviscapus (Vant) Hand-Mazz, a traditional Chinese medicine, is often coprescribed with clopidogrel for the treatment of ischemic vascular disease. Scutellarin is the representative bioactive flavonoid isolated from this herb. In vitro studies using rat liver microsomes showed that scutellarin could significantly inhibit the metabolism of clopidogrel in a concentration-dependent manner, with an IC50 value of 2.1μM. The effect of scutellarin on the pharmacokinetics of clopidogrel was investigated in male rats. After pretreatment with scutellarin, significant increases were observed in the clopidogrel AUC(0-infinity) (0.9±0.4 versus 1.7±0.6ng·hour/mL; P<0.05) and Cmax (0.4±0.1 versus 0.9±0.1 ng/mL; P<0.05), whereas the active metabolite of clopidogrel exhibited significant decreases in AUC(0-infinity) (18.2±5.6 versus 11.4±3.7 ng·hour/mL; P <0.05) and Cmax (8.2±1.2 versus 4.3±0.3 ng/mL; P<0.05).51 The pharmacokinetic changes in clopidogrel and its active metabolite may be due to the inhibitory effects of scutellarin on drug metabolism and/or P-gp transport of clopidogrel.
Implications and risk management
Potential herb-drug interaction between scutellarin and clopidogrel should be taken into consideration in clinical use to avoid the reduced antiplatelet effect, and further study is needed to explore the underlying mechanism for the DDI.
Fluoxetine and clopidogrel
DDI and risk description
Patients receiving antiplatelet treatment to prevent recurrent acute myocardial infarction are often also prescribed a selective serotonin reuptake inhibitor to treat coexisting depression. Both clopidogrel and fluoxetine are among the most widely sold products throughout the world. An open-label crossover study assessed the effect of fluoxetine on the pharmacological activity of clopidogrel in eight healthy volunteers who received a single 600 mg loading dose of clopidogrel followed by 20 mg of fluoxetine for 4 days and then 20 mg of fluoxetine plus 600 mg of clopidogrel on day 5. After coadministration of fluoxetine, the AUC and Cmax of the active metabolite of clopidogrel were 20.6% and 25.3% lower, respectively. The bioavailability of clopidogrel decreased by 23%, with lower percentage maximum platelet aggregation values and platelet reactivity index.52 The underlying mechanism for these changes may include two possibilities. Fluoxetine and norfluoxetine (the major active metabolite) block the formation of the active metabolite of clopidogrel by strongly inhibiting CYP2C19, CYP2C9, and CYP3A4, so potentially reduce the efficacy of clopidogrel, and there is also the possibility of a pharmacodynamic interaction.
There was a study assessing the DDI between ticagrelor and the antidepressant venlafaxine. Healthy subjects received a single 180 mg oral dose of ticagrelor on days 1 and 9 and oral doses of venlafaxine on day 4 (37.5 mg twice daily) and days 5–10 (75 mg twice daily). Ticagrelor had no effect on the area under the time concentration curve over the dosing interval (AUC(0-tau)) of venlafaxine, or the AUC(0-tau) and Cmax of O-desmethylvenlafaxine. Venlafaxine had no effect on the Cmax and AUC(0-infinity) of ticagrelor and its metabolite AR-C124910XX. Ticagrelor and venlafaxine were well tolerated in combination.53
Implications and risk management
Combination use of fluoxetine and clopidogrel should be avoided. Further studies are needed to determine whether DDIs exist between clopidogrel and other antidepressants. Venlafaxine appears to have a low potential for pharmacokinetic interactions with concomitant drugs that are substrates of P-gp and various CYP enzymes,54 and thus may be an alternative antidepressant when there is a need to comedicate with clopidogrel. Combination therapy of ticagrelor and venlafaxine is an alternative regimen for antidepressant and antiplatelet treatment.
Azole antifungal agents and clopidogrel
DDI and risk description
In a randomized crossover study, healthy subjects received a loading dose of prasugrel 60 mg or clopidogrel 300 mg, followed by five daily maintenance doses (15 mg and 75 mg, respectively) with or without the potent CYP3A inhibitor ketoconazole (400 mg/day). Ketoconazole did not affect exposure to the active metabolite of prasugrel (R-138727) or inhibition of platelet aggregation by prasugrel; however, it decreased the AUC(0–24h) of the active metabolite of clopidogrel by 22%–29% and reduced inhibition of platelet aggregation by 28%–33%.55 The underlying mechanism of the different DDI strength may be that inhibition of CYP3A by ketoconazole affects formation of the active metabolite of clopidogrel but not that of prasugrel.
A study by Suh et al revealed that patients with the CYP3A5 non-expressor genotype were vulnerable to DDIs between clopidogrel and CYP3A inhibitors. Clopidogrel was administered to 16 healthy volunteers with the CYP3A5 nonexpressor genotype (*3 allele) and 16 who had the CYP3A5 expressor genotype (*1 allele) with and without pretreatment with itraconazole, a potent CYP3A inhibitor. The change in platelet aggregation after administration of clopidogrel and pretreatment with itraconazole was greater among subjects with the CYP3A5 expressor genotype than in the non-expressor genotype carriers at 4 hours, 24 hours, and 7 days (P<0.01).56
Implications and risk management
Combination of clopidogrel and azole antifungal agents like ketoconazole and itraconazole should be avoided. Prasugrel is an alternative to comedicate with azole antifungal agents. Patients with the CYP3A5 non-expressor genotype are more vulnerable to DDIs between clopidogrel and CYP3A inhibitors than those with the CYP3A5 expressor genotype,56 so pharmacogenetic screening for CYP3A5*3 seems necessary prior to initiating combination therapy of clopidogrel and CYP3A inhibitors.
Calcium channel blockers and clopidogrel
DDI and risk description
CYP2C19 and CYP3A4/5 are the main enzymes involved in the conversion of clopidogrel into the active metabolite. As some calcium channel blockers (CCBs) are inhibitors of CYP3A4, concomitant use of these drugs might play a role in the wide interindividual variability in response to clopidogrel. Meanwhile, some CCBs also have strong inhibitory effects on P-gp, which may cause decreased intestinal efflux of clopidogrel, thereby increasing plasma concentrations of clopidogrel and counteracting the impairing effect of CCBs on the metabolic activation of clopidogrel. Harmsze et al evaluated the effect of coadministration of P-gp-inhibiting CCBs (verapamil, nifedipine, diltiazem, barnidipine) or non-P-gp-inhibiting CCBs (amlodipine) on the on-treatment platelet reactivity (OPR) of clopidogrel in patients on dual antiplatelet therapy after elective PCI. Only the use of amlodipine was significantly associated with a 2.3-fold increased risk of poor response to clopidogrel,57 demonstrating that the DDI between clopidogrel and amlodipine might be more clinically.
Seo et al assessed changes in antiplatelet activity in patients receiving clopidogrel and CCBs for at least 2 months prior to enrollment in the study. The antiplatelet activity of clopidogrel was measured in the same patients while medicated with CCBs and at 8 weeks after discontinuation of CCBs. After discontinuation of the CCBs, angiotensin receptor blockers were newly administered to the patients or dosed up for control of blood pressure. PRU values significantly decreased after discontinuation of CCBs, indicating that CCBs could inhibit the antiplatelet activity of clopidogrel.58
Park et al investigated the relationship between CYP3A4 genotype and the inhibitory effect of CCBs on clopidogrel responsiveness in 1,247 consecutive patients with drug-eluting stent implantation. In total, 332 (26.6%) CCB users had higher clopidogrel OPR compared with 915 (73.4%) non-CCB users. The distribution of the CYP3A4 (IVS10+12G>A) genotype was 63.6%, 32.6%, and 3.8% for the GG, GA, and AA genotypes, respectively. The effects of CCB use, expressed as Δ PRU, were as follows: +8 PRU (patients with 0 A-allele, P=0.210), +24 PRU (patients with 1 A-allele, P=0.012), +50 PRU (patients with 2 A-allele, P=0.025), and +24 PRU (patients with GA/AA genotypes, P=0.005). Furthermore, CCB use significantly increased the risk for high OPR (odds ratio 1.84, P=0.010) only among the GA/AA-genotype carriers, strongly indicating that the number of CYP3A4 (IVS10+12G>A) A-alleles may increase vulnerability to the effects of CCBs on variation in clopidogrel response.59
Implications and risk management
Combination use of clopidogrel and amlodipine should be avoided. When concurrent therapy of clopidogrel and a CCB is initiated, close therapeutic monitoring should be performed to prevent a potentially poor response to clopidogrel, especially in patients with a mutant CYP3A4*1G gene.
Sulfonylureas and clopidogrel
DDI and risk description
Harmsze et al evaluated the relationship between sulfonylurea (oral hypoglycemic) drugs and the clopidogrel OPR in patients with type 2 diabetes mellitus undergoing elective coronary stent implantation. The clopidogrel OPR was significantly higher in patients in the presence of sulfonylureas compared with patients without concomitant sulfonylurea treatment (adjusted odds ratio 2.0, P<0.05).60 The influence of sulfonylureas on the clopidogrel OPR is comparable with that of CYP2C19*2, the genetic variant shown to be of great clinical importance in clopidogrel-treated patients.61 The underlying mechanism might be competitive inhibition of CYP2C9-mediated metabolic activation of clopidogrel by sulfonylureas, which are extensively metabolized by CYP2C9. A randomized, double-blind, two-period, crossover study in healthy volunteers showed insignificant DDI between ticagrelor and tolbutamide. Ticagrelor 180 mg twice daily for 9 days had no effect on the steady-state pharmacokinetics of tolbutamide, and tolbutamide had no effect on the pharmacokinetics of either ticagrelor or its active metabolite. Coadministration of ticagrelor and tolbutamide was well tolerated. The underlying mechanism may be that ticagrelor undergoes extensive CYP3A4-mediated metabolism and thus is unlikely to be susceptible to the presence of sulfonylureas.62
Implications and risk management
Ticagrelor should be considered and clopidogrel should be avoided when there is a need to initiate combination therapy of sulfonylureas and P2Y12 receptor inhibitors.
Ritonavir and clopidogrel
DDI and risk description
A review published in June 2014 pointed out that clopidogrel does not appear to have clinically significant interactions with protease inhibitors.7 However, Metzger and Momary reported a case of clopidogrel non-responsiveness in a patient with human immunodeficiency virus, latent tuberculosis, cardiovascular disease, and a history of myocardial infarction.63 The case involved a significant DDI between clopidogrel, isoniazid, and ritonavir. The underlying mechanism might be that isoniazid and ritonavir could impair the activation of clopidogrel since isoniazid is a potent inhibitor of CYP2C19/CYP3A and ritonavir is a potent CYP3A inhibitor. Ancrenaz et al assessed the effect of ritonavir 100 mg on the pharmacokinetics of the active metabolite of prasugrel in healthy volunteers. In the presence of ritonavir, CYP3A4 activity as reflected by the midazolam metabolic ratio was significantly impaired. The Cmax and AUC of the active metabolite of prasugrel were decreased by 45% (P<0.01) and 38% (P<0.01), respectively.64 The underlying mechanism may be a potent simultaneous inhibition of bioactivation of prasugrel via CYP2B6 and CYP3A by ritonavir.65 The prasugrel-ritonavir DDI might lead to a significant reduction in the efficacy of prasugrel in patients with human immunodeficiency virus infection and acute coronary syndrome.
Implications and risk management
Clinicians should underscore the importance of a detailed DDI screening in patients with an infectious disease who are taking complex medication regimens containing clopidogrel or prasugrel. Management of these interactions should be as follows: avoiding combinations such as ritonavir-clopidogrel and ritonavir-prasugrel (CYP3A4-mediated inhibition by ritonavir and potential lower antiplatelet effect) and clopidogrel-efavirenz (CYP2B6-mediated inhibition by clopidogrel and potential increased efavirenz toxicity);34 choosing a combination such as efavirenz-ticagrelor or ritonavir-ticagrelor since ticagrelor does not require metabolic activation; and selecting antiretroviral medications with a lower DDI potential. Nevirapine is an inducer of CYP3A and CYP2B6, and is usually combined with other antiretroviral medications to avoid resistance to anti-human immunodeficiency virus treatment.7 Theoretically, nevirapine may enhance the metabolic activation of clopidogrel and offset the adverse DDI between clopidogrel and antiretroviral medications with inhibitory effects on CYP3A4. Further studies are necessary to confirm this assumption.
DDIs potentially augmenting the antiplatelet effects of clopidogrel comedicated as an object drug
Aspirin and clopidogrel
A study showed that aspirin treatment induced expression of multidrug-resistance protein 1 (MDR1) in human epithelial colorectal (Caco-2) cells in vitro and in the rat intestine in vivo. Along with upregulation of MDR1 by aspirin, absorption of clopidogrel significantly decreased in aspirin-treated Caco-2 cells and in the rat intestine.66 A clinical study evaluated the effect of aspirin coadministration on the pharmacokinetics/pharmacodynamics of clopidogrel in humans. A single 75 mg dose of clopidogrel was orally administered before and after 2 and 4 weeks of administration of aspirin100 mg once daily in 18 healthy volunteers. After aspirin pretreatment, the P-gp microRNA miR-27a increased by up to 7.67-fold (P=0.004) and the clopidogrel AUC decreased by 14%, but the AUC of the active metabolite remained unchanged and relative platelet inhibition increased by up to 15% (P=0.002).67 Another study confirmed that concentrations of the active metabolite of clopidogrel 1 hour post loading dose (300 or 600 mg) were similar in patients taking different doses of aspirin (325 or 81 mg).68 These findings indicate that coadministration of low-dose aspirin may decrease the bioavailability of clopidogrel but does not decrease its efficacy.
Taubert et al confirmed a correlation of the MDR1 C3435T genotype with the Cmax and AUC of clopidogrel and its active metabolite after a single oral loading dose of 300 or 600 mg in patients with coronary artery disease who underwent PCI. In the 300 mg and 600 mg groups, the Cmax and AUC values for clopidogrel and its active metabolite were lower in subjects homozygous for the MDR1 3435T variant compared with subjects with the 3435C/T and 3435C/C genotypes.69 Chen et al observed that administration of aspirin 50 mg/day for 7 days and 14 days induced in vivo activity of CYP2C19 in healthy volunteers.70 The exact mechanism for this induction remains unclear. Concomitant aspirin might induce CYP2C19-mediated bioactivation of clopidogrel; this assumption is worthy of further investigation. Paraoxonase-1 (PON1) is also a key factor in the bioactivation and clinical activity of clopidogrel.71 In mice, aspirin can cause a two-fold increase in plasma PON1 activity and significant induction of PON1 gene expression in the liver.72 Coadministered aspirin might further enhance the pharmacodynamics of clopidogrel by induction of CYP2C19 and PON1.
Collectively, we assume that the influence of aspirin on the pharmacodynamics of clopidogrel is comprehensive. In addition to a purely pharmacodynamic synergistic effect (ie, low-dose aspirin enhances the antithrombotic action of clopidogrel by blocking the formation of thromboxane A2), it may also involve the balance of aspirin’s “advantageous” inducible effects on CYP2C19 and PON1 and “disadvantageous” inducible effects on P-gp. Evidence from pharmacokinetic DDI studies seems to provide a new basis for dual therapy with clopidogrel and aspirin, which is currently the gold standard treatment after PCI.
Curcumin and clopidogrel
Curcumin, the major component of turmeric, has a variety of potentially beneficial health effects. Oral administration of curcumin 100 mg/kg for 7 days significantly increased the AUC and Cmax of clopidogrel carboxylic acid by 1.61 and 1.81, respectively, in Wistar rats. However, a combination of curcumin and clopidogrel had no significant effect on the maximum platelet aggregation rate in rats compared with the use of clopidogrel alone.73 In vivo and ex vivo studies revealed that oral intake of curcumin could markedly activate CYP3A4. Moreover, curcumin is a P-gp inhibitor. Coadministration of curcumin significantly increased the plasma concentration of talinolol (a typical P-gp probe substrate) in healthy volunteers and the effect of curcumin on talinolol was associated with ABCB1 genotypes (C3435T).74 Given that clopidogrel is a substrate of CYP3A4 and P-gp and that curcumin has inhibitory effects on these two biomacromolecules, DDIs between curcumin and clopidogrel may exist. Curcumin may be a potential booster of clopidogrel efficacy. Further clinical studies need to be done to confirm the assumption.
St John’s wort and clopidogrel
Hypericum perforatum, more commonly known as St John’s wort (SJW), is a popular medicinal herb used for the treatment of depression. It affects the pharmacokinetics of many drugs by inducing CYP3A4, CYP2C19, CYP2C9, and P-gp. DDI between H. perforatum and clopidogrel seems to have benefits (eg, an increase in clopidogrel responsiveness).75
Trana et al confirmed that SJW might represent a valid option to improve clopidogrel responsiveness in patients with stable coronary artery disease. Stable angina patients who were non-responders to clopidogrel 600 mg were randomized to SJW (n=15) or placebo (n=8). SJW (300 mg three times daily) was administered for 2 weeks after PCI. Platelet reactivity was assessed before (baseline) and 2 (T1) and 4 weeks (T2) after PCI. PRU changes from baseline were higher at T1 in the SJW group than in the placebo group (P=0.0033), with no differences between the groups at T2.75 Residual platelet reactivity improved with SJW during the first month post-PCI, which is the most vulnerable time window for the patient in terms of increased thrombotic risk. Lau et al evaluated whether SJW enhances the pharmacodynamic response of clopidogrel. Hyporesponsive volunteers (n=10) received SJW (300 mg three times daily) for 14 days followed by a single 300 mg dose of clopidogrel. SJW decreased platelet aggregation at 2, 4, and 6 hours (P<0.05) and increased CYP3A4 activity (P=0.002). It was also observed that SJW significantly decreased platelet reactivity and increased platelet inhibition in post-coronary stent patients with hyporesponsiveness to clopidogrel.76 The available evidence indicates that SJW may be a future therapeutic option to increase the antiplatelet effect of clopidogrel in hyporesponders.
Cyclosporine and clopidogrel
In clinical practice, antiplatelet agents may be given to patients who are also receiving cyclosporine, a P-gp inhibitor. Therefore, it is necessary to conduct the relevant DDI study. Coadministration of cyclosporine (10 mg/kg) significantly increased the AUC and Cmax of clopidogrel carboxylic acid in rats.77,78 However, in dogs, the plasma concentrations of clopidogrel carboxylic acid were not considerably changed by coadministration of cyclosporine.78 The pharmacokinetic DDI between ticagrelor and cyclosporine was assessed in healthy volunteers who were randomized to receive cyclosporine (600 mg, as a single oral dose) plus ticagrelor (180 mg, as a single oral dose), cyclosporine alone, or ticagrelor alone. Coadministration of cyclosporine with ticagrelor resulted in statistically significant increases in the AUC of ticagrelor and that of its active metabolite AR-C124910XX (183% and 33%, respectively), but had no effect on the pharmacokinetics of cyclosporine.79
The findings of the cyclosporine-clopidogrel DDI research suggest that the risk of DDI differs between animal models. A follow-up clinical study is needed to explore the relevance of these remarkable species differences in P-gp-mediated interaction. The magnitude of the effect of cyclosporine on the pharmacokinetics of ticagrelor does not warrant dose adjustment of the latter.
Rifampicin and clopidogrel
DDI and risk description
A study addressed the pharmacokinetic/pharmacodynamic DDI between rifampicin and clopidogrel. Healthy volunteers received a 600 mg loading dose of clopidogrel followed by 75 mg daily for 7 days. After a washout period, they received treatment with rifampicin (300 mg twice a day) and the same regimen of clopidogrel. Rifampicin increased the AUC of the active metabolite of clopidogrel (clopidogrel alone 89±22 ng·hour/mL, clopidogrel + rifampicin 335±86 ng·hour/mL, P<0.0001) enhanced P2Y12 blockade (unblocked receptors: clopidogrel 48±24, clopidogrel + rifampicin 4±2, P<0.0001), and reduced platelet aggregation (P<0.01).80
In contrast, coadministration with rifampicin could reduce ticagrelor exposure and resulted in a more rapid offset of ticagrelor-mediated inhibition of platelet aggregation.81 Healthy volunteers received a single 180 mg oral dose of ticagrelor on days 1 and 15, and rifampicin 600 mg once daily on days 4–17. Comedicated rifampicin significantly decreased the Cmax of ticagrelor from 1,091 to 297.8 ng/mL, the AUC of ticagrelor from 6,225 to 864.0 ng·hour/mL, and ticagrelor plasma half-life from 8.4 to 2.8 hours. The Cmax of AR-C124910XX was unaffected by rifampicin, whereas the AUC was significantly decreased by 46%, and the metabolite to parent ratio for AUC increased four-fold. Although the maximal inhibition of platelet aggregation was unaffected, the offset of ticagrelor-mediated inhibition of platelet aggregation was more rapid in the presence of rifampicin. The underlying mechanism may be that ticagrelor is predominantly metabolized by CYP3A and both the parent compound and its active metabolite (AR-C124910XX) are substrates of P-gp, whereas rifampicin is a strong inducer of CYP3A and P-gp.
Farid et al assessed the DDI between rifampicin and prasugrel in healthy male subjects. In the first treatment period, the subjects received prasugrel as an oral 60 mg loading dose on the first day followed by ten oral 10 mg daily maintenance doses. After a 2-week washout period, subjects received oral rifampicin alone (600 mg once daily) for 8 days, followed by treatment with oral rifampicin and the same regimen of prasugrel. Coadministration of rifampicin did not affect exposure to the active metabolite of prasugrel (R-138727), indicating that formation of R-138727 was not affected by potent enzyme induction with rifampicin. Coadministration of rifampicin with prasugrel causes only a small reduction in inhibition of platelet aggregation, and such an interaction is unlikely to be clinically significant.82
Implications and risk management
Combination use of rifampicin can augment the antiplatelet efficacy of clopidogrel; however, it is necessary to know the increased risk of clopidogrel-associated bleeding. Coadministration of rifampicin with ticagrelor should be discouraged. Dose adjustment is not necessary when prasugrel is administered with rifampicin.
Angiotensin-converting enzyme inhibitors and clopidogrel
DDI and risk description
Most angiotensin-converting enzyme inhibitors (ACEIs) are metabolized by hepatic carboxylesterase 1 (CES1), a metabolizing enzyme that plays a significant role in the metabolism of clopidogrel. Kristensen et al examined the effects of ACEIs on the bioactivation of clopidogrel in vitro and linked the results with a pharmacoepidemiological study. All tested ACEIs increased the bioactivation of clopidogrel in S9 human liver fractions. Coincubation of clopidogrel with trandolapril or enalapril significantly increased formation of the active metabolite of clopidogrel. In 70,934 patients with myocardial infarction, hazard ratios for clinically significant bleeding in ACEI-treated patients comedicated with or without clopidogrel were 1.10 and 0.90, respectively, as compared with patients who did not receive ACEIs. The difference between these two hazard ratios, expressed as the hazard rate ratio, was statistically significant (hazard rate ratio 1.27; 95% confidence interval 1.09–1.49; P=0.002).83
Implications and risk management
Combination therapy of clopidogrel and ACEI may be favorable in hyporesponsive patients receiving treatment with clopidogrel due to enhanced antiplatelet effects; however, vigilance should be exercised regarding the elevated risk of bleeding events. Aspirin, prasugrel, and ticagrelor are not metabolized by CES1 (aspirin and prasugrel are hydrolyzed by CES2, a distinct CES predominantly present in the intestine, and ticagrelor is not a prodrug) and are therefore not subject to CES1-mediated DDI.
Tobacco smoking and clopidogrel
Cigarette smoking remains highly prevalent in most countries, and may affect drug therapy by both pharmacokinetic and pharmacodynamic mechanisms. The drug interaction between clopidogrel and cigarette smoking is interesting. Yousef et al reported that smoking was a significant factor affecting the pharmacokinetics of clopidogrel following administration of a single 75 mg dose in young healthy volunteers. Smokers had a lower AUC(0-infinity) (6.24±2.32 μg hour/mL versus 8.93±3.80 μg hour/mL, respectively; P<0.001) and a shorter half-life (5.46±2.99 versus 8.43±4.26, P=0.001) for clopidogrel than non-smokers, whereas smoking behavior had no influence on the Cmax and Tmax of clopidogrel. That study supports recommendations regarding smoking cessation.84 However, the investigators did not evaluate the influence of smoking on the pharmacokinetics of the active metabolite of clopidogrel or its clinical effects. Hochholzer et al confirmed that smoking does not impact on platelet reactivity in patients after a loading dose or on maintenance doses of clopidogrel.85
Park et al reported an interesting finding, ie, there was a genotype-dependent effect of smoking on clopidogrel responsiveness, ie, an enhanced clopidogrel response in smokers, known as the smokers’ paradox, which was not universal and only observed in carriers of the CYP1A2 (-163C>A) A-allele.86 The clopidogrel OPR was measured in 1,431 consecutive patients who underwent coronary angiography. Two hundred and forty-nine (17%) smokers had lower OPR compared with 1,182 (83%) non-smokers (227.6±76.0 versus 244.9±79.7, P=0.001). CYP1A2 showed a genotype-dependent change in the effect of cigarette smoking on OPR. After adjustment for possible confounding factors, smoking was associated with a lower OPR (odds ratio 0.48; 95% confidence interval 0.31–0.74) in patients with the AA or CA genotype but not in those with the CC genotype.
Gurbel et al evaluated the interaction between smoking and clopidogrel or prasugrel in patients with stable coronary artery disease. Clopidogrel therapy (75 mg daily) was associated with lower calculated inhibition of platelet aggregation, higher PRU, vasodilator-stimulated phosphoprotein phosphorylation, and platelet reactivity index in non-smokers than in smokers (all P-values <0.05), demonstrating lower clopidogrel active metabolite exposure and weaker pharmacodynamic effects of clopidogrel in non-smokers relative to smokers. Prasugrel 10 mg daily was associated with greater active metabolite exposure and pharmacodynamic effects than clopidogrel regardless of smoking status (P<0.001).87
The mechanism involved in most interactions between cigarette smoking and drugs involves the induction of metabolism (eg, CYP1A). CYP1A2 plays a minor role in metabolic activation of clopidogrel in humans, as indicated by an in vitro CYP reaction phenotyping test and insignificant DDI between clopidogrel and CYP1A2 substrate theophylline in volunteers indicate CYP1A2 plays a minor role in metabolic activation of clopidogrel in human.12,88 Thus, the underlying mechanism regarding the interaction between smoking and clopidogrel is currently unclear. For smokers with the CYP1A2 (-163CC) genotype, the smokers’ paradox does not exist and prasugrel may be a better choice than clopidogrel in this population.
Food and clopidogrel
Fed condition versus fasted condition
Interactions between food and drugs may affect drug therapy. Nirogi et al observed that food could substantially enhance the bioavailability of clopidogrel in healthy subjects. The Cmax, and AUC0-infinity of clopidogrel increased 6.1-fold and 9.2-fold, respectively, in the fed condition compared with the fasted condition. The elimination half-life increased from 2.5 hours in the fasted state to 5.0 hours in the fed state.89 However, their study did not address changes in the pharmacokinetics of the active metabolite of clopidogrel or the antiplatelet effects. Hurbin et al characterized the effects of a high-fat or standard breakfast on adenosine diphosphate-induced platelet aggregation and exposure to clopidogrel and the active metabolite following treatment with clopidogrel (300 mg loading dose, 75 mg/day for 4 days) in 72 healthy men, and confirmed that clopidogrel can be taken with or without food.90 Administration of clopidogrel with a standard breakfast resulted in a 3.32-fold increase in the AUC(0–24h) of clopidogrel, but with a nonsignificant decrease in the AUC(0–24h) of the active metabolite. The numerical increase in maximum platelet aggregation in the fed state versus fasted state was small. Also, food had no statistically significant effect on the pharmacokinetic profile of ticagrelor or prasugrel.91,92
Grapefruit juice and clopidogrel
CYP2C19 and CYP3A4 participate in the bioactivation of clopidogrel whereas grapefruit juice constituents potently inactivate intestinal CYP3A4 and also inhibit CYP2C19.12 In a randomized crossover study in 14 healthy volunteers, grapefruit juice (200 mL three times daily for 3 days) reduced the Cmax of the active metabolite of clopidogrel to 13% of the control value (P<0.001) and the AUC(0–3h) to 14% of the control value (P<0.001) following a single dose of 600 mg, but had no significant effect on the parent clopidogrel. Moreover, grapefruit juice markedly decreased the platelet-inhibiting effect of clopidogrel.93
In contrast, a randomized crossover study reported that grapefruit juice could increase ticagrelor exposure by more than two-fold, leading to an enhanced and prolonged antiplatelet effect of ticagrelor.94 The mechanism of this clinically relevant interaction involves the inhibitory effect of grapefruit juice on intestinal metabolism and transport of ticagrelor, a substrate of CYP3A4 and P-gp. Therefore, grapefruit juice is best avoided during clopidogrel therapy. Clinicians and patients should be aware of the enhanced antiplatelet effect of ticagrelor in the presence of grapefruit juice to avoid the risk of potential bleeding events.
Summary of information for clopidogrel DDIs
A summary of clopidogrel-associated DDIs was presented in Table 1. The factors determining the degree of pharmacokinetic DDI with clopidogrel include genetic status, ie, CYP2B6*6 (sibutramine), CYP2C19 polymorphism (omeprazole), CYP3A5*3 (CYP3A inhibitors), CYP3A4*1G (CCBs), and CYP1A2-163C>A (smoking), species differences (clopidogrel-cyclosporine), and clopidogrel dose strength (load dosing versus maintenance dosing) regarding DDIs with omeprazole, esomeprazole, and rosuvastatin. The DDI risk with clopidogrel does not exhibit a class effect, eg, the effects of clopidogrel on cerivastatin versus other statins, the effects of PPIs on clopidogrel (omeprazole and esomeprazole versus pantoprazole and rabeprazole), the effects of rifampicin on clopidogrel versus other P2Y12 receptor inhibitors (ticagrelor and prasugrel) and the effects of CCBs on clopidogrel (amlodipine versus P-gp-inhibiting CCBs). The mechanism for clopidogrel-associated DDIs involves modulation of CYPs (eg, CYP2B6, CYP2C8, CYP2C19, and CYP3A4), PON1, CES1, and drug transporters like P-gp and OATP1B1. Some DDIs of clopidogrel involve a balance of “advantageous” and “disadvantageous” effects (eg, DDIs of aspirin-clopidogrel and P-gp-inhibiting CCBs-clopidogrel).
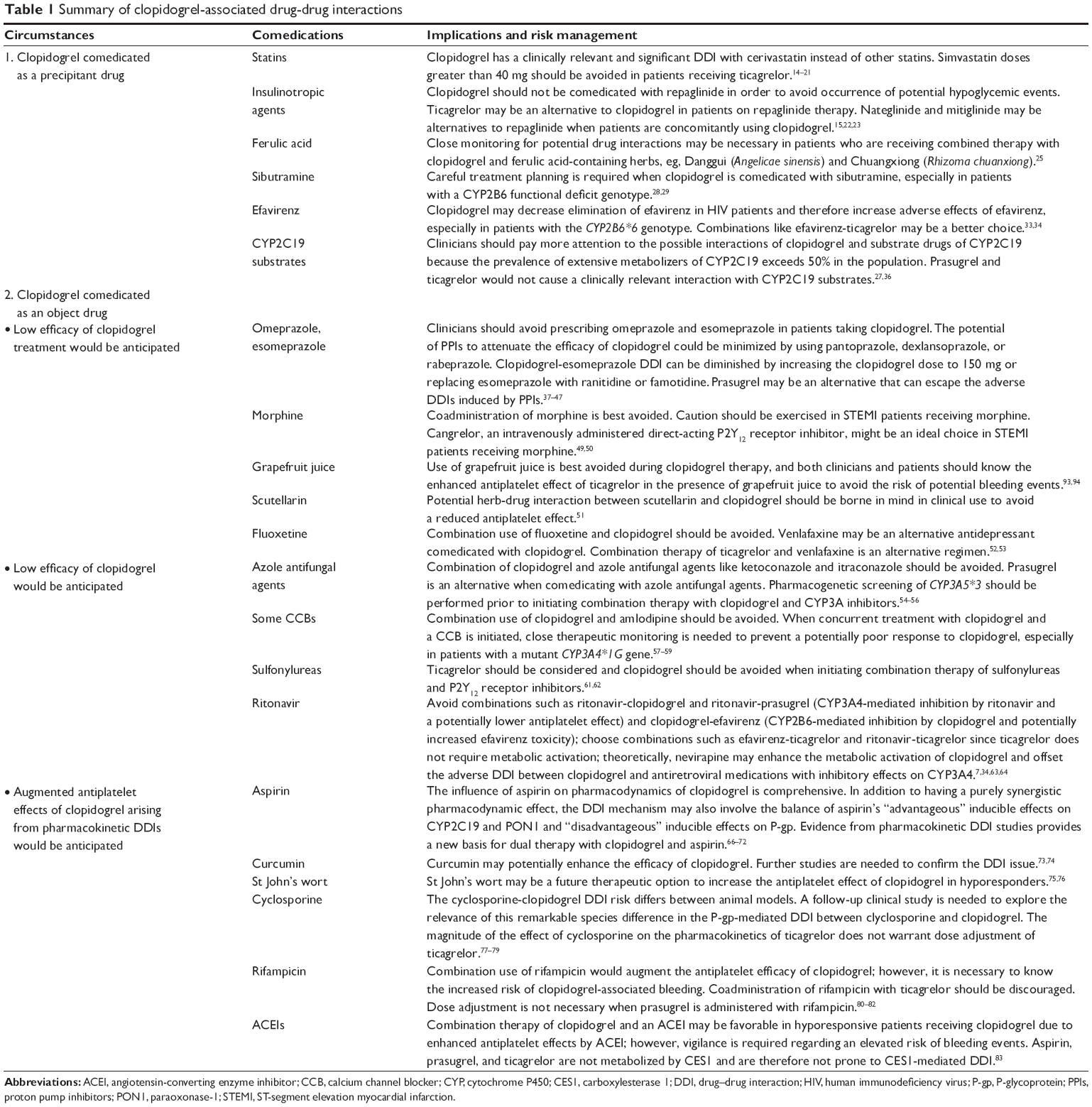 | Table 1 Summary of clopidogrel-associated drug-drug interactions |
Further research opportunities
Further studies are needed. First, not all the DDIs of clopidogrel have been studied in humans, either from a pharmacokinetic or a clinical perspective. Second, the clinical validity or relevance of the DDIs between clopidogrel and some comedications should be further addressed in patients receiving long-term regimens. Third, it needs to be confirmed whether some theoretically feasible drug combinations genuinely do have a lower risk of clinically relevant DDIs. Fourth, comparative studies of DDIs associated with P2Y12 receptor inhibitors should be encouraged. Finally, there are still many circumstances under which clopidogrel and other drugs are concomitantly used but whether DDIs exist between them has not been investigated. For example, clopidogrel hypersensitivity affects up to 6% of treated patients, most commonly in the form of a pruritic rash. It can be successfully treated using short-course corticosteroids and antihistamines without interrupting drug therapy.95 In addition, a paradox phenomenon has occurred, ie, recurrent acute stent thrombosis due to allergic reaction secondary to clopidogrel therapy has been reported when clopidogrel is given to prevent stent thrombosis.96,97 The management of such complex patients may need to focus on treatment of the allergic or hematologic reaction itself in addition to ensuring effective antiplatelet therapy. It would be worthwhile to investigate the DDIs between clopidogrel and antiallergy agents in the future.
Integrated care is essential for complex patients. In order to enable even more synergy between health care professionals in the care of complex patients, pharmacists should focus on management of medication and play a key role in collaborative practice. Around the scientific question about the DDIs of clopidogrel as well as new P2Y12 receptor inhibitors (prasugrel, ticagrelor, and cangrelor), physicians and pharmacists can do a lot of things.
This review has some limitations. We did not present our paper in the form of a Cochrane-style systematic review or meta-analysis, which can resolve limitations due to sample size, although controversial conclusions derived from clopidogrel-associated DDI studies are rare. Also, other databases like EMBASE could not be used due to inaccessibility in the People’s Republic of China. However, a new horizon might be opened up by this updated review with plenty of new findings.
Conclusion
In this review, we specifically addressed the pharmacokinetic DDIs of clopidogrel, presenting interesting research findings from recent years, such as the circumstances under which DDIs occur when clopidogrel is the precipitant drug, the factors determining the degree of DDI, and the corresponding risk management. Effective and safe combination therapy using clopidogrel can be achieved by increasing the awareness of potential changes in therapeutic efficacy and adverse drug reactions, rationally prescribing alternatives, tailoring drug therapy based on genotype, checking the appropriateness of physician orders, and performing pharmacotherapeutic monitoring.
Acknowledgments
This work was supported by grants from the Zhejiang Provincial Bureau of Education (N20140209), the National Natural Science Foundation of China (81373488), and the National Major Projects of China (2012ZX09506001-004).
Disclosure
The authors report no conflicts of interest in this work.
References
Li W, Zeng S, Yu LS, Zhou Q. Pharmacokinetic drug interaction profile of omeprazole with adverse consequences and clinical risk management. Ther Clin Risk Manag. 2013;9:259–271. | ||
Zhou YT, Yu LS, Zeng S, Huang YW, Xu HM, Zhou Q. Pharmacokinetic drug–drug interactions between 1,4-dihydropyridine calcium channel blockers and statins: factors determining interaction strength and relevant clinical risk management. Ther Clin Risk Manag. 2014;10:17–25. | ||
Joint Commission International. Accreditation Standards for Hospitals. 5th ed. Oak Brook, IL, USA: Joint Commission Resources; 2013. | ||
Wikipedia. Clopidogrel. Available from: http://en.wikipedia.org/wiki/Clopidogrel. Accessed January 2, 2014. | ||
Kounis NG, Grapsas N. Pathways of platelet activation and unexplained clopidogel variability: causes of poor response to clopidogrel. Thromb Res. 2013;132:312. | ||
Ferri N, Corsini A, Bellosta S. Pharmacology of the new P2Y12 receptor inhibitors: insights on pharmacokinetic and pharmacodynamic properties. Drugs. 2013;73:1681–1709. | ||
Egan G, Hughes CA, Ackman ML. Drug interactions between antiplatelet or novel oral anticoagulant medications and antiretroviral medications. Ann Pharmacother. 2014;48:734–740. | ||
Shah BS, Parmar SA, Mahajan S, Mehta AA. An insight into the interaction between clopidogrel and proton pump inhibitors. Curr Drug Metab. 2012;13:225–235. | ||
Scott SA, Owusu Obeng A, Hulot JS. Antiplatelet drug interactions with proton pump inhibitors. Expert Opin Drug Metab Toxicol. 2014;10:175–189. | ||
Siller-Matula JM, Trenk D, Krähenbühl S, Michelson AD, Delle-Karth G. Clinical implications of drug–drug interactions with P2Y12 receptor inhibitors. J Thromb Haemost. 2014;12:2–13. | ||
Tabassome S, Celine V, Murielle MK, et al. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med. 2009;360:363–375. | ||
Kazui M, Nishiya Y, Ishizuka T, et al. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos. 2010;38:92–99. | ||
Fareed J, Jeske W, Thethi I. Metabolic differences of current thienopyridine antiplatelet agents. Expert Opin Drug Metab Toxicol. 2013;9:307–317. | ||
Floyd JS, Kaspera R, Marciante KD, et al. A screening study of drug–drug interactions in cerivastatin users: an adverse effect of clopidogrel. Clin Pharmacol Ther. 2012;91:896–904. | ||
Tornio A, Filppula AM, Kailari O, et al. Glucuronidation converts clopidogrel to a strong time-dependent inhibitor of CYP2C8: a phase II metabolite as a perpetrator of drug–drug interactions. Clin Pharmacol Ther. 2014;96:498–507. | ||
Tamraz B, Fukushima H, Wolfe AR, et al. OATP1B1-related drug–drug and drug-gene interactions as potential risk factors for cerivastatin-induced rhabdomyolysis. Pharmacogenet Genomics. 2013;23:355–364. | ||
Schmidt M, Johansen MB, Maeng M, et al. Concomitant use of clopidogrel and statins and risk of major adverse cardiovascular events following coronary stent implantation. Br J Clin Pharmacol. 2012;74:161–170. | ||
Farid NA, Small DS, Payne CD, et al. Effect of atorvastatin on the pharmacokinetics and pharmacodynamics of prasugrel and clopidogrel in healthy subjects. Pharmacotherapy. 2008;28:1483–1494. | ||
Ayalasomayajula SP, Vaidyanathan S, Kemp C, Prasad P, Balch A, Dole WP. Effect of clopidogrel on the steady-state pharmacokinetics of fluvastatin. J Clin Pharmacol. 2007;47:613–619. | ||
Pinheiro LF, França CN, Izar MC, et al. Pharmacokinetic interactions between clopidogrel and rosuvastatin: effects on vascular protection in subjects with coronary heart disease. Int J Cardiol. 2012;158:125–129. | ||
Teng R, Mitchell PD, Butler KA. Pharmacokinetic interaction studies of co-administration of ticagrelor and atorvastatin or simvastatin in healthy volunteers. Eur J Clin Pharmacol. 2013;69:477–487. | ||
Scheen AJ. Drug–drug and food-drug pharmacokinetic interactions with new insulinotropic agents repaglinide and nateglinide. Clin Pharmacokinet. 2007;46:93–108. | ||
Yu L, Lu S, Lin Y, Zeng S. Carboxyl-glucuronidation of mitiglinide by human UDP-glucuronosyltransferases. Biochem Pharmacol. 2007;73:1842–1851. | ||
Zhou D, Andersson TB, Grimm SW. In vitro evaluation of potential drug–drug interactions with ticagrelor: cytochrome P450 reaction phenotyping, inhibition, induction, and differential kinetics. Drug Metab Dispos. 2011;39:703–710. | ||
Li Y, Liu C, Zhang Y, Mi S, Wang N. Pharmacokinetics of ferulic acid and potential interactions with Honghua and clopidogrel in rats. J Ethnopharmacol. 2011;137:562–567. | ||
Bae SK, Cao S, Seo KA, et al. Cytochrome P450 2B6 catalyzes the formation of pharmacologically active sibutramine (N-{1-[1-(4-chlorophenyl)cyclobutyl]-3-methylbutyl}-N,N-dimethylamine) metabolites in human liver microsomes. Drug Metab Dispos. 2008;36:1679–1688. | ||
Nishiya Y, Hagihara K, Kurihara A, et al. Comparison of mechanism-based inhibition of human cytochrome P450 2C19 by ticlopidine, clopidogrel, and prasugrel. Xenobiotica. 2009;39:836–843. | ||
Bae JW, Jang CG, Lee SY. Effects of clopidogrel on the pharmacokinetics of sibutramine and its active metabolites. J Clin Pharmacol. 2011;51:1704–1711. | ||
Pan W, Bae SK, Shim EJ, et al. Effects of clopidogrel and clarithromycin on the disposition of sibutramine and its active metabolites M1 and M2 in relation to CYP2B6*6 polymorphism. Xenobiotica. 2013;43:211–218. | ||
Peeters PA, Crijns HJ, Tamminga WJ, Jonkman JH, Dickinson JP, Necciari J. Clopidogrel, a novel antiplatelet agent, and digoxin: absence of pharmacodynamic and pharmacokinetic interaction. Semin Thromb Hemost. 1999;25 Suppl 2:51–54. | ||
Teng R, Butler K. A pharmacokinetic interaction study of ticagrelor and digoxin in healthy volunteers. Eur J Clin Pharmacol. 2013;69:1801–1808. | ||
RxList®. BRILINTA® (ticagrelor) tablets. New York, NY, USA. Available from: http://www.rxlist.com/brilinta-drug/clinical-pharmacology.htm. Accessed January 1, 2014. | ||
Abdelhady AM, Desta Z, Jiang F, Yeo CW, Shin JG, Overholser BR. Population pharmacogenetic-based pharmacokinetic modeling of efavirenz, 7-hydroxy- and 8-hydroxyefavirenz. J Clin Pharmacol. 2014;54:87–96. | ||
Jiang F, Desta Z, Shon JH, et al. Effects of clopidogrel and itraconazole on the disposition of efavirenz and its hydroxyl metabolites: exploration of a novel CYP2B6 phenotyping index. Br J Clin Pharmacol. 2013;75:244–253. | ||
Xu C, Ogburn ET, Guo Y, Desta Z. Effects of the CYP2B6*6 allele on catalytic properties and inhibition of CYP2B6 in vitro: implication for the mechanism of reduced efavirenz metabolism and other CYP2B6 substrates in vivo. Drug Metab Dispos. 2012;40:717–725. | ||
Chen BL, Chen Y, Tu JH, et al. Clopidogrel inhibits CYP2C19-dependent hydroxylation of omeprazole related to CYP2C19 genetic polymorphisms. J Clin Pharmacol. 2009;49:574–581. | ||
Angiolillo DJ, Gibson CM, Cheng S, et al. Differential effects of omeprazole and pantoprazole on the pharmacodynamics and pharmacokinetics of clopidogrel in healthy subjects: randomized, placebo-controlled, crossover comparison studies. Clin Pharmacol Ther. 2011;89:65–74. | ||
Frelinger AL 3rd, Lee RD, Mulford DJ, et al. A randomized, 2-period, crossover design study to assess the effects of dexlansoprazole, lansoprazole, esomeprazole, and omeprazole on the steady-state pharmacokinetics and pharmacodynamics of clopidogrel in healthy volunteers. J Am Coll Cardiol. 2012;59:1304–1311. | ||
Johnson DA, Chilton R, Liker HR. Proton-pump inhibitors in patients requiring antiplatelet therapy: new FDA labeling. Postgrad Med. 2014;126:239–245. | ||
Harmsze AM, van Werkum JW, Taubert D, Hackeng CM, Deneer VH. Esomeprazole but not pantoprazole is associated with lower plasma concentrations of clopidogrel’s active metabolite. Ann Pharmacother. 2011;45:542–543. | ||
Funck-Brentano C, Szymezak J, Steichen O, et al. Effects of rabeprazole on the antiplatelet effects and pharmacokinetics of clopidogrel in healthy volunteers. Arch Cardiovasc Dis. 2013;106:661–671. | ||
Wu J, Jia LT, Shao LM, et al. Drug–drug interaction of rabeprazole and clopidogrel in healthy Chinese volunteers. Eur J Clin Pharmacol. 2013;69:179–187. | ||
Ferreiro JL, Ueno M, Capodanno D, et al. Pharmacodynamic effects of concomitant versus staggered clopidogrel and omeprazole intake: results of a prospective randomized crossover study. Circ Cardiovasc Interv. 2010;3:436–441. | ||
Moceri P, Doyen D, Cerboni P, Ferrari E. Doubling the dose of clopidogrel restores the loss of antiplatelet effect induced by esomeprazole. Thromb Res. 2011;128:458–462. | ||
Collet JP, Hulot JS, Abtan J, et al. Prasugrel but not high dose clopidogrel overcomes the lansoprazole neutralizing effect of P2Y12 inhibition: results of the randomized DOSAPI study. Eur J Clin Pharmacol. 2014;70:1049–1057. | ||
Small DS, Farid NA, Li YG, et al. Effect of ranitidine on the pharmacokinetics and pharmacodynamics of prasugrel and clopidogrel. Curr Med Res Opin. 2008;24:2251–2257. | ||
Ohbuchi M, Noguchi K, Kawamura A, Usui T. Different effects of proton pump inhibitors and famotidine on the clopidogrel metabolic activation by recombinant CYP2B6, CYP2C19 and CYP3A4. Xenobiotica. 2012;42:633–640. | ||
Small DS, Farid NA, Payne CD, et al. Effects of the proton pump inhibitor lansoprazole on the pharmacokinetics and pharmacodynamics of prasugrel and clopidogrel. J Clin Pharmacol. 2008;48:475–484. | ||
Hobl EL, Stimpfl T, Ebner J, et al. Morphine decreases clopidogrel concentrations and effects: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2014;63:630–635. | ||
Parodi G, Valenti R, Bellandi B, et al. Comparison of prasugrel and ticagrelor loading doses in ST-segment elevation myocardial infarction patients: RAPID (Rapid Activity of Platelet Inhibitor Drugs) primary PCI study. J Am Coll Cardiol. 2013;61:1601–1606. | ||
Chen X, Jin J, Chen Y, et al. Effect of scutellarin on the metabolism and pharmacokinetics of clopidogrel in rats. Biopharm Drug Dispos. 2015;36:64–68. | ||
Delavenne X, Magnin M, Basset T, et al. Investigation of drug–drug interactions between clopidogrel and fluoxetine. Fundam Clin Pharmacol. 2013;27:683–689. | ||
Teng R, Kujacic M, Hsia J. Evaluation of the pharmacokinetic interaction between ticagrelor and venlafaxine, a cytochrome P-450 2D6 substrate, in healthy subjects. Clin Ther. 2014;36:1217–1225. | ||
Oganesian A, Shilling AD, Young-Sciame R, et al. Desvenlafaxine and venlafaxine exert minimal in vitro inhibition of human cytochrome P450 and P-glycoprotein activities. Psychopharmacol Bull. 2009;42:47–63. | ||
Farid NA, Payne CD, Small DS, et al. Cytochrome P450 3A inhibition by ketoconazole affects prasugrel and clopidogrel pharmacokinetics and pharmacodynamics differently. Clin Pharmacol Ther. 2007;81:735–741. | ||
Suh JW, Koo BK, Zhang SY, et al. Increased risk of atherothrombotic events associated with cytochrome P450 3A5 polymorphism in patients taking clopidogrel. CMAJ. 2006;174:1715–1722. | ||
Harmsze AM, Robijns K, van Werkum JW, et al. The use of amlodipine, but not of P-glycoprotein inhibiting calcium channel blockers is associated with clopidogrel poor response. Thromb Haemost. 2010;103:920–925. | ||
Seo KD, Kim YD, Yoon YW, Kim JY, Lee KY. Antiplatelet effect of clopidogrel can be reduced by calcium channel blockers. Yonsei Med J. 2014;55:683–688. | ||
Park JJ, Park KW, Kang J, et al. CYP3A4 genetic status may be associated with increased vulnerability to the inhibitory effect of calcium-channel blockers on clopidogrel. Circ J. 2013;77:1289–1296. | ||
Harmsze AM, Van Werkum JW, Moral F, et al. Sulfonylureas and on-clopidogrel platelet reactivity in type 2 diabetes mellitus patients. Platelets. 2011;22:98–102. | ||
Harmsze AM, van Werkum JW, Bouman HJ, et al. Besides CYP2C19*2, the variant allele CYP2C9*3 is associated with higher on-clopidogrel platelet reactivity in patients on dual antiplatelet therapy undergoing elective coronary stent implantation. Pharmacogenet Genomics. 2010;20:18–25. | ||
Teng R, Mitchell P, Butler K. Evaluation of the pharmacokinetic interaction between ticagrelor and tolbutamide, a cytochrome P450 2C9 substrate, in healthy volunteers. Int J Clin Pharmacol Ther. 2013;51:305–312. | ||
Metzger NL, Momary KM. A patient with HIV and tuberculosis with diminished clopidogrel response. Int J STD AIDS. 2013;25:532–534. | ||
Ancrenaz V, Déglon J, Samer C, et al. Pharmacokinetic interaction between prasugrel and ritonavir in healthy volunteers. Basic Clin Pharmacol Toxicol. 2013;112:132–137. | ||
Daali Y, Ancrenaz V, Bosilkovska M, Dayer P, Desmeules J. Ritonavir inhibits the two main prasugrel bioactivation pathways in vitro: a potential drug–drug interaction in HIV patients. Metabolism. 2011;60:1584–1589. | ||
Jung KH, Chu K, Lee ST, et al. Prolonged use of aspirin alters human and rat intestinal cells and thereby limits the absorption of clopidogrel. Clin Pharmacol Ther. 2011;90:612–619. | ||
Oh J, Shin D, Lim KS, et al. Aspirin decreases systemic exposure to clopidogrel through modulation of P-glycoprotein but does not alter its antithrombotic activity. Clin Pharmacol Ther. 2014;95:608–616. | ||
Liang Y, Hirsh J, Weitz JI, et al. Active metabolite concentration of clopidogrel in patients taking different doses of aspirin: results of the Interaction Trial. J Thromb Haemost. December 30, 2014. [Epub ahead of print]. | ||
Taubert D, von Beckerath N, Grimberg G, et al. Impact of P-glycoprotein on clopidogrel absorption. Clin Pharmacol Ther. 2006;80:486–501. | ||
Chen XP, Tan ZR, Huang SL, Huang Z, Ou-Yang DS, Zhou HH. Isozyme-specific induction of low-dose aspirin on cytochrome P450 in healthy subjects. Clin Pharmacol Ther. 2003;73:264–271. | ||
Bouman HJ1, Schömig E, van Werkum JW, et al. Paraoxonase-1 is a major determinant of clopidogrel efficacy. Nat Med. 2011;17:110–116. | ||
Jaichander P, Selvarajan K, Garelnabi M, Parthasarathy S. Induction of paraoxonase 1 and apolipoprotein A-I gene expression by aspirin. J Lipid Res. 2008;49:2142–2148. | ||
Liu AC, Zhao LX, Lou HX. Curcumin alters the pharmacokinetics of warfarin and clopidogrel in Wistar rats but has no effect on anticoagulation or antiplatelet aggregation. Planta Med. 2013;79:971–977. | ||
He X, Mo L, Li ZY, Tan ZR, Chen Y, Ouyang DS. Effects of curcumin on the pharmacokinetics of talinolol in human with ABCB1 polymorphism. Xenobiotica. 2012;42:1248–1254. | ||
Trana C, Toth G, Wijns W, Barbato E. Clopidogrel undergoing percutaneous coronary intervention: a single-center randomized open-label trial (St John’s Trial). J Cardiovasc Transl Res. 2013;6:411–414. | ||
Lau WC, Welch TD, Shields T, Rubenfire M, Tantry US, Gurbel PA. The effect of St John’s wort on the pharmacodynamic response of clopidogrel in hyporesponsive volunteers and patients: increased platelet inhibition by enhancement of CYP3A4 metabolic activity. J Cardiovasc Pharmacol. 2011;57:86–93. | ||
Lee JH, Shin YJ, Kim HJ, Oh JH, Jang YP, Lee YJ. Danshen extract does not alter pharmacokinetics of docetaxel and clopidogrel, reflecting its negligible potential in P-glycoprotein- and cytochrome P4503A-mediated herb-drug interactions. Int J Pharm. 2011;410:68–74. | ||
Lee JH, Shin YJ, Oh JH, Lee YJ. Pharmacokinetic interactions of clopidogrel with quercetin, telmisartan, and cyclosporine A in rats and dogs. Arch Pharm Res. 2012;35:1831–1837. | ||
Teng R, Kujacic M, Hsia J. Pharmacokinetic interaction study of ticagrelor and cyclosporine in healthy volunteers. Clin Drug Investig. 2014;34:529–536. | ||
Judge HM, Patil SB, Buckland RJ, Jakubowski JA, Storey RF. Potentiation of clopidogrel active metabolite formation by rifampicin leads to greater P2Y12 receptor blockade and inhibition of platelet aggregation after clopidogrel. J Thromb Haemost. 2010;8:1820–1827. | ||
Teng R, Mitchell P, Butler K. Effect of rifampicin on the pharmacokinetics and pharmacodynamics of ticagrelor in healthy subjects. Eur J Clin Pharmacol. 2013;69:877–883. | ||
Farid NA, Jakubowski JA, Payne CD, et al. Effect of rifampin on the pharmacokinetics and pharmacodynamics of prasugrel in healthy male subjects. Curr Med Res Opin. 2009;25:1821–1829. | ||
Kristensen KE, Zhu HJ, Wang X, et al. Clopidogrel bioactivation and risk of bleeding in patients cotreated with angiotensin-converting enzyme inhibitors after myocardial infarction: a proof-of-concept study. Clin Pharmacol Ther. 2014;96:713–722. | ||
Yousef AM, Arafat T, Bulatova NR, et al. Smoking behaviour modulates pharmacokinetics of orally administered clopidogrel. J Clin Pharm Ther. 2008;33:439–449. | ||
Hochholzer W, Trenk D, Mega JL, et al. Impact of smoking on antiplatelet effect of clopidogrel and prasugrel after loading dose and on maintenance therapy. Am Heart J. 2011;162:518–526. | ||
Park KW, Park JJ, Jeon KH, et al. Enhanced clopidogrel responsiveness in smokers: smokers’ paradox is dependent on cytochrome P450 CYP1A2 status. Arterioscler Thromb Vasc Biol. 2011;31:665–671. | ||
Gurbel PA, Bliden KP, Logan DK, et al. The influence of smoking status on the pharmacokinetics and pharmacodynamics of clopidogrel and prasugrel: the PARADOX study. J Am Coll Cardiol. 2013;62:505–512. | ||
Caplain H, Thebault JJ, Necciari J. Clopidogrel does not affect the pharmacokinetics of theophylline. Semin Thromb Hemost. 1999;25 Suppl 2:65–68. | ||
Nirogi RV, Kandikere VN, Mudigonda K. Effect of food on bioavailability of a single oral dose of clopidogrel in healthy male subjects. Arzneimittelforschung. 2006;56:735–739. | ||
Hurbin F, Boulenc X, Daskalakis N, et al. Clopidogrel pharmacodynamics and pharmacokinetics in the fed and fasted state: a randomized crossover study of healthy men. J Clin Pharmacol. 2012;52:1506–1515. | ||
Teng R, Mitchell PD, Butler K. Lack of significant food effect on the pharmacokinetics of ticagrelor in healthy volunteers. J Clin Pharm Ther. 2012;37:464–468. | ||
Small DS, Farid NA, Payne CD, et al. Effect of intrinsic and extrinsic factors on the clinical pharmacokinetics and pharmacodynamics of prasugrel. Clin Pharmacokinet. 2010;49:777–798. | ||
Holmberg MT, Tornio A, Neuvonen M, Neuvonen PJ, Backman JT, Niemi M. Grapefruit juice inhibits the metabolic activation of clopidogrel. Clin Pharmacol Ther. 2014;95:307–313. | ||
Holmberg MT, Tornio A, Joutsi-Korhonen L, et al. Grapefruit juice markedly increases the plasma concentrations and antiplatelet effects of ticagrelor in healthy subjects. Br J Clin Pharmacol. 2013;75:1488–1496. | ||
Campbell KL, Cohn JR, Fischman DL, et al. Management of clopidogrel hypersensitivity without drug interruption. Am J Cardiol. 2011;107:812–816. | ||
Karabay CY, Can MM, Tanboğa IH, Ahmet G, Bitigen A, Serebruany V. Recurrent acute stent thrombosis due to allergic reaction secondary to clopidogrel therapy. Am J Ther. 2011;18:e119–e122. | ||
Kounis NG, Soufras GD. Coronary stent thrombosis: beware of an allergic reaction and of Kounis syndrome. Indian Heart J. 2014; 66:153–155. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.