Back to Journals » Drug Design, Development and Therapy » Volume 11
Pharmacokinetic and pharmacodynamic evaluation according to absorption differences in three formulations of ibuprofen
Authors Shin D ![]() , Lee SJ
, Lee SJ ![]() , Ha Y, Choi YS, Kim JW, Park SR, Park MK
, Ha Y, Choi YS, Kim JW, Park SR, Park MK ![]()
Received 6 September 2016
Accepted for publication 1 November 2016
Published 4 January 2017 Volume 2017:11 Pages 135—141
DOI https://doi.org/10.2147/DDDT.S121633
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Georgios Panos
Dongseong Shin,1 Sook Joung Lee,2 Yu-Mi Ha,3 Young-Sim Choi,3 Jae-Won Kim,3 Se-Rin Park,3 Min Kyu Park3
1Clinical Trials Center, Gachon University Gil Medical Center, Incheon, 2Department of Rehabilitation Medicine, 3Department of Clinical Pharmacology and Therapeutics, Dong-A University College of Medicine and Hospital, Busan, South Korea
Objective: Prostaglandin E2 (PGE2) synthesis is modulated by COX2. Changes in PGE2 could be used to quantify the COX2 inhibition after ibuprofen administration. This study investigated the pharmacokinetic and pharmacodynamic relationships for COX2 inhibition according to three formulations of ibuprofen in healthy male subjects.
Materials and methods: A randomized, open-label, single-dose, three-treatment, six-sequence crossover study was performed in 36 healthy South Korean male volunteers. Enrolled subjects received the following three 200 mg ibuprofen formulations: ibuprofen arginine, solubilized ibuprofen capsule, and standard ibuprofen. Pharmacokinetic and pharmacodynamic blood samples were collected for 16 hours following treatment. For pharmacodynamic evaluations, lipopolysaccharide (LPS)-induced PGE2 inhibition at each time point compared to predose was measured. Noncompartmental analysis was used for pharmacokinetic assessment, and time-weighted average inhibition (WAI) of PGE2 was applied to the pharmacodynamic evaluation.
Results: After a single oral dose of the ibuprofen formulations, the median times to maximum concentration were 0.42, 0.5, and 1.25 hours in ibuprofen arginine, solubilized ibuprofen capsule, and ibuprofen, respectively. The maximum observed plasma concentration was lower in ibuprofen, and the area under the plasma concentration–time curve was comparable among the three formulations. A significant difference was observed between fast-acting formulations and standard ibuprofen tablets for both maximum concentration and time taken to reach it. Individual formulations had an effect on PGE2 WAI during the 8 hours following treatment, resulting in significantly lower WAI in standard ibuprofen: ibuprofen arginine 18.4%, solubilized ibuprofen capsule 18.4%, and standard ibuprofen 11.6%.
Conclusion: Rapid absorption and higher peak concentration were observed in ibuprofen arginine and the solubilized ibuprofen capsule. Additionally, fast-acting formulations had more predominant inhibitory activity on the COX2 enzyme.
Keywords: ibuprofen, different formulation, COX2-inhibitory effects
Introduction
The severity of pain is influenced by several factors, including the amount of damage and emotional and environmental factors, and pharmacological treatment, interventional techniques, and behavioral therapies are available for pain management.1 Oral pharmacologic agents have been the primary option, and the use of drugs has increased exponentially. Nonsteroidal anti-inflammatory drugs (NSAIDs) have generally been effective for musculoskeletal pain.2,3 For pain control, earlier absorption of ibuprofen could lead to early pain relief with a similar adverse-event profile as occurs upon later absorption.4,5
Ibuprofen is one of the most commonly used NSAIDs and a potent inhibitor of prostaglandin (PG) synthesis that can manage various pain types and has anti-inflammatory activity.6,7 The S+ enantiomer has the majority of the pharmacological activity of ibuprofen, and similarly inhibits COX1 and COX2 activity.6 After oral administration, the absolute bioavailability of ibuprofen is almost complete; then, ibuprofen undergoes enantiomeric inversion and hepatic oxidative metabolism via CYP 2C9. Finally, it is excreted in the form of a glucuronide-conjugate metabolite through urine.6,8 Pharmacokinetic characteristics, such as absorption rate, of ibuprofen are reported to be different according to the formulation, while apparent bioavailability is equivalent among the dosage forms.9,10 Faster, higher maximum plasma concentrations have been observed in fast-acting formulations than in standard formulations.4 However, the treatment effectiveness of COX2 inhibition according to the ibuprofen formulation and the pharmacokinetics of different formulations have not previously been established.
In this clinical study, the pharmacokinetic and pharmacodynamic profiles of three ibuprofen formulations were evaluated at a dose of 200 mg, the usual prescribed dose for an analgesic effect, and the pharmacodynamic effects were compared based on time–COX inhibition relationships.11
Materials and methods
Study population and design
This clinical trial was a randomized, open-label, single-dose, three-treatment, six-sequence crossover study performed on 36 healthy South Korean male volunteers (age 19–50 years). Participants were enrolled according to the following inclusion criteria: body mass index of 18.5–27 kg/m2, weight ≥50 kg, lack of clinically significant medical history, physical examination findings, 12-lead electrocardiogram readings, or clinical laboratory-testing results, including hematology, serum chemistry, infectious serology, and urinalysis. The objective and contents were fully explained, and written informed consent was obtained. The institutional review board of Dong-A University Hospital approved this study, which was conducted in accordance with the Declaration of Helsinki and Korean Good Clinical Practice.
The 36 participants were randomly assigned to one of six sequences of the three treatments. Participants received a single oral dose of 200 mg ibuprofen in each treatment period of the Carol-F tablet (ibuprofen arginine), Advil Liqui-Gels® (solubilized ibuprofen capsule), or Brufen tablet (ibuprofen). Participants received the study drugs with 150 mL of water after an overnight fast of 10 hours and were discharged 24 hours following the treatment. A washout interval of 7 days was required from the last dose of the previous treatment period.
Pharmacokinetic and pharmacodynamic assessment
Blood samples for pharmacokinetic evaluation were obtained before dosing (0 hour) and 0.17, 0.25, 0.33, 0.42, 0.5, 0.58, 0.67, 0.75, 0.83, 1, 1.25, 1.5, 2, 3, 4, 6, 8, 10, 12, and 16 hours after study-drug administration in each period. At the time points for pharmacokinetic evaluation, 6 mL of blood was drawn into a sodium heparin tube and centrifuged at 3,000 rpm for 10 minutes at 4°C. Separated plasma samples were frozen and stored at −70°C until analysis. Pharmacokinetic samples were analyzed using positive-ion liquid chromatography tandem mass spectrometry (LC-MS/MS) (Biosuntek Laboratory Co Ltd, Seongnam, South Korea).
For pharmacodynamic evaluation of PGE2, blood samples were collected at 0, 0.17, 0.33, 0.5, 0.67, 0.83, 1, 1.25, 1.5, 2, 3, 4, 6, 8, 10, 12, and 16 hours. Blood (10 mL) was contained in K2-ethylenediaminetetraacetic acid tubes and stored in a 36°C incubator after treatment with 10 ng/mL lipopolysaccharide (LPS) for 24 hours. Plasma was separated by centrifugation at 3,000 rpm for 10 minutes at 4°C. Separated plasma was frozen at −70°C until PGE2 evaluation. The PGE2 level in plasma was determined using an enzyme immunoassay (EIA; Department of Pharmacology, Dong-A University College of Medicine).
Bioanalytical methods
The plasma concentration of ibuprofen was estimated using positive-ion LC (Agilent 1200 series; Agilent Technologies, Santa Clara, CA, USA) and LC-MS/MS (Agilent 6410 triple-quadrupole LC/MS system). The plasma specimens were mixed with methanol in the presence of an internal standard (ibuprofen-d3). Chromatography was performed at 45°C on a Unison UK-C8 column (75×2 mm, 3 μm) with mobile phase A (0.1% acetic acid in 1 mM ammonium acetate) and mobile phase B (methanol). The flow rate was 0.3 mL/min. The calibration curves were linear within the range of 0.1–60 μg/mL. Coefficients of variation of intra- and interassay accuracy and precision were less than 10%.
PGE2 is synthesized and released into the extracellular space when cells are activated or exogenous free arachidonate is supplied. PGE2 is rapidly converted to an inactive metabolite (13,14-dihydro-15-keto PGE2) by the PG15-dehydrogenase pathway. COX2 activity (PGE2 level) in plasma was determined using commercially available EIA kits (Cayman Chemical, Ann Arbor, MI, USA). Both samples and standards were assayed in parallel. The detection limit of PGE2 quantification was 15 pg/mL. The reaction solution consisted of 50 μL of human plasma sample per well with 50 μL of EIA, PGE2 AChE tracer, and PGE2 monoclonal antibody. This assay typically develops in 1.5 hours, and is measured in a 96-well plate spectrophotometer at 405 nm (SpectraMax 340; Molecular Devices LLC, Sunnyvale, CA, USA).
Pharmacokinetic and pharmacodynamic analysis
Pharmacokinetic parameters for ibuprofen were calculated using the noncompartmental analysis of WinNonlin® 6.4 (Certara, Princeton, NJ, USA). The time to the maximum concentration observed in plasma (Tmax) and maximum observed plasma concentration (Cmax) were directly obtained from the plasma concentration–time profiles. The area under the plasma concentration–time curve (AUC0–t) after the administration of the study drug was calculated by the linear-up and log-down trapezoidal method. The terminal elimination half-life (t½) was estimated as ln (2)/λz, and the elimination rate constant (λz) was the slope of the terminal log-linear phase.
For the pharmacodynamic assessment, COX2 inhibition was considered the percentage change from baseline (predose) in LPS-induced PGE2 at each time point. The time-weighted average inhibition (WAI) for PGE2 was calculated from the AUC (AUC0–8) on the linear trapezoidal method up to 8 hours after individual ibuprofen administration. The observed Tmax was assessed from the PGE2-inhibition time course.
Statistical analysis
SPSS 22.0 (IBM, Armonk, NY, USA) was used for statistical analyses. Using a mixed-effect model, analysis of variance was performed to compare the 90% confidence intervals (CIs) for the geometric mean ratios of AUC0–t and Cmax pharmacokinetic values. Also, to assess the treatment effect, differences in the WAI for PGE2 were evaluated based on the 90% CI between the treatments. In the mixed-effect model, the sequence, period, and treatment were considered fixed effects, and the subject nested within a sequence was used as a random effect. The Tmax values for the plasma concentration of ibuprofen and inhibition of PGE2 synthesis were compared using the Mann–Whitney U test, which was followed by Bonferroni correction. If the P-value was less than 0.025, the Tmax was considered significantly different between two treatments.
Results
Demographic characteristics
Among the 37 healthy male subjects who were enrolled, a total of 33 subjects completed the study and were included in pharmacokinetic and pharmacodynamic evaluation. With the exception of one subject who experienced a pretreatment adverse event, six subjects started each treatment. However, one subject in sequence A and two in sequence D withdrew from the study. Mean ± standard deviation (SD) values for age, weight, height, and body mass index were 25±3 years, 70.3±7.9 kg, 175.1±6.1 cm, and 22.9±2.2 kg/m2, respectively. There were no significant differences in the demographic characteristics between the sequences.
Pharmacokinetics
Pharmacokinetic profiles (Tmax, Cmax, and AUC0–t) are described in Table 1 and Figure 1. The median Tmax of ibuprofen arginine, solubilized ibuprofen capsule, and ibuprofen were 0.42, 0.5, and 1.25 hours, respectively, and there was a significant difference between ibuprofen arginine and ibuprofen (P<0.001) and between solubilized ibuprofen capsule and ibuprofen (P<0.001). After the oral intake of 200 mg of ibuprofen, when evaluating the systemic exposures of solubilized ibuprofen capsule and ibuprofen compared to ibuprofen arginine, the Cmax was lower in ibuprofen and the AUC0–t was equivalent. In the elimination process, the t½ and apparent clearance were not significantly different among the three treatments (P=0.813 and P=0.906, respectively).
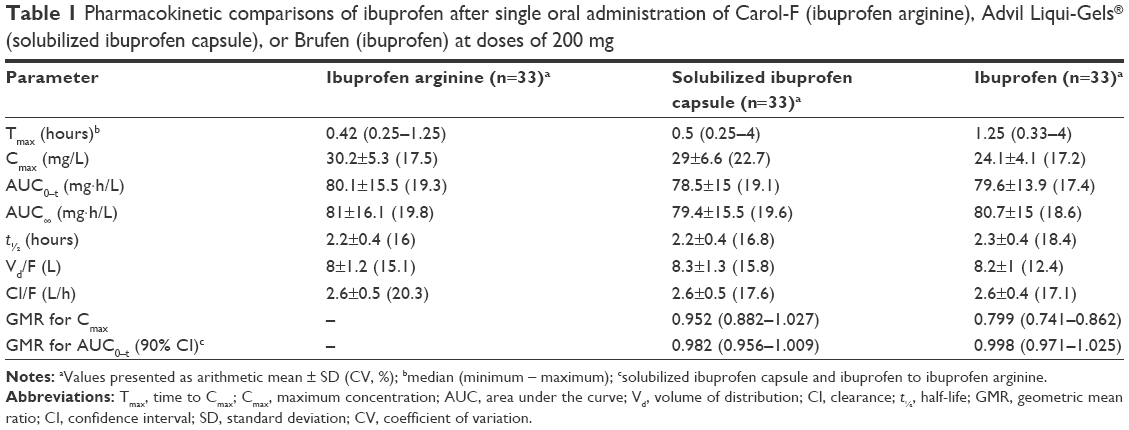 | Table 1 Pharmacokinetic comparisons of ibuprofen after single oral administration of Carol-F (ibuprofen arginine), Advil Liqui-Gels® (solubilized ibuprofen capsule), or Brufen (ibuprofen) at doses of 200 mg |
 | Figure 1 Mean plasma concentration–time profiles (on a log scale) of ibuprofen after single administration of Carol-F (ibuprofen arginine), Advil Liqui-Gels® (solubilized ibuprofen capsule), and Brufen (ibuprofen). |
Pharmacodynamics
COX2 inhibition, based on changes in the LPS-induced PGE2 level from the pretreatment baseline, reached a maximum level at a median Tmax of 0.83, 2, and 0.67 hours with ibuprofen arginine, solubilized ibuprofen capsule, and ibuprofen, respectively (Figure 2). Ibuprofen arginine and ibuprofen had faster COX2-inhibition effects than solubilized ibuprofen capsules (both P=0.001). The maximum inhibition (Imax) and WAI of PGE2 after three ibuprofen formulations at doses of 200 mg are displayed in Table 2. The individual formulations produced a similar Imax for PGE2 following single oral administration of ibuprofen (200 mg). Conversely, the formulations affected the WAI in the 8 hours following treatment and decreased the WAI in ibuprofen compared to the other two formulations (both P=0.002) (Figure 3). The mean differences (90% CI) between formulations compared to ibuprofen arginine were as follows: 0 (−3.1% to 3.1%) for solubilized ibuprofen capsule and −6% (−9.1% to −2.8%) for ibuprofen.
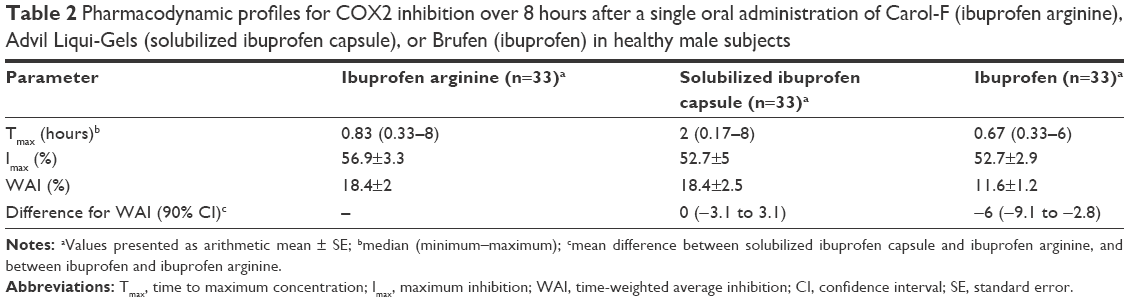 | Table 2 Pharmacodynamic profiles for COX2 inhibition over 8 hours after a single oral administration of Carol-F (ibuprofen arginine), Advil Liqui-Gels (solubilized ibuprofen capsule), or Brufen (ibuprofen) in healthy male subjects |
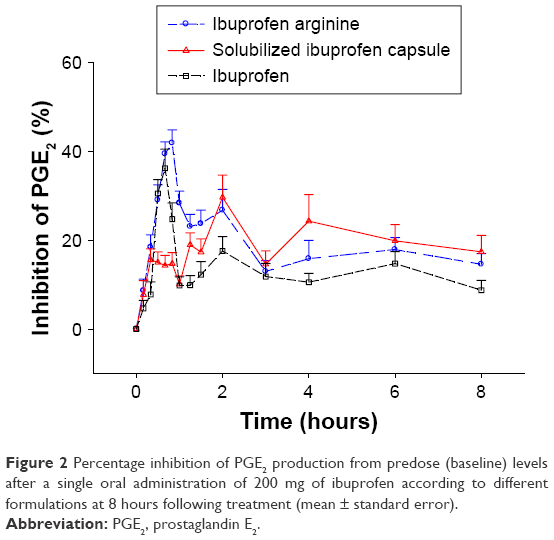 | Figure 2 Percentage inhibition of PGE2 production from predose (baseline) levels after a single oral administration of 200 mg of ibuprofen according to different formulations at 8 hours following treatment (mean ± standard error). |
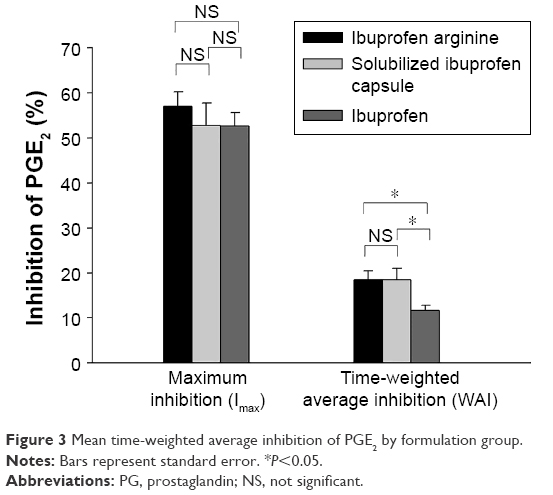 | Figure 3 Mean time-weighted average inhibition of PGE2 by formulation group. |
Safety and tolerability
There were no adverse events related to the study drugs. Clinically meaningful physical examination findings, laboratory abnormalities, vital signs, and electrocardiogram results were not reported. None of the subjects discontinued in this study due to adverse events.
Discussion
In this six-sequence, three-treatment crossover study of healthy subjects, direct comparison of PGE2-synthesis inhibition was performed. Pharmacodynamic profiles were demonstrated as a mediator between pharmacokinetic characteristics and clinical efficacy after a single dose of three ibuprofen formulations. Two formulations are fast-acting, ibuprofen arginine and solubilized ibuprofen capsule, and the other was a standard ibuprofen-formulation tablet. A dose of 200 mg was selected based on consideration of the common clinical initial dosage for managing pain. A validated method using changes in PGE2 production from predose level in LPS-stimulated whole blood was chosen as an index of COX2 enzyme activities.12,13
The absorption rate and extent depend on multiple factors, including the administration route, physiological status, site of absorption, and physicochemical properties of compounds.14 Because ibuprofen is a relatively weak acid with a pKa of 4.4 and aqueous solubility under acidic conditions is low, dissolution and water solubility might be the key factors influencing drug absorption.5,15 To improve the aqueous solubility and dissolution rate, salt formation is a commonly applied method that does not change the chemical structure or biological properties.16 In a previous report, the fast-dissolving ibuprofen formulation had faster absorption, resulting in a greater Cmax and shorter Tmax.15 Based on the results of our clinical study, the Cmax values of ibuprofen arginine and solubilized ibuprofen capsule were 125.1% and 119.1%, respectively, compared to standard ibuprofen after the oral administration of a 200 mg single dose. Also, the median Tmax for ibuprofen was significantly delayed compared to those of the other formulations. Conversely, the individual formulations had little effect on the AUC0–t, and the 90% CIs of geometric mean ratios between formulations were within the range of 0.8–1.25. With respect to distribution and elimination, the volume of distribution and apparent clearance were comparable among ibuprofen formulations. Consequently, the change of formulations into ibuprofen arginine or a solubilized ibuprofen capsule led to a rapid absorption process, including a higher Cmax and earlier Tmax, conserving other pharmacokinetic processes, such as distribution or elimination.
Inhibition of PGE2 synthesis by NSAIDs was based on blocking the COX-mediated transformation of arachidonic acid to thromboxane and PGs, resulting in anti-inflammatory, analgesic, and antipyretic effects.8 An analgesic effect was reported to have a positive relationship with the plasma concentration of NSAIDs, and the COX2-inhibition mechanism was time-dependent and irreversible, in contrast to the instantaneous and competitively reversible inhibition of COX1.17,18 In this clinical trial, direct comparison of pharmacodynamic parameters was conducted using an ex vivo assay of COX2 inhibition. PGE2 production with LPS stimulation was more effectively suppressed in ibuprofen arginine and the solubilized ibuprofen capsule than in standard ibuprofen. The average inhibition of COX2 was similar between ibuprofen arginine and solubilized ibuprofen capsules. Generally, better or faster pain relief for acute pain management was possible in soluble or rapidly absorbed formulations, and delays in the absorption or reduction of the drug concentration may cause treatment failure.4,18 However, the significant difference in the COX2-inhibition activities of various NSAIDs did not represent superior clinical efficacy in the long-term management of rheumatoid arthritis pain and inflammation when comparing diclofenac to celecoxib and diclofenac to etoricoxib.19–21
From this pharmacokinetic–pharmacodynamic study, fast-acting ibuprofen arginine provided a shorter Tmax and remarkable inhibition of PGE2 formation. Also, the solubilized ibuprofen capsule inhibited PGE2 better than standard ibuprofen. These results were consistent with a previous report that ibuprofen formulations with early pharmacokinetic profiles achieved pharmacodynamic advantages.4 Considering that the effect of ibuprofen on COX2 depends on time and there are therapeutic advantages of NSAIDs with early pharmacokinetic characteristics, the Tmax and Imax in the time course of COX2 inhibition did not have a major impact on the clinical efficacy or therapeutic end point. Furthermore, in terms of clinically effective analgesic activity, full PGE2 inhibition is not required. If plasma ibuprofen concentration reaches the minimum level required to inhibit COX2 to achieve a clinical effect, pain relief is possible in the early phase following drug administration, regardless of the Tmax or Imax.22,23
Although these results clearly describe the pharmacokinetic–pharmacodynamic relationship of ibuprofen according to different formulations, there were some limitations in this clinical study. Because this study was a single study with a relatively low dose, considering the sigmoidal concentration–effect curves, complete or full-range COX2 inhibition was not routinely observed.24 Because this study was conducted on a small number of healthy, young, male volunteers, the result could not fully reflect the clinical pathophysiology of pain or inflammatory disease. A direct comparison between COX2 inhibition and clinical efficacy, including pain relief or anti-inflammation biomarkers, will be helpful for understanding the mechanistic process of pain control and achieving therapeutic goals.
Conclusion
These data indicated that fast-acting ibuprofen formulations act on the absorption phase and inhibit PGE2 in healthy subjects. Based on the rapid absorption, the fast-acting formulations were able to induce stronger, more persistent inhibition of PGE2 synthesis. Collectively, the pharmacokinetic and pharmacodynamic advantages of fast-acting formulations can be considered favorable for early, potent, and more sustainable therapeutic success.
Acknowledgment
This study was sponsored by Ildong Pharmaceutical Co Ltd, South Korea.
Disclosure
The authors report no conflicts of interest in this work.
References
Turk DC, Wilson HD, Cahana A. Treatment of chronic non-cancer pain. Lancet. 2011;377(9784):2226–2235. | ||
Martin BI, Deyo RA, Mirza SK, et al. Expenditures and health status among adults with back and neck problems. JAMA. 2008;299(6):656–664. | ||
Roelofs PD, Deyo RA, Koes BW, Scholten RJ, van Tulder MW. Non-steroidal anti-inflammatory drugs for low back pain. Cochrane Database Syst Rev. 2008;(1):CD000396. | ||
Moore RA, Derry S, Straube S, Ireson-Paine J, Wiffen PJ. Faster, higher, stronger? Evidence for formulation and efficacy for ibuprofen in acute pain. Pain. 2014;155(1):14–21. | ||
Cattaneo D, Clementi E. Clinical pharmacokinetics of ibuprofen arginine. Curr Clin Pharmacol. 2010;5(4):239–245. | ||
Davies NM. Clinical pharmacokinetics of ibuprofen: the first 30 years. Clin Pharmacokinet. 1998;34(2):101–154. | ||
McGettigan P, Henry D. Use of non-steroidal anti-inflammatory drugs that elevate cardiovascular risk: an examination of sales and essential medicines lists in low-, middle-, and high-income countries. PLoS Med. 2013;10(2):e1001388. | ||
Evans AM. Comparative pharmacology of S(+)-ibuprofen and (RS)-ibuprofen. Clin Rheumatol. 2001;20 (Suppl 1):S9–S14. | ||
Gillespie WR, DiSanto AR, Monovich RE, Albert KS. Relative bioavailability of commercially available ibuprofen oral dosage forms in humans. J Pharm Sci. 1982;71(9):1034–1038. | ||
Mendes GD, Mendes FD, Domingues CC, et al. Comparative bioavailability of three ibuprofen formulations in healthy human volunteers. Int J Clin Pharmacol Ther. 2008;46(6):309–318. | ||
Awa K, Satoh H, Hori S, Sawada Y. Prediction of time-dependent interaction of aspirin with ibuprofen using a pharmacokinetic/pharmacodynamic model. J Clin Pharm Ther. 2012;37(4):469–474. | ||
Van Hecken A, Schwartz JI, Depre M, et al. Comparative inhibitory activity of rofecoxib, meloxicam, diclofenac, ibuprofen, and naproxen on COX-2 versus COX-1 in healthy volunteers. J Clin Pharmacol. 2000;40(10):1109–1120. | ||
Brideau C, Kargman S, Liu S, et al. A human whole blood assay for clinical evaluation of biochemical efficacy of cyclooxygenase inhibitors. Inflamm Res. 1996;45(2):68–74. | ||
Martinez MN, Amidon GL. A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. J Clin Pharmacol. 2002;42(6):620–643. | ||
Schettler T, Paris S, Pellett M, Kidner S, Wilkinson D. Comparative pharmacokinetics of two fast-dissolving oral ibuprofen formulations and a regular-release ibuprofen tablet in healthy volunteers. Clin Drug Invest. 2001;21(1):73–78. | ||
Lee T, Wang YW. Initial salt screening procedures for manufacturing ibuprofen. Drug Dev Ind Pharm. 2009;35(5):555–567. | ||
Jamali F, Aghazadeh-Habashi A. Rapidly dissolving formulations for quick absorption during pain episodes: ibuprofen. Int J Clin Pharmacol Ther. 2008;46(2):55–63. | ||
Hawkey CJ. COX-2 inhibitors. Lancet. 1999;353(9149):307–314. | ||
Schwartz JI, Dallob AL, Larson PJ, et al. Comparative inhibitory activity of etoricoxib, celecoxib, and diclofenac on COX-2 versus COX-1 in healthy subjects. J Clin Pharmacol. 2008;48(6):745–754. | ||
Cannon CP, Curtis SP, FitzGerald GA, et al. Cardiovascular outcomes with etoricoxib and diclofenac in patients with osteoarthritis and rheumatoid arthritis in the Multinational Etoricoxib and Diclofenac Arthritis Long-term (MEDAL) programme: a randomised comparison. Lancet. 2006;368(9549):1771–1781. | ||
Emery P, Zeidler H, Kvien TK, et al. Celecoxib versus diclofenac in long-term management of rheumatoid arthritis: randomised double-blind comparison. Lancet. 1999;354(9196):2106–2111. | ||
Fenner H. Differentiating among nonsteroidal antiinflammatory drugs by pharmacokinetic and pharmacodynamic profiles. Semin Arthritis Rheum. 1997;26 (6 Suppl 1):28–33. | ||
Ehrich EW, Dallob A, De Lepeleire I, et al. Characterization of rofecoxib as a cyclooxygenase-2 isoform inhibitor and demonstration of analgesia in the dental pain model. Clin Pharmacol Ther. 1999;65(3):336–347. | ||
Giagoudakis G, Markantonis SL. Relationships between the concentrations of prostaglandins and the nonsteroidal antiinflammatory drugs indomethacin, diclofenac, and ibuprofen. Pharmacotherapy. 2005;25(1):18–25. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.