Back to Journals » Drug Design, Development and Therapy » Volume 10
Pharmacoinformatic and molecular docking studies reveal potential novel antidepressants against neurodegenerative disorders by targeting HSPB8
Authors Sehgal SA, Mannan S, Ali S
Received 6 December 2015
Accepted for publication 2 February 2016
Published 6 May 2016 Volume 2016:10 Pages 1605—1618
DOI https://doi.org/10.2147/DDDT.S101929
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Sheikh Arslan Sehgal,1–4 Shazia Mannan,1 Sannia Ali1
1Department of Bioscience, COMSATS Institute of Information Technology, Sahiwal, Pakistan; 2State Key Laboratory of Biomembrane and Membrane Biotechnology, Institute of Zoology, Chinese Academy of Sciences, 3University of Chinese Academy of Sciences, Beijing, People’s Republic of China; 4Department of Bioinformatics and Biotechnology, International Islamic University, Islamabad, Pakistan
Abstract: Charcot–Marie–Tooth (CMT) disease is an inherited peripheral neuromuscular disorder characterized by length-dependent and progressive degeneration of peripheral nerves, leading to muscular weakness. Research has shown that mutated HSPB8 may be responsible for depression, neurodegenerative disorders, and improper functioning of peripheral nerves, resulting in neuromuscular disorders like CMT. In the current work, a hybrid approach of virtual screening and molecular docking studies was followed by homology modeling and pharmacophore identification. Detailed screening analyses were carried out by 2-D similarity search against prescribed antidepressant drugs with physicochemical properties. LigandScout was employed to ascertain novel molecules and pharmacophore properties. In this study, we report three novel compounds that showed maximum binding affinity with HSPB8. Docking analysis elucidated that Met37, Ser57, Ser58, Trp60, Thr63, Thr114, Lys115, Asp116, Gly117, Val152, Val154, Leu186, Asp189, Ser190, Gln191, and Glu192 are critical residues for ligand–receptor interactions. Our analyses suggested paroxetine as a potent compound for targeting HSPB8. Selected compounds have more effective energy scores than the selected drug analogs. Additionally, site-directed mutagenesis could be significant for further analysis of the binding pocket. The novel findings based on an in silico approach may be momentous for potent drug design against depression and CMT.
Keywords: bioinformatics, computer-aided drug design, HSPB8, heat shock protein (HSP), molecular docking, modeling, pharmacoinformatics, virtual screening, neurodegenerative disorders, antidepressants
Introduction
The superfamily of ten human small HSPs (sHSPs) has been characterized on the basis of a conserved C-terminal part called the α-crystallin domain.1,2 This domain is involved in hetero- and homodimer formation, which is observed in many HSPs. Large oligomer formations may also occur, based on the interactions of sHSP dimer amino-terminal regions.3,4 The formation of oligomers by sHSP has been linked with their chaperoning functional properties.5 Some sHSPs are restricted to specific cell-type expression, eg, HSPB9 and HSPB10 are present in male germ cells, while HSPB4 (αA-crystallin) is associated with the eye lens. HSPB5 (αB-crystallin), HSPB1 (HSP27), and to a lesser extent HSPB3, HSPB7 (cvHSP), HSPB2 (MKBP) and HSPB8 (HSP22) are expressed in a variety of cell tissues and types. The latter seven sHSPs are abundant in muscles.6–10
sHSPs, a large protein family with low molecular weight (12–43 kDa), are ubiquitously expressed in all phyla.11 This protein family contains a 90- to 100-residue-long α-crystallin domain, which is present at the C-terminal part of the molecule.12,13 sHSPs tend to change in composition and size of oligomers, with flexible structure.14,15 This significant feature obscures sHSP-structure investigation for the long, stable oligomer structure of Hsp16.5 of Archaea Methanococcus16 and Hsp16.917 of wheat. To date, structures of certain mammalian members of sHSPs have been revealed.18–20 Ten sHSPs are encoded in the human genome.1,2 It is supposed that human sHSPs may play a role in the accumulation of harmful protein aggregates by interacting with partially unfolded proteins.12,13 HSPs maintain the soluble state of unfolded proteins and prevent their precipitation.21 This family is present in animals,22 plants,23 fungi, eukaryotes,24 bacteria,25 and Archaea.16 Multiple homologues of sHSPs are present in the organisms: up to 15 in plants,26 13 in zebrafish,27 ten in humans,27 and only a few in yeast and bacteria.24 In higher eukaryotes, sHSPs are tissue specific and distributed subcellularly.6,28 Mutations in sHSPs are associated with some severe pathologies, such as neurodegenerative diseases, cataract, Charcot–Marie–Tooth (CMT) disease, distal hereditary motor neuropathy (HMN), and desmin-related myopathy.29 Prominent sHSP-specific activity is mediated by binding of other proteins and protecting them from aggregation. This property of sHSPs may inhibit the aggregation of many pathogenic proteins, responsible for Huntington’s disease, dementia with Lewy bodies, Alzheimer’s disease, and Parkinson’s disease. Moreover, filament formation in the mutated and wild-type Parkinson’s forms of α-synuclein can be stopped in the presence of HSPB8.30
Stressful life events are considered a major predisposing risk factor for the development of depression. Depressive individuals usually show profound neuroendocrine alterations (hyper- or hypocortisolism) arising from dysregulation of the hypothalamic–pituitary–adrenocortical axis. The hippocampus has a significant role in stress-related mood disorders. Acute versus chronic stress effects on the integrity of hippocampal circuitry in influencing the vulnerability to or resilience against neuronal injury are poorly understood. Filipović et al concluded that chronic isolation stress induces profound alterations in the functionality of the hypothalamic–pituitary–adrenocortical axis. In contrast to acute stress, chronic isolation strongly induces a reduction in parvalbumin immunoreactivity in γ-aminobutyric acidergic interneurons in all regions of the hippocampus.31 The efficacy of antidepressant drugs used in short-term major depression treatment considered as under the control of genetic variations. Numerous pharmacogenetic predictors of antidepressant efficacy have been studied in the last two decades, but reliability is still contradictory. An association between HSP70-coding genes and antidepressant response in mood disorders has been reported, such that genetic variants within the genes coding for HSP70-family proteins may affect the action of antidepressants and thus their therapeutic efficacy.32
HSPB8 is linked with breast cancer cells, glioblastoma, protection from apoptosis in melanoma, and cell proliferation.33–35 However, HSPB8 antiapoptotic activity may differ in cancer types. The overexpression of HSPB8 in melanocytes enhances the transformation of tumor during the G1 cell-cycle stage. Alternatively, HSPB8 opposes Sam68 in glioblastoma cells and facilitates cell-cycle progression from G1 to S by enhanced expression of cell-cycle regulatory A (PCNA) and cyclin E proteins.34 HSPB8 activates the Akt–PI3K pathway in cardiac cells and supports their survival and growth.36 The level of HSPB8 in some tumor tissues is lower than in normal cells, while restoring normal concentration by cell transfection and DNA demethylation leads to apoptosis induction in p38 MAPK- and caspase-dependent pathways.37 Therefore, an HSPB8-specific role cannot be considered simply anti- or proapoptotic.
The early expression of sHSP orthologues (HSPB1, HSPB2, HSPB5, HSPB6, and HSPB8) during heart development has been reported in a wide range of animal species, including human,38,39 pig,7 mouse,40 zebrafish,41 sea squirt,42 and Drosophila melanogaster.43 The expression of HSPB8 is upregulated by ischemic stress.44,45 It has been inferred in experimental transgenic mice that HSPB8 is involved in cardioprotection via a nitric acid-dependent mechanism in an ischemia context.46 HSPB8-specific protective activity is similar to ischemic preconditioning, which involves metabolic adaptation, inhibition of apoptosis, upregulation of HSPs, and activation of cell-cycling pathways.36 Overall, HSPB8 upregulation is correlated with increased expression of the inducible isoform of nitric oxide synthase.44 HSPB8 is found in the hypertrophic heart, and its upregulation in the transgenic mouse heart results in cardiac hypertrophy. Knockout HSPB8 mice under stress conditions and pressure overload show various abnormalities, comprising increased mortality, faster transition into heart failure, accumulation of interstitial collagen, increased cardiomyocyte length, impaired contractile function, ventricular dilation, and hypertrophy.47
HSPB8 is expressed in the brain, where its expression is not limited but concentrated into the neurogenic niche of the hippocampus (www.brain-map.org; Hspb8-sagittal-b04-0153). The expression of HSPB8 is upregulated in response to hypoxia48 and also expressed in cultured hippocampal neurons.49 The expression of HSPB8 is in the hippocampus of adult rats and mice, but HSPB8 messenger RNA expression depends on the developmental stage.49,50 Its expression in adults increases considerably, whereas at postnatal and embryonic stages it is modestly or not at all expressed in the hippocampus of rats.49 It is also expressed in blood vessels and neurons.51 It is also associated with neuronal survival by interacting with Bag3 to induce macroautophagic removal of misfolded proteins.52,53 HSPB8 mutations are involved in the hereditary peripheral neuropathy of CMT neuropathy type 2.54,55 The missense mutations occur in HSPB8 crystalline-domain neurodegeneration in cultured motor neurons without inducing cell death.55 Moreover, alterations in the crystalline domain lead to CMT neuropathy type 2 and distal HMN. However, the effect of missense mutation is specific only to motor neurons.55
CMT is an inherited peripheral nerve disorder that is characterized by length-dependent and progressive degeneration of peripheral nerves, leading to weakness of muscles and wasting in distal hands, feet, and limbs. Neuropathological and neurophysiological defects in sensory and motor nerves create sensory deficits, wheelchair dependence, walking disabilities, and foot deformities. Depending on the severity of sensory and motor deficiency, CMT is grouped with hereditary sensory and autonomic neuropathies and distal hereditary motor neuropathies. Mutations in HSPB8 have been reported to induce CMT2L (OMIM 60867354), neuronopathy, and distal HMN2A (OMIM 15859056).
The present work demonstrates pharmacophore-based virtual screening to reveal novel inhibitors. Pharmacophore-based molecular libraries were screened by 2-D similarity search against recommended US Food and Drug Administration-approved antidepressants (CMT and HMN2A drugs). The inclusive in silico analyses may provide evidence for a reliable framework that could assist medicinal chemists in the design and development of lead molecules and targeting potential antidepressant drugs.
Materials and methods
In the present work, structure prediction, molecular dynamic (MD) simulation, sequence comparison, library screening, pharmacoinformatics, and docking analyses were performed on an HP workstation. The amino acid sequence of HSPB8 (196 residues) was retrieved for homology modeling, as the gene is a suspected candidate of depression, neurodegenerative disorders, HMN2A,56 and CMT2L.54 The sequence was retrieved in FASTA format from UniProt Knowledgebase with the accession number Q9UJY1. The retrieved sequence of HSPB8 was subjected to a protein–protein Basic Local Alignment Search Tool search against the Protein Data Bank57 for the identification of a suitable template. A 3-D structure of the α-crystallin domain of human HSPB1 and HSPB6 (ID 3Q9P) at a resolution of 2.0 Å was selected as a suitable template, with 55% identity, 37% query coverage, and E-value of 2e-22. The automated protein-modeling program Modeller 9v1058 was utilized to predict the 3-D structure of HSPB8 by satisfying spatial restraints. The threading approach was also utilized to generate an effective 3-D structure. Geometry optimization followed by energy minimization was performed by utilizing UCSC Chimera 1.659 for a total of 750 steps (step size 0.02 Å), employing a conjugate-gradient method followed by protonation of wild-type histidines using the Amber ff98 method.60 The structure was heated to 303 K and simulated for 30 nanoseconds using Gromacs 4.6.5. For input parameters to Gromacs, HSPB8 was provided and MD simulation performed with total steps of 15,000,000 with 0.002 time steps at an average temperature of 303.3 K. The trajectory file was written for every 1,000 MD steps. The ensemble used for MD simulation was NTP (constant number of atoms, temperature, and pressure), with an average grid for the x-axis, y-axis, and z-axis to 8.965 Å. For analysis of MD trajectories, the Visual Molecular Dynamics tool was utilized. Root mean square deviation (RMSD), RMS fluctuation (RMSF), and average RMSD per frame was calculated by loading HSPB8 with a trajectory file. Graphs for RMSD, RMSF, and average RMSD per frame were plotted for better induction of results and analysis of MD simulation. The evaluation tools Anolea,61 Errat,62 Rampage,63 and ProCheck64 were applied to assess the quality of the predicted HSPB8 model. The predicted structure was further evaluated by the MolProbity65 server. Finally, Ramachandran outliers and poor rotamers were corrected by the WinCoot66 tool.
Numerous online servers and tools (ChemDraw,67 Mcule,68 Vega ZZ,69 Osiris Property Explorer,70 Molinspiration,71 Cresset, PyMol, Discovery Studio, AutoDockTools,72 and UCSF Chimera 1.6) were utilized to design novel compounds that might potentially inhibit HSPB8 by interacting with a 3-D model. Docking analyses were performed by AutoDock 4.273 and AutoDock Vina. The hydrogen polar atoms were added to the receptor molecule. The total docking runs were set to 100 for each docking experiment. The grid size was set at 60×60×60 Å in the x-, y, and z-axis, respectively, with 0.575 Å grid spacing to cover the whole receptor. The genetic algorithm implemented in AutoDock 4.2 was employed as the main search protocol, while other parameters were set to default values. The number of H-bond acceptors, rotatable bonds, and H-bond donors were obtained using Molinspiration, Mcule, and PubChem.74 The online tool Osiris Property Explorer was utilized to estimate the possible mutagenic risks, reproductive or tumorigenic behavior, and to calculate the drug-like properties of novel designed molecules. Lipinski’s rule of five was analyzed by Mcule and Cresset servers. Drug-likelihood values calculated by Osiris software are positive when fragments of designed molecules are frequently present in the approved drugs. The Osiris program was used to estimate the mutagenesis of novel molecules, and no mutagenic risks were detected. The properties of already reported drugs (fluoxetine, paroxetine, fluvoxamine, and ethacrynic acid) of depressant, neurodegenerative disorders HMN2A and CMT2L were used for library screening and designing novel molecules. The energy minimization and geometry optimization of 3-D structures of designed molecules were performed by ChemDraw Ultra, Vega ZZ, and LigandScout.75 The absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties were analyzed by the AdmetSAR server.76
Pharmacophoric screening of compounds was performed by employing LigandScout. The three compound libraries (ZINC, Drug, and DrugLike) were screened against HSPB8 structure. The procedure of screening was performed using the LigandScout’s default parameters. The compounds with high pharmacophore score (>70) were extracted, and docking studies were performed on the top hits. The interacting residues were elucidated by employing AutoDock 4.2. The grid box was used to define the screening site. Three pharmacophore-based compounds with optimum binding energies were selected and visualized by LigPlot,76 Discovery Studio 4.0, and UCSF Chimera 1.6. Results were analyzed by using Discovery Studio, LigPlot, UCSF Chimera 1.5.2, and AutoDock 4.2.
Results and discussion
The objective of the study under consideration was based on HSPB8’s relation to depression and the neurodegenerative disorders HMN2A and CMT2L, and its computational analysis for designing, identifying, and evaluating novel inhibitors. The top five optimally aligned templates with query coverage, maximum identity, E-values, and total scores are given in Table 1. The 3Q9P template showed better evaluation results. The 3-D model of HSPB8 was generated by homology modeling. Query coverage and identity were not satisfactory for the generation of 3-D structure by comparative modeling. The evaluation tools also validated that the predicted structures had errors and were not satisfactory for further analyses.
 | Table 1 Five BLAST aligned templates of HSPB8 with identity, query coverage, and E-values |
The threading approach and multiple-template comparative modeling approach were utilized for 3-D structure prediction of HSPB8. To build the 3-D structures, various web servers (3D-Jigsaw, Swiss Model, M4T, and I-TASSER) and Modeller 9.10 were employed. As a result, various evaluation tools were utilized for the comparison of predicted structures and suitable optimal predicted model of HSPB8 was selected. The HSPB8 3-D structure was comparatively modeled using the threading approach.
The predicted model of HSPB8 was subjected to MD simulation by employing appropriate solvent molecules, ensemble, and temperature. Energy minimization was performed on the scrutinized structure to remove steric constraints by relaxing the system. The gradual velocities of the system were gradually increased through heating to the 303.3 K temperature. In 30 nanoseconds’ simulation of protein, it was observed that the protein changed its structure dramatically. The changes in structure were analyzed through RMSD, RMSF, and average RMSD per frame of protein. According to the RMSD graph, the protein seemed to attain higher RMSD at the start of the simulation.
RMSD increased to 14 Å in the first 8 nanoseconds, while stabilization of the protein was observed afterward till the end of the simulation. The line of linearity fell at 9.6476 Å, which depicted high RMSD attainment during simulation (Figure 1). The overall RMSD change according to the Pearson product-moment coefficient was 0.5242 Å, demonstrating protein stabilization after abrupt change in structure.
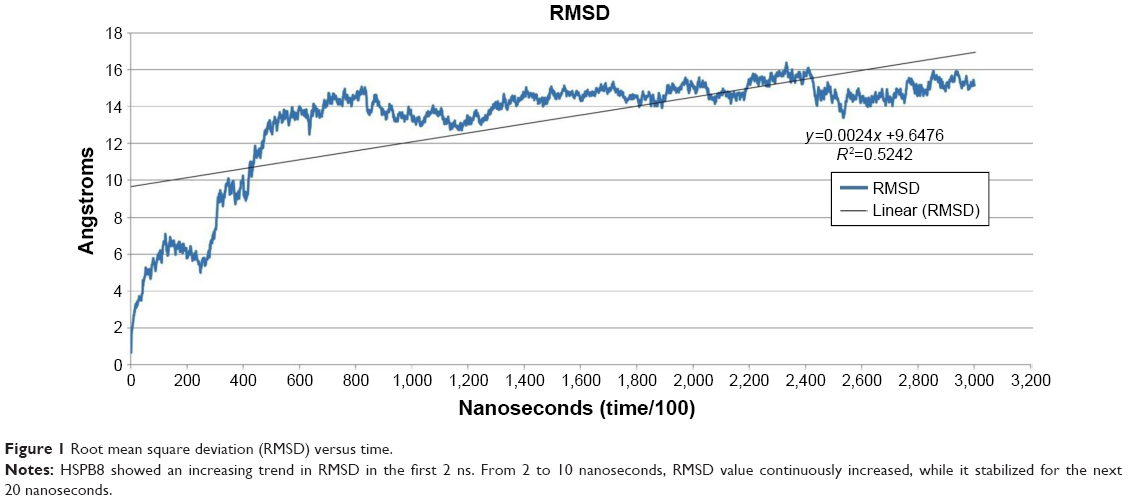 | Figure 1 Root mean square deviation (RMSD) versus time. |
The RMSF graph demonstrated per-residue fluctuations throughout simulation. The observed RMSF values showed that residue fluctuations were less while fluctuation observed in residues from 1–17 and 167–196 that can also seen in protein structure at last nanoseconds (Figure 2). Overall protein residues did not show high fluctuation, fluctuations lasted only up to 8 nanoseconds, and afterwards the protein was stabilized with minimum fluctuation, as can be observed in the RMSD graph.
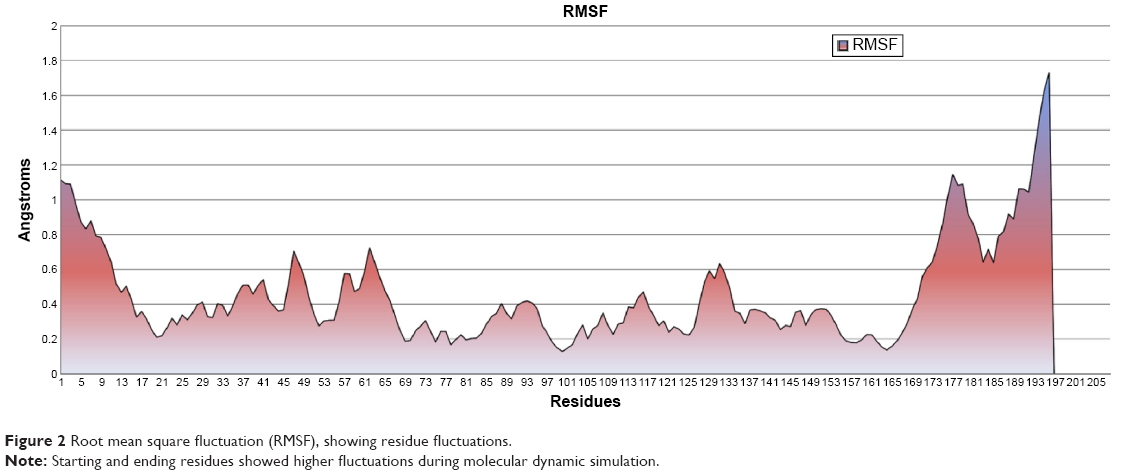 | Figure 2 Root mean square fluctuation (RMSF), showing residue fluctuations. |
Average RMSD per frame demonstrated per-residue fluctuation, including side chains, to give an overall RMSD of a frame. Average RMSD is different from RMSD in the sense that RMSD provides information for fluctuations of only backbone atoms from reference coordinates of protein, while average RMSD calculates changes of coordinates of protein from the reference structure, including complete residues with side chains. While average RMSD per frame values were being compared with RMSD and RMSF values, it could be analyzed that the protein showed higher fluctuation only at the start of the simulation, while fluctuation decreased toward the end of the simulation. Based on average RMSD, starting frames of simulation showed much higher RMSD values, while afterward stabilization of the protein was observed (Figure 3). The simulated minimized 3-D structure of HSPB8 was visualized by UCSF Chimera 1.5.2 (Figure 4).
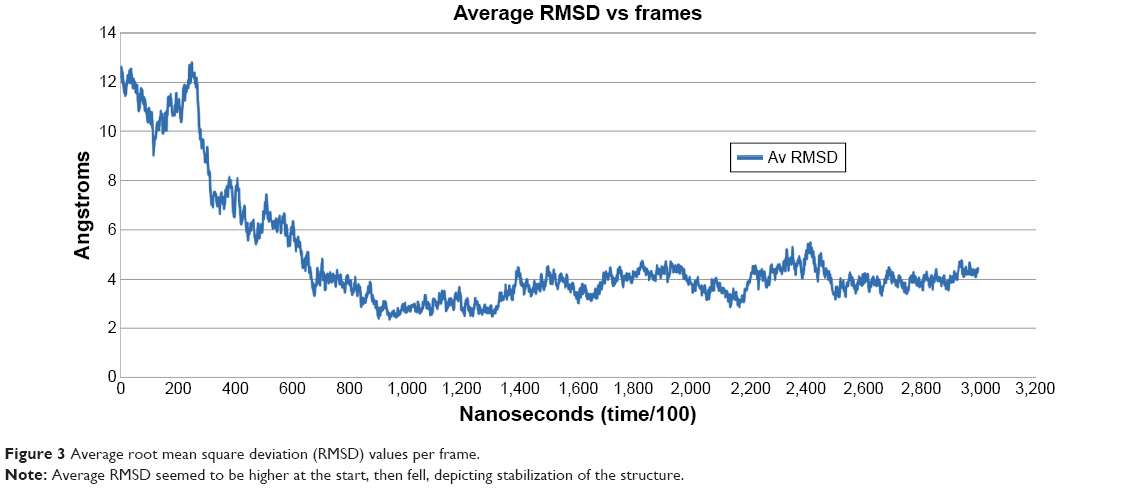 | Figure 3 Average root mean square deviation (RMSD) values per frame. |
 | Figure 4 Predicted HSPB8 structure simulated at 30 nanoseconds. |
The utilized evaluation tools indicated the reliability and efficacy of predicted HSPB8 structure. The Ramachandran plot showed the presence of 93.3% residues in favored region, and only 15 of 196 residues were observed in the outlier region. Errat showed an overall quality factor of 77.444 for structure.
Subsequently, poor rotamers and outliers were corrected to refine the HSPB8 predicted model. Experimental analysis elucidated that selected drugs used in this study (Figure 5) exhibited significant abilities (Table 2) to cure depression, neurodegenerative disorders, and CMT. However, our docking studies of selected drugs exposed variations in their binding energies. Initially, docking studies were performed with 100 runs and 15 poses saved, of which the best pose with the lowest binding energy was selected for each compound. Our results indicated that the four selected compounds (fluoxetine, paroxetine, fluvoxamine, and ethacrynic acid) efficiently bound to HSPB8 (Table 2).
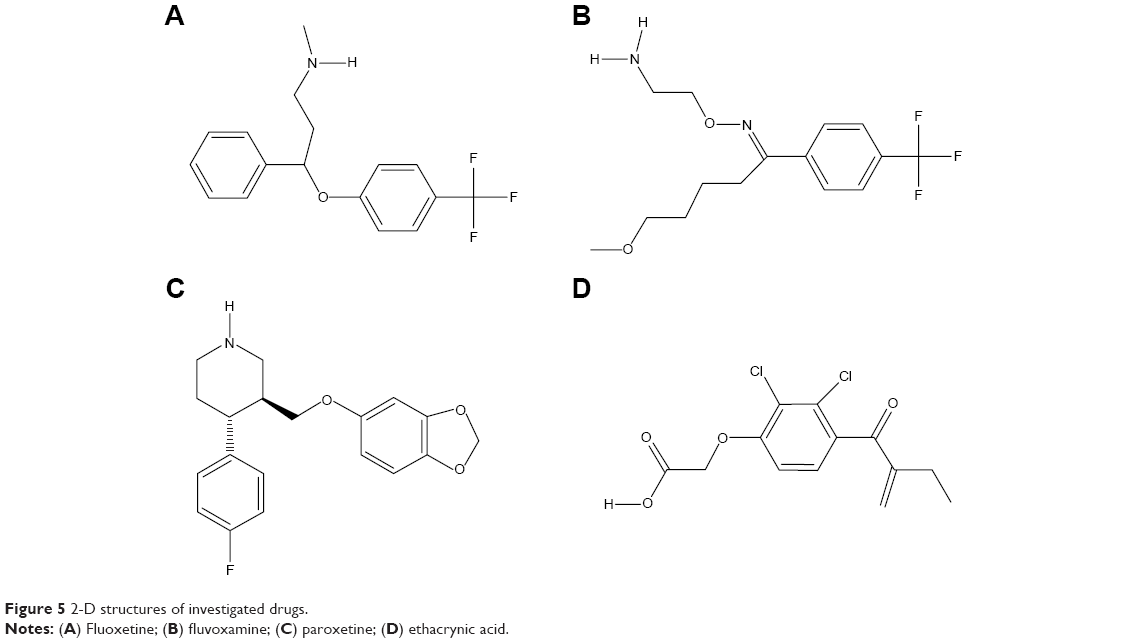 | Figure 5 2-D structures of investigated drugs. |
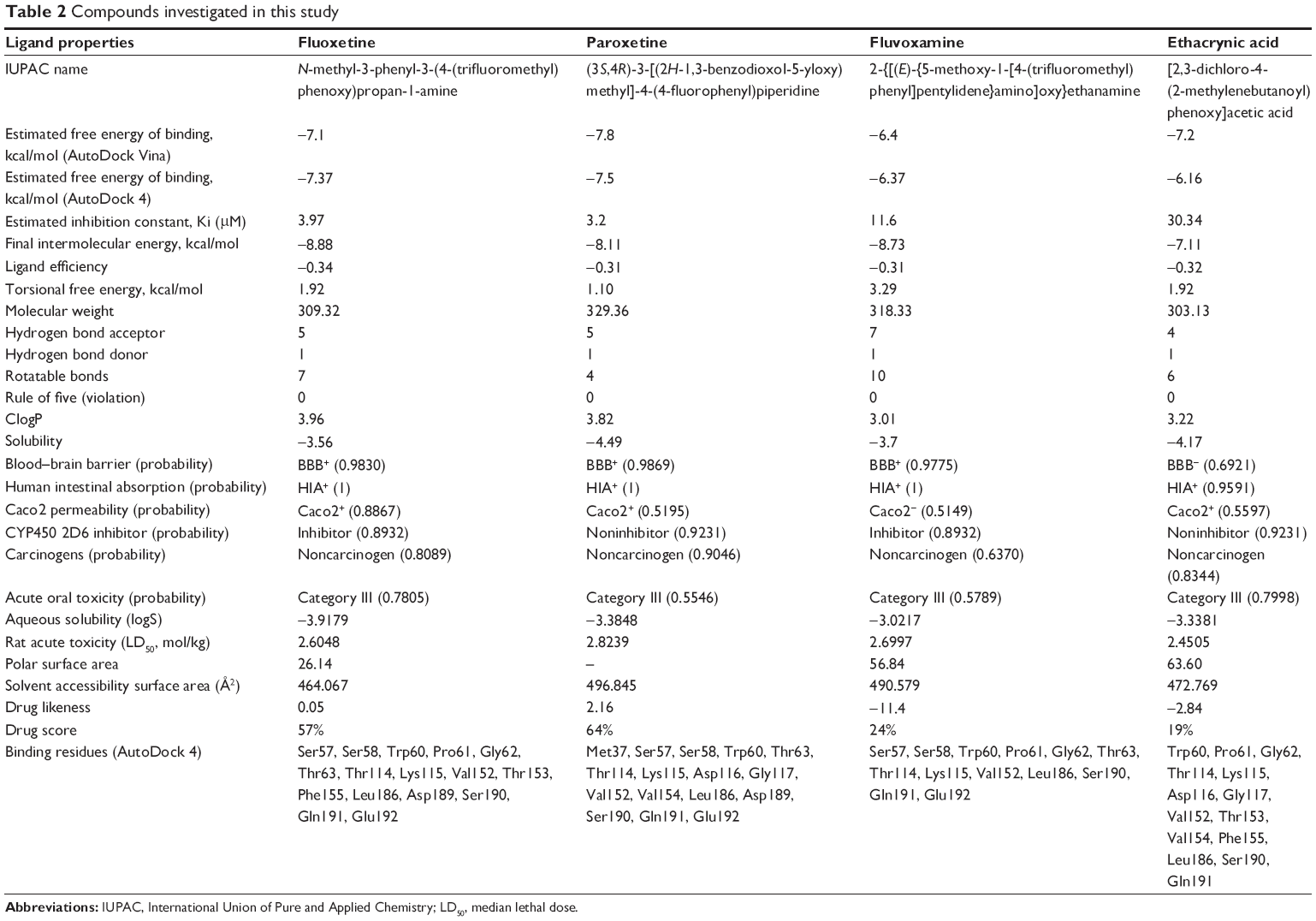 | Table 2 Compounds investigated in this study |
Subsequently, four selected drugs were analyzed on the basis of binding energy values, and ADMET and drug properties (Table 2) indicated that the selected drugs exhibited significant biological properties. Therefore, these drugs may be considered potential agents against depression, neurodegenerative disorders, and CMT. The lowest binding energy value (−7.8 kcal/mol by AutoDock 4 with Vina) was observed for paroxetine. These four drugs were used to generate the pharmacophore.
Libraries were screened by LigandScout, and the 21 compounds with the lowest pharmacophore scores (>70) were selected for further analyses. AutoDock 4 and AutoDock Vina were employed for docking analyses, and the three complexes with the lowest binding energy were chosen (Figure 6). The current work gave reliable results and conclusions by docked analyses of selected compounds against HSPB8. In an effort to investigate the novel hits, three scrutinized molecules from a combination of selected libraries were revealed (Table 3). It was found that the majority of compounds were bound at the binding residues between Ser57 and Ser190. The docking analyses revealed that known (Figure 7) and novel molecules bound at the same types of residues, sharing a quite similar binding pocket. Another observation was that most molecules occupied the space between Ser57 and Gly117.
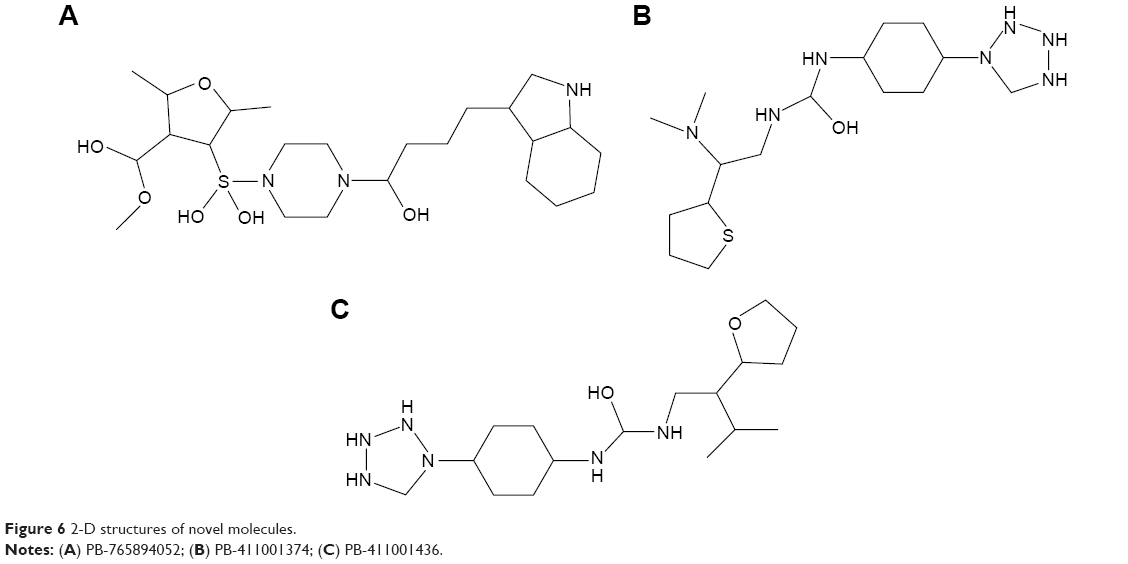 | Figure 6 2-D structures of novel molecules. |
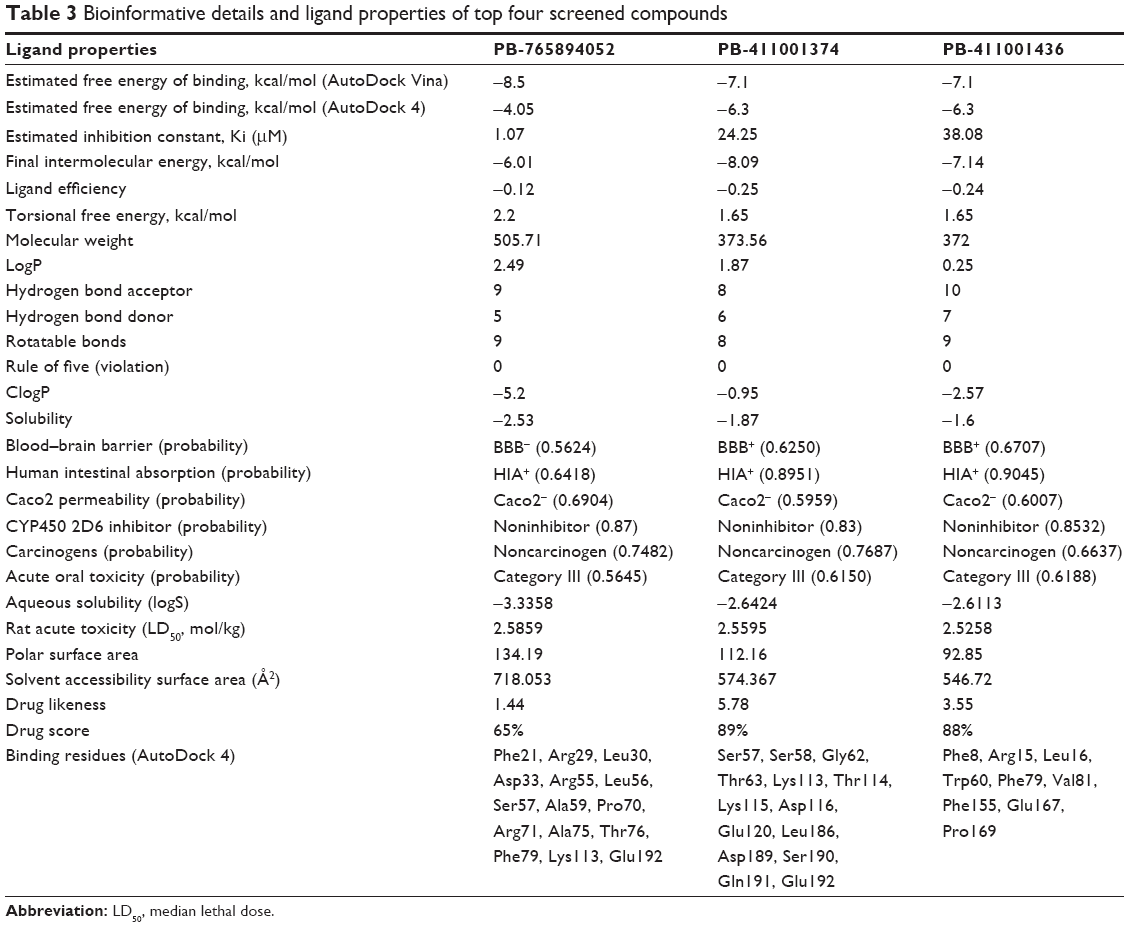 | Table 3 Bioinformative details and ligand properties of top four screened compounds |
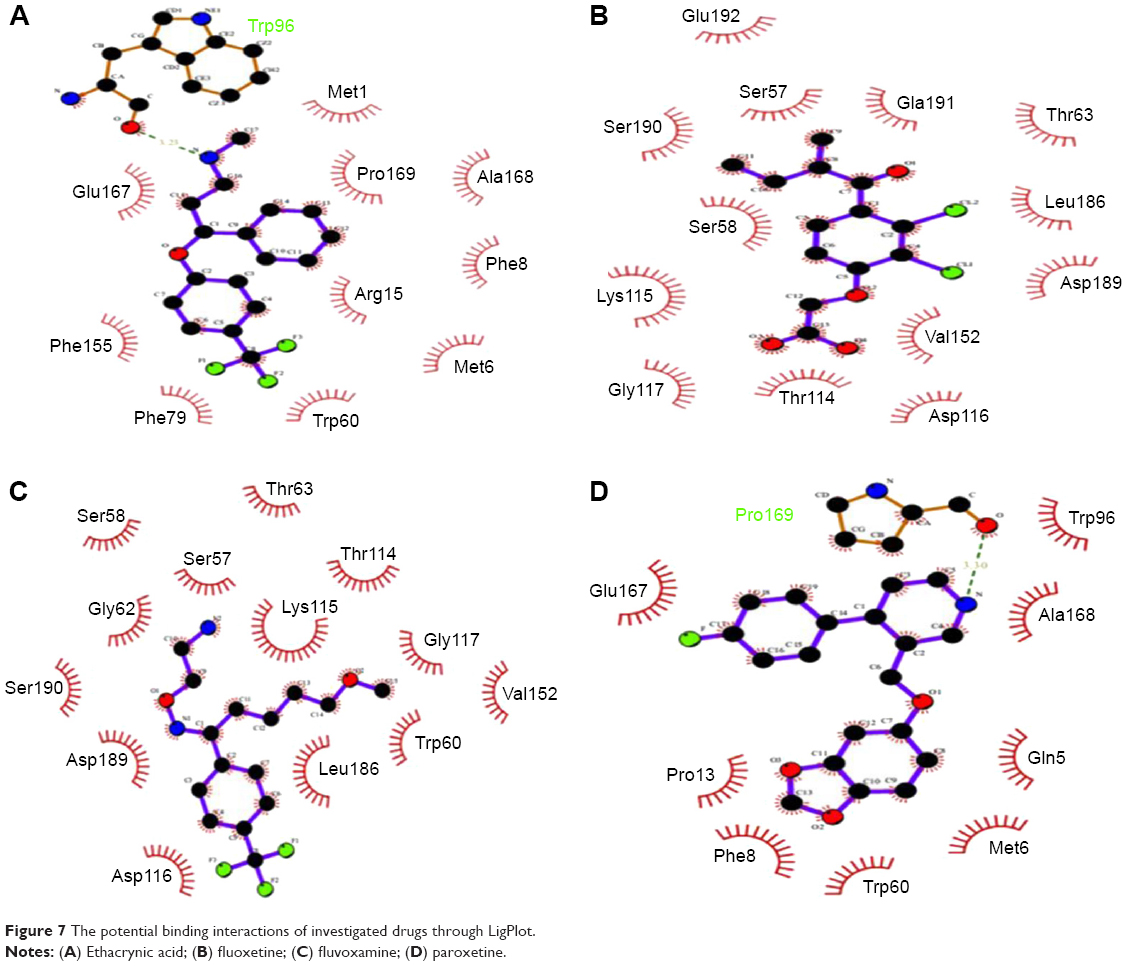 | Figure 7 The potential binding interactions of investigated drugs through LigPlot. |
The highest-ranked hits with the lowest binding energy values from all screened libraries were elucidated, namely PB-765894052. It was observed that Met37, Ser57, Ser58, Trp60, Thr63, Thr114, Lys115, Asp116, Gly117, Val152, Val154, Leu186, Asp189, Ser190, Gln191, and Glu192 of HSPB8 exhibited high binding affinity with all the docked ligands. In an attempt to evaluate the occurrence of detailed interactions among HSPB8-active sites and analyzed ligands, plots of ligand and residue interactions were generated by utilizing UCSF Chimera 1.5.2 (Figure 8).
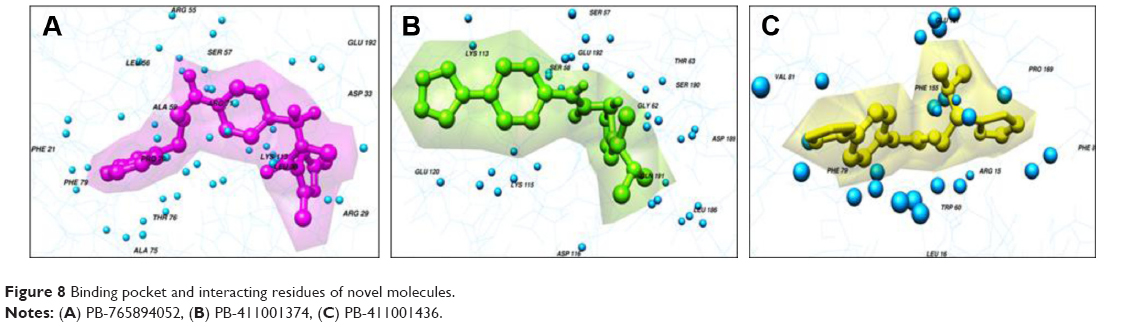 | Figure 8 Binding pocket and interacting residues of novel molecules. |
The HSP family members share significant features: a highly dynamic structure that plays a chaperone-like role, increased expression under stressful conditions, into large oligomers consisting in many cases of subunits, monomeric molecular mass range (12–43 kDa), and formation of the conserved α-crystallin domain structure with about 80–100 residues in the C-terminus.77 HSPB8 is highly demanding, due to its significant role in pathological and physiological processes. The mutations link with the development of CMT and distal motor neuropathy type II.55,78 HSPB8 has also been detected in hereditary cerebral hemorrhage with amyloidosis of the Dutch type and in cerebral amyloid angiopathy and senile plaques in patients with Alzheimer’s disease.50,79,80 HSPB8 has a conserved α-crystallin domain.
The α-crystallin domain extends from residues 86 to 176, while the N-terminus extends from residues 1 to 85. The HSPB8 has 21.6 kDa mass.9,77,81 Fluvoxamine is a widely used drug for depression treatment. The analgesic effects of fluvoxamine have been documented for diabetic neuropathic pain in rats, and are limited due to its administration as a single dose.82 Fluvoxamine has effectively used for ameliorates cardiac hypertrophy, σ1-receptor and dysfunction via σ1-receptor stimulation. Patients in old age usually receive statin drugs for the secondary and primary prevention of cerebrovascular and cardiovascular events.
Patients commonly receive selective serotonin-reuptake inhibitors (SSRIs) as antidepressant drugs for the treatment of anxiety, depression, and other conditions. SSRIs are linked with pharmacokinetics through drug interactions linked to cytochrome CYP450 metabolic pathway inhibition. SSRIs’ specific effect and statin metabolism on CYP450 enzymes suggest that paroxetine, citalopram, and escitalopram are safe with statins. Fluoxetine is a commonly used medication for the treatment of depression. Fluvoxamine used with the combination of lovastatin, simvastatin, or atorvastatin, and the risk can be minimized by monitoring patients and utilizing lower doses of statin.82 Recent research has suggested a possible effect of fluoxetine on bone. This drug may improve the quantity of bone regeneration in calvarial defects of rats.83 Gene-expression changes of β1- and β2-adrenoceptors were studied in the left and right ventricles and atria after treatment with fluoxetine in stress-induced depression in adult male rats. Fluoxetine treatment led to increased expression of β1-adrenoceptor messenger RNA, which suggests that therapy with fluoxetine plays a significant role in animals with heightened sympathetic nervous activity.84 It was investigated whether paroxetine contained adverse effects in comparison with other antidepressant agents, and no clear evidence has yet been reported that paroxetine is worse or better in comparison to antidepressant agents give response to increase the treatment.85 Patients with depression, neurodegenerative disorders, or CMT disease lose quality of life and feel mentally depressed. Mentally relaxed patients recover from CMT earlier compared to depressed patients. The drugs analyzed are used as antidepressant agents, and the identified novel hits may be considered better antidepressants on the basis of overall analysis by targeting HSPB8.
Bioinformatics utilizes in silico analyses of biological problems by using mathematical and statistical approaches,86 and structural bioinformatics predicts and analyzes 3-D structures of macromolecules. Structural bioinformatics has solved numerous biological problems and led to novel computer-aided designed molecules against neurological disorders87,88 and cancer.89–91 Computer-aided drug design helps scientists to design effective active molecules against drugs. Drug design and development is not only a costly procedure but also time-consuming. Therefore, we applied computational approaches and methodologies for pharmacophore generation in a drug-discovery procedure. This emerging tendency has colossal significance in reducing the time phase, by amplifying the design of pharmacophore molecules with minimal side effects for specific targets of disease and better biological activity.
Chemical structures are assessed for better oral bioavailability and to be efficacious drug-like compounds subjected to Lipinski’s rule of five.92 According to Lipinski’s rule of five, molecules should have not violated more than one of the following conditions: logP no higher than 5, molecular weight no more than 500, no more than ten hydrogen-bond acceptors, and no more than five hydrogen-bond donors.93 ADMET properties of molecules within an organism were predicted for selected compounds by utilizing the AdmetSAR online server. Eight mathematical models (blood–brain barrier penetration, aqueous solubility [logS], carcinogens, acute oral toxicity, Caco2 permeability, cytochrome P450 2D6 inhibition, rat acute toxicity, and human intestinal absorption) were utilized to predict the ADMET characteristics of molecular chemical structures. A variety of toxicities were predicted that are often used in drug design (Table 3). These toxicities help in evaluating pollutants, metabolites, and intermediates, along with adjusting the dose range for animal assays.
Polar surface area (PSA) has an inverse relationship with human intestinal absorption. Therefore, cell-wall permeability through a relationship between permeability and PSA has been revealed. PSA values for massive molecular transport through the membrane showed lower PSA at a standard limit of 140 Å2. The prediction of aqueous solubility (defined water at 25°C) showed that selected compounds are soluble in water. LogP is the lipophilicity measurement, which is the solubility ratio of a compound in octanol compared to its solubility in water. It was observed that novel compounds showed lower logP values compared to known drugs, and followed Lipinski’s rule of five, showing better oral bioavailability. The process of excretion that purges the compounds from the human body also depends on logP.92 Orally administered drugs and novel compounds must be absorbed by the human intestine. The analyzed human intestinal absorption results showed that all the selected drugs and novel compounds can be easily absorbed by the intestine. From the selected four compounds, paroxetine was found to be a noninhibitor of cytochrome P450 2D6, which indicated that it may be well metabolized in phase I metabolism. It was also observed that all the identified novel compounds showed noninhibitor properties of cytochrome P450 2D6 and could be metabolized in phase I metabolism. Cytochrome P450 2D6 is one of the significant enzymes involved in drug metabolism.92 Toxicity-risk assessment and carcinogenicity was performed for the selected drugs and novel compounds, and showed that all the compounds were noncarcinogenic.
In the current study, in silico analyses were carried out, and the 3-D structure of HSPB8 was modeled and simulated at 30 nanoseconds. The simulated model had a good degree of accuracy, especially at the active site of the protein. Comparative molecular docking studies were analyzed by AutoDock 4 and AutoDock Vina, which revealed the ligand–receptor interactions of the most representative drugs. The selected drugs showed binding in the binding domain and critical binding residues (Met37, Ser57, Ser58, Trp60, Thr63, Thr114, Lys115, Asp116, Gly117, Val152, Val154, Leu186, Asp189, Ser190, Gln191, Glu192), observed in molecular docking studies of selected drugs with HSPB8. It was found that these residues also interacted with novel compounds (Figure 9), revealed by the binding region. The novel compounds also showed effective drug likeness and drug score compared with selected drugs. The molecular docking studies suggested that effective ligand molecule must have the least binding energy and also it has to satisfy the drug-likeness parameters. Given this, and the satisfaction of the parameters of drug likeness, ADMET properties, drug score, and lowest docking energy, it is suggested that paroxetine and novel molecules are potential drug molecules. It stands to reason that the discovered compounds have the propensity to be good candidate molecules.
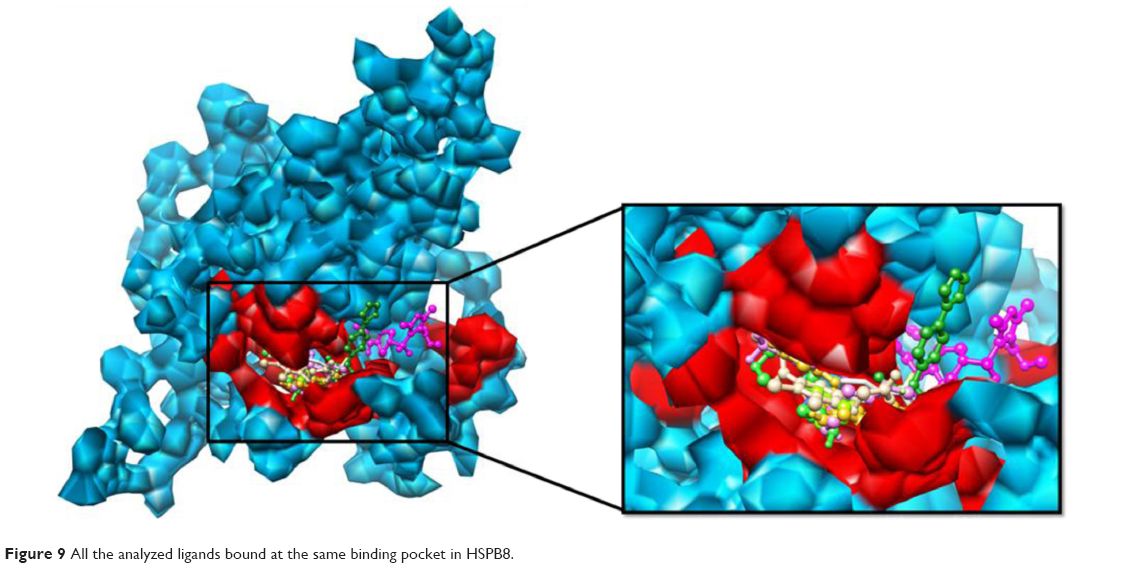 | Figure 9 All the analyzed ligands bound at the same binding pocket in HSPB8. |
In conclusion, the present study suggested that novel compounds and selected drugs are effective in depression, neurodegenerative disorder, and CMT treatment. Though various divergences exist among trail-methodology studies, baseline population- and bioinformatics-based pharmacoanalysis seem to be sufficient to conclude that paroxetine may be the better option for treatment. Further studies and synthesis of novel compounds considering these findings may expect similar response rates.
Acknowledgments
The work was sponsored by a CAS-TWAS President’s Fellowship for International PhD Students. The authors are thankful to Mirza A Hammad, and Rana Adnan Tahir, Bioinformatics Research Laboratory, Department of Bioscience, COMSATS Institute of Information Technology, Sahiwal, Pakistan, for their kind guidance in interpreting and suggestions for improving the results. The project was funded by a grant from the National Natural Sciences Foundation of China (NSFC31370028 to Lei Du).
Disclosure
The authors report no conflicts of interest in this work.
References
Fontaine JM, Rest JS, Welsh MJ, Benndorf R. The sperm outer dense fiber protein is the 10th member of the superfamily of mammalian small stress proteins. Cell Stress Chaperones. 2003;8:62–69. | ||
Kappé G, Franck E, Verschuure P, Boelens WC, Leunissen JA, De Jong WW. The human genome encodes 10 α-crystallin-related small heat shock proteins: HspB1-10. Cell Stress Chaperones. 2003;8:53–61. | ||
Lambert H, Charette SJ, Bernier AF, Guimond A, Landry J. HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J Biol Chem. 1999;274:9378–9385. | ||
Liu C, Welsh MJ. Identification of a site of Hsp27 binding with Hsp27 and αB-crystallin as indicated by the yeast two-hybrid system. Biochem Biophys Res Commun. 1999;255:256–261. | ||
Rogalla T, Ehrnsperger M, Preville X, et al. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor α by phosphorylation. J Biol Chem. 1999;274:18947–18956. | ||
Verschuure P, Tatard C, Boelens WC, Grongnet JF, David JC. Expression of small heat shock proteins HspB2, HspB8, Hsp20 and cvHsp in different tissues of the perinatal developing pig. Eur J Cell Biol. 2003;82:523–530. | ||
Tallot P, Grongnet JF, David JC. Dual perinatal and developmental expression of the small heat shock proteins (FC12) αB-crystallin and Hsp27 in different tissues of the developing piglet. Biol Neonate. 2003;83:281–288. | ||
Sugiyama Y, Suzuki A, Kishikawa M, et al. Muscle develops a specific form of small heat shock protein complex composed of MKBP/HSPB2 and HSPB3 during myogenic differentiation. J Biol Chem. 2010;275:1095–1104. | ||
Carra S, Sivilotti M, Chávez Zobel AT, Lambert H, Landry J. HspB8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum Mol Genet. 2005;14:1659–1669. | ||
Krief JF, Faivre P, Robert B, et al. Identification and characterization of cvHsp: a novel human small stress protein selectively expressed in cardiovascular and insulin-sensitive tissues. J Biol Chem. 1999;274:36592–36600. | ||
Kriehuber T, Rattei T, Weinmaier T, Bepperling A, Haslbeck M, Buchner J. Independent evolution of the core domain and its flanking sequences in small heat shock proteins. FASEB J. 2010;24:3633–3642. | ||
Vos MJ, Hageman J, Carra S, Kampinga HH. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry. 2008;47:7001–7011. | ||
Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–846. | ||
Horwitz J. Alpha-crystallin. Exp Eye Res. 2003;76:145–153. | ||
Haley DA, Bova MP, Huang QL, McHaourab HS, Stewart PL. Small heat-shock protein structures reveal a continuum from symmetric to variable assemblies. J Mol Biol. 2000;298:261–272. | ||
Kim KK, Kim R, Kim SH. Crystal structure of a small heat-shock protein. Nature. 1998;394:595–599. | ||
van Montfort RL, Basha E, Friedrich KL, Slingsby CL, Vierling E. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat Struct Biol. 2001;8:1025–1030. | ||
Bagnéris C, Bateman OA, Naylor CE, et al. Crystal structures of α-crystallin domain dimers of αB-crystallin and Hsp20. J Mol Biol. 2009;392:1242–1252. | ||
Laganowsky A, Benesch JL, Landau M, et al. Crystal structures of truncated αA and αB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Protein Sci. 2010;19:1031–1043. | ||
Jehle S, Vollmar BS, Bardiaux B, et al. N-terminal domain of αB-crystallin provides a conformational switch for multimerization and structural heterogeneity. Proc Natl Acad Sci U S A. 2011;108:6409–6414. | ||
Mymrikov EV, Seit-Nebi AS, Gusev NB. Large potentials of small heat shock proteins. Physiol Rev. 2011;91:1123–1159. | ||
Stamler R, Kappé G, Boelens W, Slingsby C. Wrapping the α-crystallin domain fold in a chaperone assembly. J Mol Biol. 2005;353: 68–79. | ||
Van Montfort R, Slingsby C, Vierling E. Structure and function of the small heat shock protein/α-crystallin family of molecular chaperones. Adv Protein Chem. 2011;59:105–156. | ||
Haslbeck M, Braun N, Stromer T, et al. Hsp42 is the general small heat shock protein in the cytosol of Saccharomyces cerevisiae. EMBO J. 2004;23:638–649. | ||
Ribeiro DA, Bem LE, Vicentini R, Ferraz LF, Murakami MT, Ottoboni LM. The small heat shock proteins from Acidithiobacillus ferrooxidans: gene expression, phylogenetic analysis, and structural modeling. BMC Microbiol. 2011;11:259. | ||
Siddique M, Gernhard S, von Koskull-Döring P, Vierling E, Scharf KD. The plant sHSP superfamily: five new members in Arabidopsis thaliana with unexpected properties. Cell Stress Chaperones. 2008;13:183–197. | ||
Elicker KS, Hutson LD. Genome-wide analysis and expression profiling of the small heat shock proteins in zebrafish. Gene. 2007;403:60–69. | ||
Willsie JK, Clegg JS. Small heat shock protein p26 associates with nuclear lamins and HSP70 in nuclei and nuclear matrix fractions from stressed cells. J Cell Biochem. 2002;84:601–614. | ||
Boncoraglio A, Minoia M, Carra S. The family of mammalian small heat shock proteins (HSPBs): implications in protein deposit diseases and motor neuropathies. Int J Biochem Cell Biol. 2012;44:1657–1669. | ||
Bruinsma IB, Bruggink KA, Kinast K, et al. Inhibition of α-synuclein aggregation by small heat shock proteins. Proteins. 2011;79:2956–2967. | ||
Filipović D, Zlatković J, Gass P, Inta D. The differential effects of acute vs. chronic stress and their combination on hippocampal parvalbumin and inducible heat shock protein 70 expression. Neuroscience. 2013;236:47–54. | ||
Pae CU, Mandelli L, Serretti A, et al. Heat-shock protein-70 genes and response to antidepressants in major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1006–1011. | ||
Yang C, Trent S, Ionescu-Tiba V, et al. Identification of cyclin D1- and estrogen-regulated genes contributing to breast carcinogenesis and progression. Cancer Res. 2006;66:11649–11658. | ||
Modem S, Chinnakannu K, Bai U, Reddy GP, Reddy TR. Hsp22 (HspB8/H11) knockdown induces Sam68 expression and stimulates proliferation of glioblastoma cells. J Cell Physiol. 2011;226: 2747–2751. | ||
Smith CC, Yu YX, Kulka M, Aurelian L. A novel human gene similar to the protein kinase (PK) coding domain of the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) codes for a serine-threonine PK and is expressed in melanoma cells. J Biol Chem. 2000;275:25690–25699. | ||
Sui X, Li D, Qiu H, Gaussin V, Depre C. Activation of the bone morphogenetic protein receptor by H11 kinase/Hsp22 promotes cardiac cell growth and survival. Circ Res. 2009;104:887–895. | ||
Gober MD, Smith CC, Ueda K, Toretsky JA, Aurelian L. Forced expression of the H11 heat shock protein can be regulated by DNA methylation and trigger apoptosis in human cells. J Biol Chem. 2003;278:37600–37609. | ||
Lutsch G, Vetter R, Offhauss U, et al. Abundance and location of the small heat shock proteins HSP25 and αB-crystallin in rat and human heart. Circulation. 1997;96:3466–3476. | ||
Shama KM, Suzuki A, Harada K, et al. Transient up-regulation of myotonic dystrophy protein kinase-binding protein, MKBP, and HSP27 in the neonatal myocardium. Cell Struct Funct. 1999;24:1–4. | ||
Gernold M, Knauf U, Gaestel M, Stahl J, Kloetzel PM. Development and tissue-specific distribution of mouse small heat shock protein Hsp25. Dev Genet. 1993;14:103–111. | ||
Tucker NR, Shelden EA. Hsp27 associates with the titin filament system in heat-shocked zebrafish cardiomyocytes. Exp Cell Res. 2009;315:3176–3186. | ||
Franck E, Madsen O, van Rheede T, et al. Evolutionary diversity of vertebrate small heat shock proteins. J Mol Evol. 2004;59:792–805. | ||
Michaud S, Marin R, Tanguay RM. Regulation of heat shock gene induction and expression during Drosophila development. Cell Mol Life Sci. 1997;53:104–113. | ||
Depre C, Kim SJ, John AS, et al. Program of cell survival underlying human and experimental hibernating myocardium. Circ Res. 2004;95:433–440. | ||
Depre C, Hase M, Gaussin V, et al. H11 kinase is a novel mediator of myocardial hypertrophy in vivo. Circ Res. 2002;91:1007–1014. | ||
Laure L, Long R, Lizano P, et al. Cardiac H11 kinase/Hsp22 stimulates oxidative phosphorylation and modulates mitochondrial reactive oxygen species production: involvement of a nitric oxide-dependent mechanism. Free Radic Biol Med. 2012;52:2168–2176. | ||
Qiu H, Lizano P, Laure L, et al. H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation. 2011;124:406–415. | ||
David JC, Boelens WC, Grongnet JF. Up-regulation of heat shock protein HSP 20 in the hippocampus as an early response to hypoxia of the newborn. J Neurochem. 2006;99:570–581. | ||
Kirbach BB, Golenhofen N. Differential expression and induction of small heat shock proteins in rat brain and cultured hippocampal neurons. J Neurosci Res. 2011;89:162–175. | ||
Quraishe S, Asuni A, Boelens WC, O’Connor V, Wyttenbach A. Expression of the small heat shock protein family in the mouse CNS: differential anatomical and biochemical compartmentalization. Neuroscience. 2008;153:483–491. | ||
Wilhelmus MM, Boelens WC, Otte-Höller I, et al. Small heat shock protein HspB8: its distribution in Alzheimer’s disease brains and its inhibition of amyloid-β protein aggregation and cerebrovascular amyloid-β toxicity. Acta Neuropathol. 2006;111:39–49. | ||
Yew EH, Cheung NS, Choy MS, et al. Proteasome inhibition by lactacystin in primary neuronal cells induces both potentially neuroprotective and proapoptotic transcriptional responses: a microarray analysis. J Neurochem. 2005;94:943–956. | ||
Gurusamy N, Lekli I, Gherghiceanu M, Popescu LM, Das DK. BAG-1 induces autophagy for cardiac cell survival. Autophagy. 2009;5:120–121. | ||
Tang D, Khaleque MA, Jones EL, et al. Expression of heat shock proteins and heat shock protein messenger ribonucleic acid in human prostate carcinoma in vitro and in tumors in vivo. Cell Stress Chaperones. 2005;10:46–58. | ||
Irobi J, Almeida-Souza L, Asselbergh B, et al. Mutant HSPB8 causes motor neuron-specific neurite degeneration. Hum Mol Genet. 2010;19:3254–3265. | ||
Irobi J, Van Impe K, Seeman P, et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 2004;36:597–601. | ||
Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. | ||
Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with Modeller. Methods Mol Biol. 2008;426:145–159. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1161. | ||
Cornell WD, Cieplak P, Bayly CI, et al. A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J Am Chem Soc. 1995;117:5179–5197. | ||
Melo F, Devos D, Depiereux E, Feytmans E. ANOLEA: a www server to assess protein structures. Proc Int Conf Intell Syst Mol Biol. 1997;97:110–113. | ||
Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. | ||
Lovell SC, Davis IW, Arendall WB 3rd, et al. Structure validation by Cα geometry: φ, ψ and Cβ deviation. Proteins. 2002;50:437–450. | ||
Laskowski RA, MacArthur MW, Moss DS, Thornton JM. ProCheck: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. | ||
Chen VB, Arendall WB 3rd, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. | ||
Emsley P, Lohkamp B, Scott W, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. | ||
Mendelsohn LD. ChemDraw 8 Ultra: Windows and Macintosh versions. J Chem Inf Comput Sci. 2004;44:2225–2226. | ||
Kiss R, Sandor M, Szalai FA. http://Mcule.com: a public web service for drug discovery. J Cheminform. 2012;4:17. | ||
Pedretti A, Villa L, Vistoli G. VEGA – an open platform to develop chemo-bio-informatics applications, using plug-in architecture and script “programming”. J Comput Aided Mol Des. 2004;18:167–173. | ||
Organic Chemistry Portal. OSIRIS property explorer. 2001. Available from: http://www.organic-chemistry.org/prog/peo. Accessed April 4, 2016. | ||
Mishra NK, Raghava GP. Prediction of specificity and cross-reactivity of kinase inhibitors. Lett Drug Des Discov. 2011;38:223–228. | ||
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. | ||
Bolton EE, Wang YL, Thiessen PA, Bryant SH. Chapter 12 – PubChem: Integrated platform of small molecules and biological activities. Annu Rep Comput Chem. 2008;4:217–241. | ||
Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005;45:160–169. | ||
Cheng F, Li W, Zhou Y, et al. AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52:3099–3105. | ||
Wallace AC, Laskowski RA, Thornton JM. LigPlot: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1996;8:127–134. | ||
Tang BS, Zhao GH, Luo W, et al. Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum Genet. 2005;116:222–224. | ||
Carra S, Sivilotti M, Chávez Zobel AT, Lambert H, Landry J. HspB8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum Mol Genet. 2005;14:1659–1669. | ||
Wilhelmus MM, Boelens WC, Kox M, et al. Small heat shock proteins associated with cerebral amyloid angiopathy of hereditary cerebral hemorrhage with amyloidosis (Dutch type) induce interleukin-6 secretion. Neurobiol Aging. 2009;30:229–240. | ||
Chowdary TK, Raman B, Ramakrishna T, Rao CM. Mammalian Hsp22 is a heat-inducible small heat-shock protein with chaperone-like activity. Biochem J. 2004;381:379–387. | ||
Kato T, Kajiyama S, Hamada H, Kawamoto M. Long-term administration of fluvoxamine attenuates neuropathic pain and involvement of spinal serotonin receptors in diabetic model rats. Hiroshima J Med Sci. 2013;62:83–89. | ||
Mortazavi SH, Khojasteh A, Vaziri H, Khoshzaban A, Roudsari MV, Razavi SH. The effect of fluoxetine on bone regeneration in rat calvarial bone defects. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;108:22–27. | ||
Spasojevic N, Gavrilovic L, Jovanovic P, Dronjak S. Fluoxetine treatment acts selectively increasing myocardial β1-adrenoceptor mRNA expression in stress-induced depression. Pharmazie. 2011;66:611–613. | ||
Purgato M, Papola D, Gastaldon C, et al. Paroxetine versus other anti-depressive agents for depression. Cochrane Database Syst Rev. 2014;4:CD006531. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. | ||
Sehgal SA, Khattak NA, Mir A. Structural, phylogenetic and docking studies of D-amino acid oxidase activator (DAOA), a candidate schizophrenia gene. Theor Biol Med Model. 2013;10:3. | ||
Sehgal SA, Hassan M, Rashid S. Pharmacoinformatics elucidation of potential drug targets against migraine to target ion channel protein KCNK18. Drug Des Devel Ther. 2014;8:571–581. | ||
Sehgal SA, Mannan S, Kanwal S, Naveed I, Mir A. Adaptive evolution and elucidating the potential inhibitor against schizophrenia to target DAOA (G72) isoforms. Drug Des Devel Ther. 2015;9:3471–3480. | ||
Tahir RA, Sehgal SA, Khattak NA, Khattak JZK, Mir A. Tumor necrosis factor receptor superfamily 10B (TNFRSF10B): an insight from structure modeling to virtual screening for designing drug against head and neck cancer. Theor Biol Med Model. 2013;10(1):38. | ||
Kanwal S, Jamil F, Ali A, Sehgal SA. Comparative Modeling, Molecular Docking, and Revealing of Potential Binding Pockets of RASSF2; a Candidate Cancer Gene. Interdiscip Rev Comput Mol Sci. 2016;18:1–10. | ||
Sehgal SA, Tahir RA, Shafique S, Hassan M, Rashid S. Molecular Modeling and Docking Analysis of CYP1A1 Associated with Head and Neck Cancer to Explore its Binding Regions. Journal of Theoretical and Computational Science. 2014;1:112. | ||
Alam S, Khan F. QSAR and docking studies on xanthone derivatives for anticancer activity targeting DNA topoisomerase IIα. Drug Des Devel Ther. 2014;8:183–195. | ||
Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76:391–396. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.