")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
Pharmacogenomics in the Management of Pulmonary Arterial Hypertension: Current Perspectives
Received 25 January 2023
Accepted for publication 19 June 2023
Published 11 July 2023 Volume 2023:16 Pages 729—737
DOI https://doi.org/10.2147/PGPM.S361222
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
James C Coons,1,2 Philip E Empey1
1Department of Pharmacy and Therapeutics, University of Pittsburgh School of Pharmacy, Pittsburgh, PA, USA; 2Department of Pharmacy, UPMC Presbyterian-Shadyside Hospital, Pittsburgh, PA, USA
Correspondence: James C Coons, Email [email protected]
Abstract: Pulmonary arterial hypertension (PAH) is a rare disease with heterogeneous causes that can lead to right ventricular (RV) failure and death if left untreated. There are currently 10 medications representative of five unique pharmacologic classes that are approved for treatment. These have led to significant improvements in overall clinical outcome. However, substantial variability in dosing requirements and treatment response is evident, leading to suboptimal outcome for many patients. Furthermore, dosing is empiric and iterative and can lead to delays in meeting treatment goals and burdensome adverse effects. Pharmacogenomic (PGx) associations have been reported with certain PAH medications, such as treprostinil and bosentan, and can explain some of the variability in response. Relevant genes associated with treprostinil include CYP2C8, CYP2C9, CAMK2D, and PFAS. CYP2C8 and CYP2C9 are the genes encoding the major metabolizing liver enzymes for treprostinil, and reduced function variants (*2, *3) with CYP2C9 were associated with lower treatment persistence. Additionally, a higher CYP2C9 activity score was associated with a significantly less risk of treatment discontinuation. Other genes of interest that have been explored with treprostinil include CAMK2D, which is associated with right ventricular dysfunction and significantly higher dose requirements. Similarly, PFAS is associated with lower concentrations of cyclic adenosine monophosphate and significantly higher dose requirements. Genes of interest with the endothelin receptor antagonist (ERA) class include GNG2 and CYP2C9. A genetic variant in GNG2 (rs11157866) was linked to a significantly increased rate of clinical improvement with ERAs. The *2 variant with CYP2C9 (encoding for the major metabolizing enzyme for bosentan) was significantly associated with a higher risk for elevations in hepatic aminotransferases and liver injury. In summary, this article reviews the relevant pharmacogenes that have been associated to date with dosing and outcome among patients who received PAH medications.
Keywords: pulmonary hypertension, pulmonary arterial hypertension, pharmacogenomics
Introduction
Pulmonary arterial hypertension (PAH) is a rare and progressive disease with a complex pathobiology that results in increased pulmonary vascular resistance. Pulmonary vasoconstriction, vessel remodeling, and thrombosis are hallmarks of the disease resulting in neointimal proliferation and vaso-occlusive lesions.1,2 Ultimately, disease progression results in right ventricular failure and death if left untreated.1,3 The main heritable risk factor is bone morphogenetic protein receptor type 2 (BMPR2) which alters transforming growth factor beta (signaling). However, many other genetic risk factors have also been identified.2 PAH can result from a heterogeneous group of disorders, including idiopathic and heritable forms, and associated conditions (ie, connective tissue diseases, infection, congenital heart disease, and drugs/toxins).3 Therapeutic targets address major pathways associated with PAH. An imbalance in vasodilators such as a deficiency of nitric oxide (NO) and prostaglandin I2 (prostacyclin) and an excess of vasoconstrictors such as endothelin-1 (ET-1) contribute to vascular dysfunction. NO and prostacyclin also inhibit platelet activation and have antiproliferative effects, whereas ET-1 exerts mitogenic effects.1,3 Pharmacotherapeutic approaches to PAH are primarily based on clinical factors, such as disease severity, and dosing is empiric. However, an evolving understanding of pharmacogenomics in this patient population offers the promise of a precision approach to treatment. Pharmacogenes of interest that influence the dosing and response to prostacyclins and endothelin receptor antagonists (ERAs) are key examples that will be highlighted.
Overview of Pharmacotherapy for PAH
Despite PAH being a rare disease, there have been more than 40 randomized clinical trials that have led to the regulatory approval of ten medications in five distinct pharmacologic classes over the past 25 years.4 Furthermore, these medications are commercially available by four routes of administration (intravenous, subcutaneous, oral, and inhaled). The nitric oxide (NO)-soluble guanylate cyclase (sGC)-cyclic guanosine monophosphate (cGMP) pathway can be enhanced through phosphodiesterase-type 5 inhibitors (PDE5i) (eg, sildenafil, tadalafil) and SGC stimulators (eg, riociguat). The endothelin-1 (ET-1) pathway can be attenuated through non-selective (eg, bosentan, macitentan) and selective (ambrisentan) endothelin receptor antagonists (ERAs). Finally, the prostacyclin pathway can be augmented through prostaglandin analogues (eg, epoprostenol, treprostinil, iloprost) and selective IP receptor agonists (eg, selexipag).
Current PAH Treatment Paradigm
The current treatment paradigm for PAH entails multiparametric risk stratification to guide pharmacotherapy.4 Several variables are assessed, such as symptom progression, right heart failure, functional class, exercise testing, 6-minute walk distance performance, hemodynamics, cardiac imaging and biomarkers. Upon risk assessment, guidelines direct clinicians to several evidence-based medication options (monotherapy or combination) yet with similar levels of evidence.3,5 With the relative lack of head-to-head clinical comparisons within a given class or between classes, clinicians often select risk-appropriate medications based on factors such as dosing convenience, cost, and other patient preferences. Certain treatment tenets apply, such as the use of upfront oral combination therapy with a PDE5i plus an ERA as a first-line approach for most low-to-intermediate risk patients who are treatment naïve.3,6 Additionally, infusion prostacyclins are recommended for patients with more advanced symptoms of PAH based on their potency.4,7
Variability in PAH Medication Response: A Basis for Pharmacogenomic Exploration
With significant advances in treatments for PAH, survival in the current era has dramatically improved in contrast to the early 1980s.8,9 However, clinical outcomes remain unsatisfactory and significant variability in response to these medications is apparent.10 The notion of “super responders” to PAH pharmacotherapy has been evident to clinicians for many years.5 While there is no consensus definition on this phenomenon, evidence for heterogeneity in response is apparent with several medication classes.5 Perhaps, the most notable example is the identification of patients (<10%) who have a positive response to long-term calcium channel blocker therapy (after meeting criteria based on vasoreactivity testing).11,12 These patients have a substantially better long-term prognosis than non-responders.12 Prostacyclins are another important example of treatment response being highly patient dependent. Furthermore, factors that predict response are insufficiently understood.5 Epoprostenol is the most potent and efficacious treatment and typically used for those with the most advanced symptoms.4 However, some patients are able to survive long term, while others advance to right heart failure, transplant, or death despite treatment.13 Still, other patients are able to successfully wean off of epoprostenol and transition to other less potent, oral therapies.14 Regardless of response, the ability to predict whether (and to what extent) an individual patient will respond to a PAH medication, such as epoprostenol, carries important prognostic and treatment implications.15
The current treatment approach to PAH is iterative and empiric and largely determined by clinical risk stratification.4 Pharmacogenomics (PGx) provides a basis for understanding how genetics influences medication response.16 Currently, pharmacogenomic studies of PAH are relatively limited. However, genomic studies that provide insight into the disease process itself are more commonplace.17–19 A precision approach to PAH pharmacotherapy offers the promise of identifying those individuals most likely to have a favorable response with minimal adverse effects.20,21 Table 1 summarizes the pharmacogenes that have been associated to date with dosing and outcome among patients who received PAH medications.
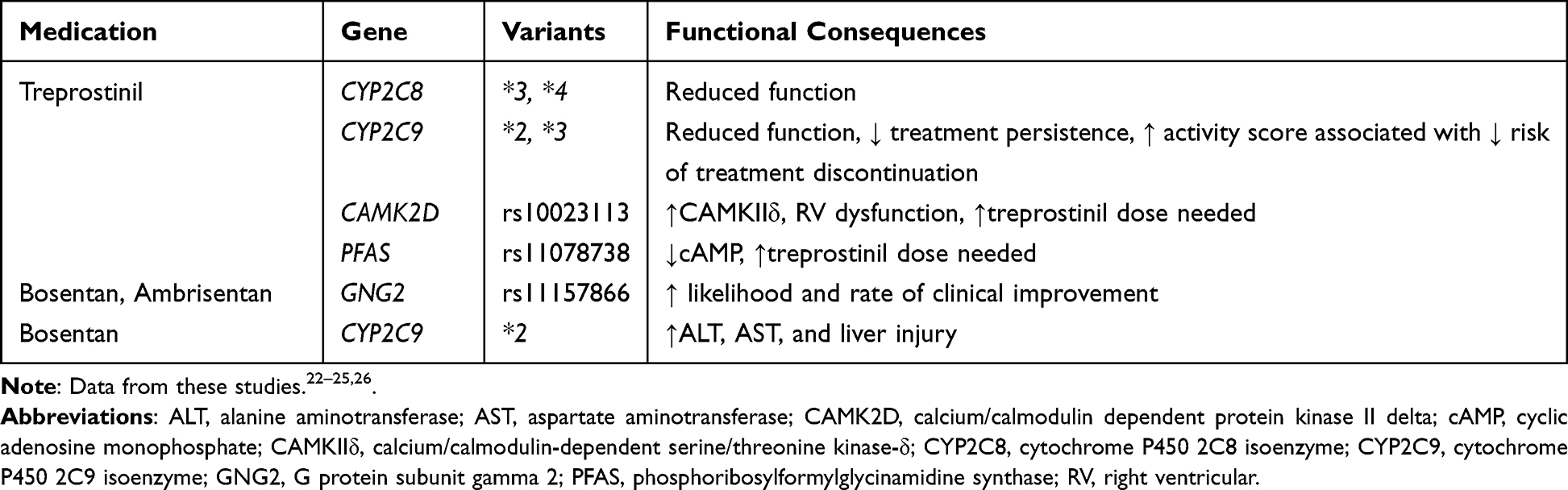 |
Table 1 Pulmonary Hypertension Medications and Pharmacogenes of Interest |
Treprostinil Metabolism and the Role of CYP2C8/CYP2C9
PGx variants that affect prostacyclin metabolism and transport have not been extensively studied. Treprostinil is a prostacyclin analogue approved for PAH and can be given subcutaneously, intravenously, inhaled, and orally.4 The hepatic cytochrome P-450 (CYP) isoenzyme system is the enzyme pathway responsible for treprostinil metabolism. Both the CYP2C8 and CYP2C9 isoenzymes are involved, although the medication is metabolized less extensively by CYP2C9. The CYP pathway is known to be polymorphic with variants in CYP2C9 (*2 and *3 alleles) having population frequencies of approximately 35% in Caucasians.27,28 These alleles are associated with reduced or no function (intermediate or poor metabolizer phenotypes). Transporters also have the potential to affect medication response. ATP-binding cassette (ABC) transporters are transmembrane proteins that use ATP for the active transport of various medications, leading to their elimination. Multidrug resistance protein 4 (MRP4 encoded by ABCC4) is highly polymorphic transporter of treprostinil.29
A single-center study explored the potential impact of PGx as a source of variability in treatment persistence and dosing with oral treprostinil.22 Treprostinil diolamine (Orenitram®) was the first oral prostacyclin approved for PAH. It can be appropriate for low-to-intermediate risk patients and in carefully selected and stable patients who seek to transition from infusion therapy.4,30 Although, like other prostacyclins, it is also associated with substantial interpatient variability in dose requirements and therapeutic response.30–33 Clinical outcomes are most optimal in those patients who can attain higher doses and remain on therapy long term; however, many develop intractable adverse effects (eg, gastrointestinal) or require a switch to other formulations.33–36 This study evaluated 15 patients who were newly started on oral treprostinil and those who switched from other treprostinil formulations (intravenous, subcutaneous, or inhaled). Variants in CYP2C8, CYP2C9, and ABCC4 were associated with treatment persistence (defined as maintenance on medication without discontinuation). For CYP2C8, variants tested included *3 (rs10509681, rs11572080) and *4 (rs1058930) alleles that have an expected frequency of 10–20% in the Caucasian population. CYP2C9 variants tested were the *2 (rs1799853, decreased function) and *3 (rs1057910, no function) alleles. Tested variants in ABCC4 included rs1751034 and rs3742106 and have expected minor allele frequencies of 20% and 40%, respectively.37 CYP2C9 predicted phenotypes were based on diplotype to activity score mappings by the Clinical Pharmacogenetics Implementation Consortium (CPIC), in which *1 = 1, *2 = 0.5, and *3 = 0.28 CYP2C8 predicted phenotypes were determined to be intermediate or poor metabolizer status when one or two decreased or loss-of-function alleles were detected, respectively.
The study population included mostly Caucasian females and the majority were on a PDE5i and/or an ERA. Nine of 15 patients were on both a PDE5i and an ERA. The study group included both de novo patients and patients who were switched from other treprostinil formulations. Oral treprostinil dosing was highly variable with a more than four-fold difference in total daily dose requirements. Forty percent of patients stopped treatment, primarily owing to intractable adverse events. About 27% of patients had a variant in CYP2C8 (*1/*3 or *1/*4), while 47% had a variant in CYP2C9 (*1/*2, *1/*3, or *2/*2). Minor allele frequencies for ABCC4 (rs1751034 and rs3742106) were 0.17 and 0.43, respectively.22 Increased CYP2C9 activity score was associated with decreased risk for treatment discontinuation (HR: 0.13; 95% CI: 0.02, 0.91; p = 0.04). In summary, patients with an estimated CYP2C9 activity score of 2 were only 13% as likely to discontinue therapy compared to those with an activity score of 1. Eighty-three percent of patients with one or more decreased or no function alleles in CYP2C9 stopped treatment due to adverse effects. In contrast, no association was seen between predicted CYP2C8 activity and outcome. However, it should be noted that CYP2C8 and CYP2C9 are in linkage disequilibrium and both enzymes metabolize treprostinil which makes it challenging to determine which gene was responsible for the effect.38 Although tested genetic variants were not associated with dose or frequency, the single lowest total daily dose was observed in the patient with CYP2C9 *2/*2 (predicted intermediate metabolizer and in the lowest CYP2C9 activity score (1) group).28 A similar trend was found between alleles that predicted decreased CYP2C8 metabolizer status (*3, *4) and dose.
In summary, this study concluded that PGx may be an important contributor to treatment persistence with oral treprostinil.22 Variants in the genes that account for treprostinil metabolism were highly prevalent. A significant association was seen between carriers of CYP2C9 decreased function alleles and lower treatment persistence. Additionally, increased CYP2C9 activity score was associated with lower risk for discontinuing treatment. Variants in the tested genes were not associated with dosing; however, this observation may have been subject to small sample size. Future studies should evaluate the impact of PGx on medication response and dosing with other PAH medications.
Other Genes Associated with Treprostinil Dose Requirements
A genome-wide association study (GWAS) was conducted by Thomeas-McEwing et al to explore predictors of a stable treprostinil dose.23 Ninety-eight adult patients of European ancestry were treated with intravenous treprostinil and confirmed to be on their ultimate stable dose, defined as the dose at which the provider determined further titration would not result in clinical improvement, and the dose remained unchanged for at least 3 months. Broad genotyping was performed on the Illumina Omni Express array (Illumina, Inc., San Diego, CA, USA). Approximately 80% of the cohort was female, and about 50% had idiopathic PAH. The median dose (IQR) was approximately 70 (38) ng/kg/min. GWAS included 715,000 single nucleotide polymorphisms (SNPs). No SNP was associated with treprostinil dose at the conventional significance threshold (p < 5 × 10−8). However, five SNPs showed suggestive associations. Two SNPs of potential significance were chosen for further evaluation.
The first SNP (rs10023113) was in the myocardial contractility-regulating gene calcium/calmodulin-dependent serine/threonine kinase-δ (CAMKIIδ) on chromosome 1. For each copy of the variant allele, the patient was found to require a dose of 26.3 ng/kg/min higher than the median dose for the cohort. CAMK2D is the gene encoding the CAMKIIδ. When upregulated, this kinase reduces cardiac myofibril contractility. The functional consequence of this variant (frequency of 14%) was hypothesized to be increased CAMKIIδ expression. This variant was explored in a separate registry by the study authors and found to be associated with greater right ventricular dysfunction as seen by higher right ventricular end-diastolic pressures and mean right atrial pressures. The authors hypothesized that this gene variant may be a marker for greater risk for right ventricular dysfunction, and that these patients might be more responsive to the pharmacologic effects of prostacyclins.
Another SNP of interest (rs11078738) was in the phosphoribosylformylglycinamidine synthase (PFAS) locus on chromosome 17. This non-synonymous variant was associated with an increased dose of 20.7 ng/kg/min above the median for each allele. PFAS encodes for an enzyme necessary for purine biosynthesis, and prostacyclins regulate vasodilation by augmenting the production of the purine cyclic adenosine monophosphate (cAMP). The functional consequence of this variant (frequency of 24%) was hypothesized to be lower cAMP production. In response to stimuli from exogenous prostacyclins, those who carry this variant produce lower concentrations of cAMP. Therefore, the authors proposed that individuals who carry this gene variant may require higher doses of prostacyclins (or more rapid titration to higher doses) to achieve the target therapeutic effect.
In summary, this study identified genetic variants associated with decreased responsiveness to prostacyclins and more severe right ventricular dysfunction. The polymorphisms associated with treprostinil dose phenotype have a direct relation to the pharmacodynamic effects of prostacyclins. Therefore, the authors surmise that this observation could be extended to other prostacyclins (epoprostenol, iloprost, beraprost) or prostacyclin receptor agonists (eg, selexipag) based on their individualized patient titration schema (with distributions of low-, medium-, and high-doses).23
Endothelin Receptor Antagonists
ERAs are a commonly used medication class to treat PAH patients who are at low-to-intermediate risk of 1-year mortality.4 ERAs also provide a compelling use case for PGx. Endothelin-1 (ET-1) expression is increased in PAH and serves as a major contributor to the pathobiologic effects of vasoconstriction and smooth muscle cell proliferation that leads to disease progression.39 ERAs improve exercise capacity, functional class, time to clinical worsening, and in some cases, a composite of PAH morbidity and mortality.3,40 However, interindividual variability in response to ERAs suggests possible genetic underpinnings.41–43 For example, the effect of ERAs is diminished in men compared to women and in Black versus White individuals.44 Multiple genetic polymorphisms in genes regulating the ET-1 pathway, corresponding receptors (ET-1 A and B), and associated G-proteins involved in subcellular signaling processes have been suggested.24
Benza et al conducted a large study (n = 715) to explore the interaction of ET-1 pathway polymorphisms and outcomes in European descent PAH patients who were treated with ERAs.24 The genes studied were ET-1 (EDN1), the A and B receptors (EDNRA and EDNRB), and guanine nucleotide binding proteins (alpha, beta, and gamma subunits). These candidate genes in the ET-1 pathway were identified from an existing GWAS (global sites participating in the STRIDE trial of sitaxsentan).45 Approximately 51% of patients received sitaxsentan (no longer available due to liver toxicity), 43% received bosentan, and 6% received ambrisentan.
A variant in the gene GNG2 (rs11157866) associated with the G-protein complex had a significantly higher rate of reaching the first clinical improvement outcome measure (composite of improvement in 6-minute walk distance and functional class) at 12, 18, and marginally at 24 months. The minor allele frequency for this variant was 12.7%. Those who were homozygous for the minor allele T/T attained the improvement faster than individuals with other genotypes. The function of GNG2 is related to proliferation, transcription, cell migration, inflammation, cellular apoptosis, and medication response. However, the specific effects in the context of ERA therapy remain uncertain. The authors concluded that ET-pathway-associated polymorphisms may help to predict the effect of ERA treatment for PAH.24
Bosentan is an oral non-selective ERA that was the first in-class ERA approved for PAH.46 However, its use is limited by a risk for liver toxicity manifested by increased aminotransferases in 10–12% of patients.46,47 Potential mechanisms of bosentan-induced hepatotoxicity are postulated to be due to a reduction in metabolism through cytochrome P450 isoenzymes (CYP) enzymes, a decrease in hepatic uptake by organic anion-transporting polypeptide (OATP) 1B1/1B3 (encoded by the SLCO1B1/1B3 genes), and intra-hepatic bile acid accumulation (a result of bosentan inhibition of hepatic efflux transporters, such as the bile salt export pump [BSEP; encoded by ABCB11]) and multidrug resistance-associated protein 2 [MRP2; encoded by ABCC2]).48–53 Therefore, Markova et al explored associations between bosentan-induced liver injury and reduced-function single-nucleotide polymorphisms (SNPs) in genes that encode for proteins involved in the drug’s disposition (CYP2C9, CYP3A4, SLCO1B1 and SLCO1B3, ABCB11, and ABCC2).25 A total of 92 ethnically diverse patients with PAH were studied; however, only the Caucasian group (n = 56) was of sufficient size for PGx analysis. In a multivariate model, CYP2C9*2 was significantly associated with increases in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) and medication-induced liver injury. This allele encodes for a variant with approximately 50% decreased function and is a common polymorphism associated with a poor metabolizer phenotype.27 The minor allele frequency for this variant is 10–15% in Caucasian and Hispanic populations, in contrast to 4% in African-Americans and 0% in Asians.54 The CYP2C9*3 variant (characterized by almost complete loss of function) was not found to have an association with liver injury in this study; however, this may have been due to sample size limitations as this variant is less common than the *2 allele.25,54 The authors concluded that CYP2C9*2 is a potential genetic marker for prediction of bosentan-induced liver injury.25
A study by Seyfarth et al evaluated variants in CYP2C9, ABCB11, and NR1HR (major transcription factor for ABCB11) as possible risk factors for bosentan-associated cholestasis.26 The study cohort included patients with (n = 23) and without (n = 25) aminotransferase elevations during bosentan therapy. The threshold for elevations in AST or ALT was >1.5 times the upper limit of normal. The investigators found no association between ABCB11 and NR1HR variants and bosentan-induced liver injury. However, they did report an allele frequency for CYP2C9*2 and CYP2C9*3 of 52% among those with increased transaminases compared to 24% for those without. Consequently, the odds ratio for bosentan-induced liver injury was 3.5 for carriers of these variants versus wild-type. In their initial analysis, they combined the *2 and *3 alleles. Upon evaluating each variant separately, they noted that only the *2 allele was associated with increased transaminases. The authors concluded that cholestatic liver injury from bosentan is a result of drug accumulation in those with the CYP2C9*2 polymorphism. Bosentan continues to be an option in the ERA class even though newer and safer alternatives have been introduced, including ambrisentan and macitentan.
Discussion
Current evidence shows that PAH mortality remains unacceptably high despite significant advances in pharmacotherapy over the past 20 years.9,55,56 Conventional PAH treatment approaches rely on multiparametric clinical risk stratification, with infusion therapies recommended for patients with advanced symptoms.3,4 As a whole, the impact of PAH pharmacotherapy on the historical PAH disease surrogate of 6-minute walk distance improvement has been relatively modest.18,57 Despite the genetic underpinnings of PAH, no current therapies target these genes and a patient’s individual molecular etiology or PGx does not determine treatment.5 Therefore, the current empiric approach to medication selection and titration remains the standard of practice.19 In these instances, prescriber and patient preference as well as convenience have dictated medication and formulation selection. Consequently, for patients with a poor response owing to adverse effects or no benefit, the time lost due to insufficient treatment leads to additional disease progression.1,10
PAH is sufficiently diverse to merit the application of PGx.58 Recent advances in the understanding of the PGx of prostacyclins and ERAs provide proof-of-concept towards predicting individual response. Substantial variability in dosing and treatment response with PAH medication classes is clinically evident, and implies an underlying role for genetic differences on the basis of medication response, PAH disease severity, or both. The identification of pharmacogenes of interest in PAH could help to enrich future studies by including those with an enhanced response (ie, “super responders”).5 The advancement of PGx approaches to PAH care was highlighted by the National Heart, Lung, and Blood Institute and will therefore be a high priority for future research.18,20
Conclusions and Future Directions
The field of PGx will continue to advance with increased applications in rare diseases such as PAH. Many of the summarized studies reported to date have yielded modest signals of association between genetic variants and clinical endpoints of interest.23 However, these findings have generally not met conventional GWAS p value thresholds for statistical significance. Instead, findings that support molecular and clinical functional associations with genetic variants may be more feasible and of equal importance. This may be especially true for rare diseases such as PAH in which enrollment of significant numbers of patients is not possible.59 Furthermore, true associations with complex phenotypes among variants with small to moderate effect sizes may be missed through GWAS.60,61 Replication, consistency of PGx association, and functional relevance are paramount to explain GWAS results.62–64 Additionally, focusing on more precise phenotyping to concentrate on a more limited contribution of genetic variants rather than the overall heritability of PAH as a complex disease itself will be important.65,66 Future investigations of PGx associations with treatment response in a wider population of real-world patients with PAH should be conducted. Prostacyclins are of high interest given their widespread use, multiple formulations, and significant cost.
Abbreviations
PAH, pulmonary arterial hypertension; PGx, pharmacogenomics.
Disclosure
James C. Coons has received investigator-initiated research funding from United Therapeutics and the University of Pittsburgh Institute for Precision Medicine. He also reports research support and consulting from Pfizer-Bristol Myers Squibb Alliance, consulting from Merck, research support from Heart Rhythm Society, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351(14):1425–1436. doi:10.1056/NEJMra040291
2. Fessel JP, Loyd JE, Austin ED. The genetics of pulmonary arterial hypertension in the post-BMPR2 era. Pulm Circ. 2011;1(3):305–319. doi:10.4103/2045-8932.87293
3. Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: association for European paediatric and congenital Cardiology (AEPC), international society for heart and lung transplantation (ISHLT). Eur Heart J. 2016;37(1):67–119. doi:10.1093/eurheartj/ehv317
4. Galie N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53(1):1801889. doi:10.1183/13993003.01889-2018
5. Halliday SJ, Hemnes AR. Identifying “super responders” in pulmonary arterial hypertension. Pulm Circ. 2017;7(2):300–311. doi:10.1177/2045893217697708
6. Galie N, Barbera JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373(9):834–844. doi:10.1056/NEJMoa1413687
7. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334(5):296–301. doi:10.1056/NEJM199602013340504
8. D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–349. doi:10.7326/0003-4819-115-5-343
9. Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL). Circulation. 2010;122(2):164–172. doi:10.1161/CIRCULATIONAHA.109.898122
10. Gomberg-Maitland M, Glassner-Kolmin C, Watson S, et al. Survival in pulmonary arterial hypertension patients awaiting lung transplantation. J Heart Lung Transplant. 2013;32(12):1179–1186. doi:10.1016/j.healun.2013.08.016
11. Rich S, Brundage BH. High-dose calcium channel-blocking therapy for primary pulmonary hypertension: evidence for long-term reduction in pulmonary arterial pressure and regression of right ventricular hypertrophy. Circulation. 1987;76(1):135–141. doi:10.1161/01.CIR.76.1.135
12. Sitbon O, Humbert M, Jais X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111(23):3105–3111. doi:10.1161/CIRCULATIONAHA.104.488486
13. Kuhn KP, Byrne DW, Arbogast PG, Doyle TP, Loyd JE, Robbins IM. Outcome in 91 consecutive patients with pulmonary arterial hypertension receiving epoprostenol. Am J Respir Crit Care Med. 2003;167(4):580–586. doi:10.1164/rccm.200204-333OC
14. Johnson RF, Loyd JE, Mullican AL, Fink CA, Robbins IM. Long-term follow-up after conversion from intravenous epoprostenol to oral therapy with bosentan or sildenafil in 13 patients with pulmonary arterial hypertension. J Heart Lung Transplant. 2007;26(4):363–369. doi:10.1016/j.healun.2007.01.022
15. McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106(12):1477–1482. doi:10.1161/01.CIR.0000029100.82385.58
16. Evans WE, McLeod HL. Pharmacogenomics--drug disposition, drug targets, and side effects. N Engl J Med. 2003;348(6):538–549. doi:10.1056/NEJMra020526
17. Soubrier F, Chung WK, Machado R, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D13–21. doi:10.1016/j.jacc.2013.10.035
18. Newman JH, Rich S, Abman SH, et al. Enhancing insights into pulmonary vascular disease through a precision medicine approach. A Joint NHLBI-Cardiovascular medical research and education fund workshop report. Am J Respir Crit Care Med. 2017;195(12):1661–1670. doi:10.1164/rccm.201701-0150WS
19. Austin ED, West J, Loyd JE, Hemnes AR. Translational advances in the field of pulmonary hypertension molecular medicine of pulmonary arterial hypertension. From population genetics to precision medicine and gene editing. Am J Respir Crit Care Med. 2017;195(1):23–31. doi:10.1164/rccm.201605-0905PP
20. Erzurum S, Rounds SI, Stevens T, et al. Strategic plan for lung vascular research: an NHLBI-ORDR workshop report. Am J Respir Crit Care Med. 2010;182(12):1554–1562. doi:10.1164/rccm.201006-0869WS
21. Pereira NL, Weinshilboum RM. Cardiovascular pharmacogenomics and individualized drug therapy. Nat Rev Cardiol. 2009;6(10):632–638. doi:10.1038/nrcardio.2009.154
22. Coons JC, Crisamore K, Adams S, et al. A pilot study of oral treprostinil pharmacogenomics and treatment persistence in patients with pulmonary arterial hypertension. Ther Adv Respir Dis. 2021;15:17534666211013688. doi:10.1177/17534666211013688
23. Thomeas-McEwing V, Psotka MA, Gamazon ER, et al. Two polymorphic gene loci associated with treprostinil dose in pulmonary arterial hypertension. Pharmacogenet Genomics. 2022;32(4):144–151. doi:10.1097/FPC.0000000000000463
24. Benza RL, Gomberg-Maitland M, Demarco T, et al. Endothelin-1 pathway polymorphisms and outcomes in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192(11):1345–1354. doi:10.1164/rccm.201501-0196OC
25. Markova SM, De Marco T, Bendjilali N, et al. Association of CYP2C9*2 with bosentan-induced liver injury. Clin Pharmacol Ther. 2013;94(6):678–686. doi:10.1038/clpt.2013.143
26. Seyfarth HJ, Favreau N, Tennert C, et al. Genetic susceptibility to hepatoxicity due to bosentan treatment in pulmonary hypertension. Ann Hepatol. 2014;13(6):803–809. doi:10.1016/S1665-2681(19)30983-4
27. Lee CR, Goldstein JA, Pieper JA. Cytochrome P450 2C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenetics. 2002;12(3):251–263. doi:10.1097/00008571-200204000-00010
28. Karnes JH, Rettie AE, Somogyi AA, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2C9 and HLA-B Genotypes and Phenytoin Dosing: 2020 Update. Clin Pharmacol Ther. 2021;109(2):302–309. doi:10.1002/cpt.2008
29. Belleville-Rolland T, Sassi Y, Decouture B, et al. MRP4 (ABCC4) as a potential pharmacologic target for cardiovascular disease. Pharmacol Res. 2016;107:381–389. doi:10.1016/j.phrs.2016.04.002
30. Chakinala MM, Feldman JP, Rischard F, et al. Transition from parenteral to oral treprostinil in pulmonary arterial hypertension. J Heart Lung Transplant. 2017;36(2):193–201. doi:10.1016/j.healun.2016.06.019
31. Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest. 2012;142(6):1383–1390. doi:10.1378/chest.11-2212
32. Tapson VF, Jing ZC, Xu KF, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial. Chest. 2013;144(3):952–958. doi:10.1378/chest.12-2875
33. White RJ, Jerjes-Sanchez C, Bohns Meyer GM, et al. Combination therapy with oral treprostinil for pulmonary arterial hypertension. A double-blind placebo-controlled clinical trial. Am J Respir Crit Care Med. 2020;201(6):707–717. doi:10.1164/rccm.201908-1640OC
34. Chin KM, Ruggiero R, Bartolome S, et al. Long-term therapy with oral treprostinil in pulmonary arterial hypertension failed to lead to improvement in important physiologic measures: results from a single center. Pulm Circ. 2015;5(3):513–520. doi:10.1086/682224
35. Coons JC, Miller T. Extended-release oral treprostinil in the management of pulmonary arterial hypertension: clinical evidence and experience. Ther Adv Respir Dis. 2018;12:1753466618766490. doi:10.1177/1753466618766490
36. Ramani G, Cassady S, Shen E, et al. Novel dose-response analyses of treprostinil in pulmonary arterial hypertension and its effects on six-minute walk distance and hospitalizations. Pulm Circ. 2020;10(3):2045894020923956. doi:10.1177/2045894020923956
37. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi:10.1038/s41586-020-2308-7
38. Yasar U, Lundgren S, Eliasson E, et al. Linkage between the CYP2C8 and CYP2C9 genetic polymorphisms. Biochem Biophys Res Commun. 2002;299(1):25–28. doi:10.1016/S0006-291X(02)02592-5
39. Giaid A, Yanagisawa M, Langleben D, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328(24):1732–1739. doi:10.1056/NEJM199306173282402
40. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809–818. doi:10.1056/NEJMoa1213917
41. Sitbon O, Badesch DB, Channick RN, et al. Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary arterial hypertension: a 1-year follow-up study. Chest. 2003;124(1):247–254. doi:10.1378/chest.124.1.247
42. Polderman KH, Stehouwer CD, van Kamp GJ, Dekker GA, Verheugt FW, Gooren LJ. Influence of sex hormones on plasma endothelin levels. Ann Intern Med. 1993;118(6):429–432. doi:10.7326/0003-4819-118-6-199303150-00006
43. Campia U, Cardillo C, Panza JA. Ethnic differences in the vasoconstrictor activity of endogenous endothelin-1 in hypertensive patients. Circulation. 2004;109(25):3191–3195. doi:10.1161/01.CIR.0000130590.24107.D3
44. Gabler NB, French B, Strom BL, et al. Race and sex differences in response to endothelin receptor antagonists for pulmonary arterial hypertension. Chest. 2012;141(1):20–26. doi:10.1378/chest.11-0404
45. Sandoval J, Torbicki A, Souza R, et al. Safety and efficacy of sitaxsentan 50 and 100 mg in patients with pulmonary arterial hypertension. Pulm Pharmacol Ther. 2012;25(1):33–39. doi:10.1016/j.pupt.2011.10.002
46. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896–903. doi:10.1056/NEJMoa012212
47. Denton CP, Pope JE, Peter HH, et al. Long-term effects of bosentan on quality of life, survival, safety and tolerability in pulmonary arterial hypertension related to connective tissue diseases. Ann Rheum Dis. 2008;67(9):1222–1228. doi:10.1136/ard.2007.079921
48. Treiber A, Schneiter R, Hausler S, Stieger B. Bosentan is a substrate of human OATP1B1 and OATP1B3: inhibition of hepatic uptake as the common mechanism of its interactions with cyclosporin A, rifampicin, and sildenafil. Drug Metab Dispos. 2007;35(8):1400–1407. doi:10.1124/dmd.106.013615
49. Burgess G, Hoogkamer H, Collings L, Dingemanse J. Mutual pharmacokinetic interactions between steady-state bosentan and sildenafil. Eur J Clin Pharmacol. 2008;64(1):43–50. doi:10.1007/s00228-007-0408-z
50. van Giersbergen PL, Halabi A, Dingemanse J. Single- and multiple-dose pharmacokinetics of bosentan and its interaction with ketoconazole. Br J Clin Pharmacol. 2002;53(6):589–595. doi:10.1046/j.1365-2125.2002.01608.x
51. Fouassier L, Kinnman N, Lefevre G, et al. Contribution of mrp2 in alterations of canalicular bile formation by the endothelin antagonist bosentan. J Hepatol. 2002;37(2):184–191. doi:10.1016/S0168-8278(02)00107-1
52. Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther. 2001;69(4):223–231. doi:10.1067/mcp.2001.114667
53. Mano Y, Usui T, Kamimura H. Effects of bosentan, an endothelin receptor antagonist, on bile salt export pump and multidrug resistance-associated protein 2. Biopharm Drug Dispos. 2007;28(1):13–18. doi:10.1002/bdd.527
54. Kirchheiner J, Brockmoller J. Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin Pharmacol Ther. 2005;77(1):1–16. doi:10.1016/j.clpt.2004.08.009
55. Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122(2):156–163. doi:10.1161/CIRCULATIONAHA.109.911818
56. Benza RL, Gomberg-Maitland M, Elliott CG, et al. Predicting survival in patients with pulmonary arterial hypertension: the REVEAL risk score calculator 2.0 and Comparison With ESC/ERS-based risk assessment strategies. Chest. 2019;156(2):323–337.
57. Peacock AJ, Naeije R, Galie N, Rubin L. End-points and clinical trial design in pulmonary arterial hypertension: have we made progress? Eur Respir J. 2009;34(1):231–242. doi:10.1183/09031936.00107108
58. Said SI, Hamidi SA. Pharmacogenomics in pulmonary arterial hypertension: toward a mechanistic, target-based approach to therapy. Pulm Circ. 2011;1(3):383–388. doi:10.4103/2045-8932.87306
59. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi:10.1038/nature08494
60. Zhong WP, Wu H, Chen JY, et al. Genomewide association study identifies novel genetic loci that modify antiplatelet effects and pharmacokinetics of clopidogrel. Clin Pharmacol Ther. 2017;101(6):791–802. doi:10.1002/cpt.589
61. Gamazon ER, Segre AV, van de Bunt M, et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet. 2018;50(7):956–967. doi:10.1038/s41588-018-0154-4
62. Maitland ML, Ratain MJ, Cox NJ. Interpreting P values in pharmacogenetic studies: a call for process and perspective. J Clin Oncol. 2007;25(29):4513–4515. doi:10.1200/JCO.2007.12.7803
63. Wheeler HE, Maitland ML, Dolan ME, Cox NJ, Ratain MJ. Cancer pharmacogenomics: strategies and challenges. Nat Rev Genet. 2013;14(1):23–34. doi:10.1038/nrg3352
64. Jones TS, Yang W, Evans WE, Relling MV. Using HapMap tools in pharmacogenomic discovery: the thiopurine methyltransferase polymorphism. Clin Pharmacol Ther. 2007;81(5):729–734. doi:10.1038/sj.clpt.6100135
65. Maranville JC, Cox NJ. Pharmacogenomic variants have larger effect sizes than genetic variants associated with other dichotomous complex traits. Pharmacogenomics J. 2016;16(4):388–392. doi:10.1038/tpj.2015.47
66. Motsinger-Reif AA, Jorgenson E, Relling MV, et al. Genome-wide association studies in pharmacogenomics: successes and lessons. Pharmacogenet Genomics. 2013;23(8):383–394. doi:10.1097/FPC.0b013e32833d7b45
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.