")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 13
Pharmacogenomic Response of Inhaled Corticosteroids for the Treatment of Asthma: Considerations for Therapy
Authors Cazzola M , Rogliani P , Calzetta L , Matera MG
Received 4 June 2020
Accepted for publication 27 July 2020
Published 4 August 2020 Volume 2020:13 Pages 261—271
DOI https://doi.org/10.2147/PGPM.S231471
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Mario Cazzola,1 Paola Rogliani,1 Luigino Calzetta,2 Maria Gabriella Matera3
1Unit of Respiratory Medicine, Department of Experimental Medicine, University of Rome “Tor Vergata”, Rome, Italy; 2Unit of Respiratory Disease and Lung Function, Department of Medicine and Surgery, University of Parma, Parma, Italy; 3Unit of Pharmacology, Department of Experimental Medicine, University of Campania “Luigi Vanvitelli”, Naples, Italy
Correspondence: Mario Cazzola Email [email protected]
Abstract: There is a large interindividual variability in response to ICSs in asthma. About 70% of the variance in ICS response is likely due at least partially to genetically determined characteristics of target genes. In this article, we examine the effects on the ICS response of gene variations in the corticosteroid pathway, and in the pharmacokinetics of corticosteroids, and also those outside the corticosteroid pathway, which have the potential to influence corticosteroid activity. Although the available evidence indicates that responses to ICSs in asthma are influenced by different genetic variants, there are still deep uncertainties as to whether a real association between these genetic variants and corticosteroid response could also possibly exist because there are difficulties in reproducing pharmacogenetic findings. This explains at least partly the insufficient use of pharmacogenomic data when treating asthmatic patients, which creates a real limitation to the proper use of ICSs in an era of precision medicine that links the right patient to the right treatment. Knowing and dealing with the genetic factors that influence the therapeutic ICS response is a fundamental condition for prescribing the right dose of ICS to the right patient at the right time.
Keywords: asthma, inhaled corticosteroids, gene polymorphisms, genome-wide association studies, precision medicine
Introduction
Inhaled corticosteroids (ICSs) are the mainstay of treatment for asthma, but are also characterized by highly variable interindividual responses among asthmatic patients, with some subjects in whom the expected therapeutic effect is not achieved, while others exhibit even severe adverse reaction and the majority show intermediate responses between the above two extreme cases.1 This between-person variability is the result of disparate causes such as disease severity, treatment compliance, concurrent illnesses, environmental and infection exposures, age, medicinal product interactions, and also the definition of response (improved lung function vs persisting symptoms vs exacerbation).1,2 However, a key role in determining interindividual variability in response to ICS seems to be played mainly by the characteristics of target genes or drug metabolizing enzymes that are under genetic influence.1,3
ICSs work by binding to the intracellular glucocorticoid receptor (GR), which is encoded by the nine-exon gene NR3C1,4 followed by translocation to the nucleus and binding to its cognate DNA sequences called glucocorticoid response elements (GREs), mediating the transcriptional activities of a wide variety of genes. Their anti-inflammatory activity is achieved through a combination of inhibition, the so-called “transrepression”, and upregulation, or “transactivation”, of gene transcription.5 The transrepression results in a reduced production of pro-inflammatory cytokines, and is due to an inhibition of genes regulating expression of many pro-inflammatory cytokines, such as interleukin (IL)-1, IL-4, IL-5, IL-6, IL-8, IL-10, IL-13, tumor necrosis factor-α (TNF-α), granulocyte–macrophage colony-stimulating factor (GM-CSF), interferon-γ (IFN-γ),4 and also that of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2).6 Alternatively, GR interferes with the activation and/or activity of other DNA-related transcription factors, mainly nuclear factor (NF)-κB, and activating protein (AP)-1, but also cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB), interferon regulatory factor 3 (IRF3), nuclear factor of activated T-cells (NFAT), signal transducer and activator of transcription (STAT), T-box expressed in T-cells (T-Bet), and GATA-3, rather than directly binding to DNA.7 In any case, it influences gene regulation.7
The transactivation results in an augmented synthesis of anti-inflammatory proteins with an upregulated expression of annexin A1, previously named lipocortin-1, which produces a direct inhibition of phospholipase A2 (PLA2) that causes a reduced prostaglandin and leukotriene production,8 an inhibition of COX-2 that occurs in a post-transcriptional manner,9 a detachment of neutrophil from the endothelium, and a reduction in neutrophil penetration through the endothelium of blood vessels.8 Furthermore, some genes that encode anti-inflammatory proteins such as inhibitor of NF-κB (IκB-α), secretory leukoprotease inhibitor, glucocorticoid-induced leucine zipper protein (GILZ), and mitogen activated kinase phosphatase-1 are induced through transactivation.10 Apparently, transactivation plays a critical role in the onset of unwanted adverse events induced by ICSs.11
About 70% of the variability in ICS response is likely due to individual characteristics of target genes.12 However, there is still notable uncertainty whether a real association between genetic variants and corticosteroid response could exist. In fact, the effects of gene variations in the corticosteroid pathway on the response to ICS therapy are often under discussion due to the frequent lack of reduplicate results.13 This is a big issue. In fact, understanding the impact of these gene variations on the response to ICSs could likely lead to identification of genetic markers that predict if an ICS treatment will work. These biomarkers would allow a personalized treatment of patients with asthma and, accordingly, a better quality of care.14
In this article, we will examine the effects on ICS response of corticosteroid pathway gene variations, those of genetic variation in the pharmacokinetics of corticosteroids, and also those of gene variations outside of the corticosteroid pathway that have the potential to influence the corticosteroid activity (Table 1). The information given, when not otherwise noted, refers to studies conducted in humans.
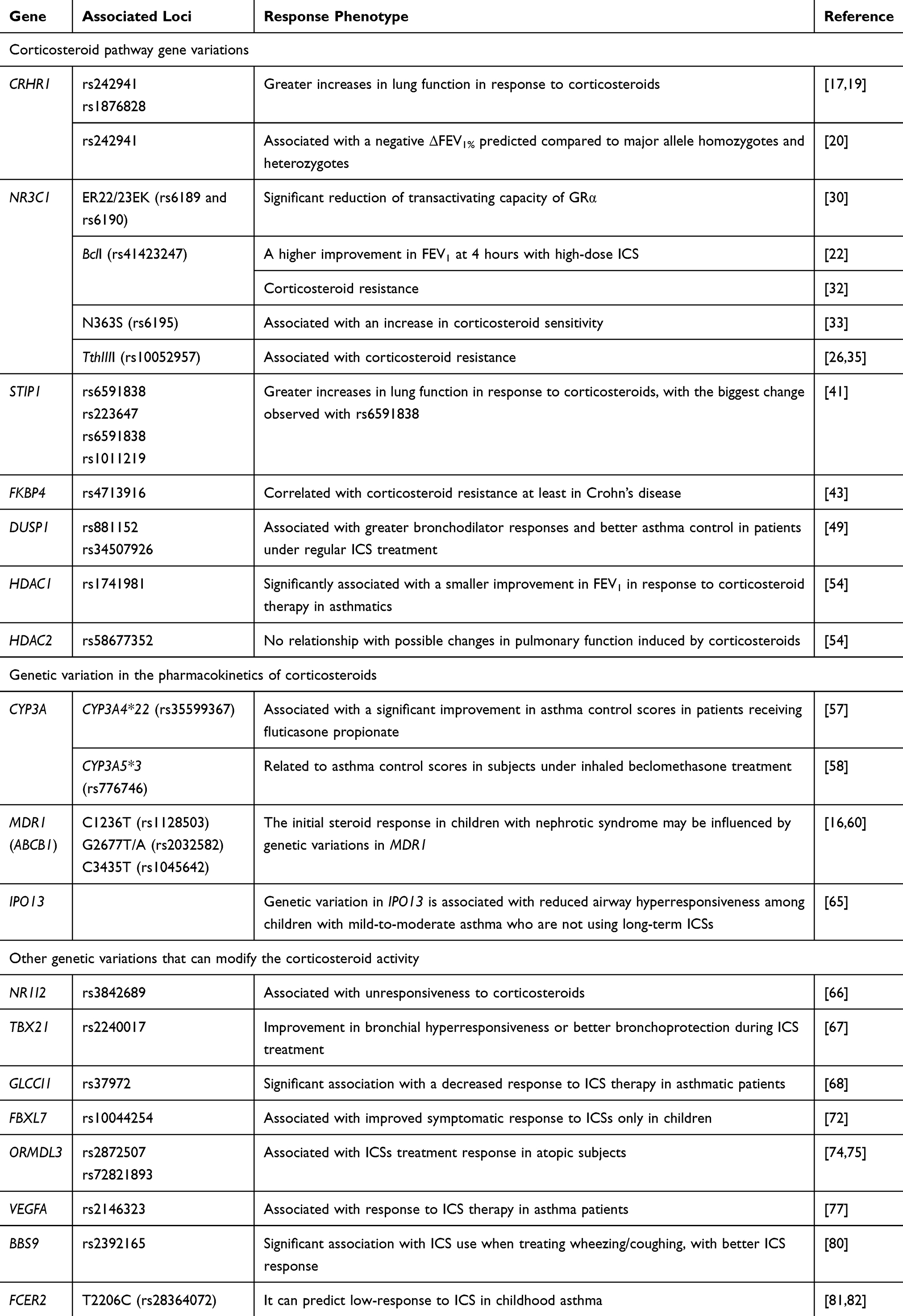 |
Table 1 Polymorphisms That Influence the Response of ICSs |
Corticosteroid Pathway Gene Variation
CRHR1 Gene Polymorphisms
Corticotrophin-releasing hormone (CRH) binds to CRH receptor 1 (CRHR1) in the pituitary gland and modulates the release of adrenocorticotropic hormone, which induces cortisol production and regulates endogenous corticosteroid levels. CRHR1, which maps on chromosome 17q12-q22, encodes for CRHR1.15 Variations in this gene could alter the response to ICS by influencing the level of endogenous corticosteroid secretion, with lower levels eliciting a better response to exogenous corticosteroids.16 However, available data are inconclusive so far.
Several intronic single nucleotide polymorphisms (SNP) in this gene have been studied. The rs242941 and rs1876828 polymorphisms of the CRHR1 gene seem to be correlated with a good response to ICSs.17 Greater increases in lung function in response to corticosteroids have been correlated with the presence of the rs242941 polymorphism.18,19 However, it was also reported that rs242941, but not rs1876828, is associated with a negative ΔFEV1%pred compared to major allele homozygotes and heterozygotes.20 Another two studies were unable to document any FEV1 improvement after ICSs treatment in the presence of these two polymorphisms.21,22
NR3C1 GR Gene Polymorphisms
The nuclear receptor subfamily 3 group C member 1 (NR3C1), which is located on chromosome 5q31–q32 and consists of nine exons, is the only known GR gene. It encodes the GR producing mRNA, which constitutes the basis for GR isoforms (GRα, GRβ, GRγ, and GRδ). Only GRα is active, whereas GRβ inhibits the signal pathway of GRα and induces an impaired sensitivity to corticosteroids,23 while the functions GRγ and GRδ, which are post-transcriptional modifications of the NR3C1 gene, are still under investigation.24 The GRα and GRβ isoforms have been subdivided into eight GRα and eight GRβ isoforms that have different post-translational modifications.25 It is likely that differential expression in various cell types might explain distinct cellular responsiveness. Within the cell, GRα cannot move into the cell nucleus because it is part of a large multiprotein complex that includes chaperone proteins (heat shock protein [HSP] 90, HSP70 and p23) and immunophilins (binding protein FK506 [FKBP] 51 and FKBP52). However, when GRα binds to the corticosteroid, it dissociates from the multiprotein complex through hyperphosphorylation and migrates to the nucleus, where it binds to the GRE.
Polymorphisms of NR3C1 cause modifications of the secondary and tertiary domain structures in the GR, and disturb transcription initiation and stability of the mRNA for the GR.26 Although many polymorphisms of this gene have been identified, we still do not know if and how really all these GR variants influence the responses to ICSs.27 In general, mutations in NR3C1 are associated with corticosteroid resistance, although some GR polymorphisms are associated with function improvements.13 They may not only inhibit the formation of the GR/corticosteroid complexes, but also reduce transcription and cause transrepression of genes that encode protein synthesis within the framework of cell response to corticosteroids. Thus, they reduce GR expression which, compromised in its structure and function, elicits a weaker response to corticosteroids.28
ER22/23EK, N363S, and BclI are the common polymorphisms of NR3C1 that influence corticosteroid treatment.16 The BclI SNP, located in intron 2 of the NR3C1, and the N363S SNPs have been associated with enhanced corticosteroid sensitivity in the general population. On the contrary, the ER22/23EK SNP, located in exon 2 of the NR3C1, is associated with relative corticosteroid resistance.29
The increase in GRα-A isoform expression induced by the ER22/23EK (rs6189 and rs6190) polymorphism that affects the GCRα-A/GCRα-B ratio and appears in 2% of the general population, is a possible mechanism explaining corticosteroid resistance because it causes a significant reduction of transactivating capacity of GRα.30
On the other side, higher methylprednisolone potency at least in vitro is associated with rs41423247, the intronic BclI polymorphism in the NR3C1 gene.31 In effect, it was documented that children with moderate-to-severe asthma exacerbation, who were homozygous carriers of the G allele of rs41423247, showed a higher improvement in FEV1 at 4 hours when treated with high-dose ICS.22 However, it has also been reported that corticosteroid resistance can be associated with BclI polymorphism.32
The N363S polymorphism (rs6195), located within exon 2 and found in about 4% of the general population, is another genetic variation associated with an increase in corticosteroid sensitivity,33 although a study was unable to detect major differences between wild-type GR and the N363S polymorphism.34
Also the TthIIII polymorphism (rs10052957), which is located in the area of the NR3C1 gene promoter, is associated with corticosteroid resistance. However, it is not functional by itself, but is linked to the ER22/23EK and the observed resistance to corticosteroid likely depends on the ER22/23EK polymorphism.26,35
Several studies have investigated the possible association of GR SNP with corticosteroid response in asthmatic patients, in addition to the two studies we have just mentioned. A Polish study showed that, contrary to what could be expected, N363S SNP, but not the ER22/23EK polymorphism, might have an important part in the development of difficult-to-treat/treatment-resistant asthma.36 Another study documented the absence of any association between GR polymorphisms (TthIIII, ER22/23EK, N363S, BclI) and the dose of ICS needed to achieve asthma control in a group of pediatric patients with asthma.37 Another two studies, one association study and one mutational analysis, concluded that NR3C1 does not contribute significantly to corticosteroid resistance.38,39
Interestingly, the frequencies of the ER22/23EK, TthIIII, and BclI polymorphisms of the NR3C1 gene were not statistically different when a comparison was made between a control group and an asthmatic group, regardless of the severity of asthma.26
Another GR SNP, the A829G polymorphism (rs2278008) in exon 5, raises the transactivation potential of GR more than eight times, but the exact mechanism for this hyperactivity is unclear.40
The Chaperone Protein Gene Polymorphisms
The stress induced phosphoprotein 1 (STIP1) is a protein that modulates the chaperone activity of HSP70 and HSP90. A number of intronic SNPs in the STIP1 gene, which is located on chromosome 11q13.1 and is composed of 14 exons, can influence lung function responses to corticosteroids after 4 (rs6591838 and rs223647) and 8 (rs6591838 and rs1011219) weeks of therapy, with the biggest change observed with rs6591838.41 Mechanisms that contribute to variation in corticosteroid response are still unknown, but interactions with HSP chaperones that influence GR function are a possibility. However, steroid-sensitive patients present a significantly lower ratio of nuclear HSP90/GR than steroid-resistant subjects,42 and no positive data exist on the association of HSP90 gene polymorphism and corticosteroid treatment.16
The FKBP4 gene, which maps on chromosome 12, at 12p13.33 and encodes for the immunophilin FKBP52, is thought to be involved in the regulation of GR signaling. It was found that the rs4713916 SNP in the FKBP4 gene correlated with corticosteroid resistance at least in Crohn’s disease.43
Usually, the clinical use of ICS in asthma does not take into account all these genetic variants. However, the intrinsic variability in the pharmacokinetics of the prescribed ICS and the effect of the disease itself affect the therapeutic dosage much more than the small differences in corticosteroid sensitivity induced by SNPs.44
Other Genetic Alterations in Corticosteroid Pathway
Phosphorylation of GRα at Ser226 reduces GR nuclear translocation and induces corticosteroid resistance.45 The involvement of serine/threonine protein phosphatase 2A regulating c-Jun N-terminal kinase (JNK) 1 and GR-Ser226 signaling appears to be a possible mechanism explaining this resistance.
Protein kinase dual-specificity phosphatases (DUSPs) dephosphorylate JNK, and influence the corticosteroid sensitivity.46 DUSP1, which is located on chromosome 5q35.1 and consists of four exons separated by three relatively small introns, encodes a protein that inactivates p38 mitogen-activated protein kinase (MAPK), thereby reducing the expression and production of proinflammatory cytokines.47 In DUSP1 knock out mice, dexamethasone is less effective in reducing the release of TNF-α and IL-6 in response to endotoxin injection.48 The DUSP1 polymorphisms rs881152, a 5′ region SNP, and rs34507926, located in intron 1, were associated with greater bronchodilator responses and better asthma control in a cohort of asthma patients under a regular treatment with an ICS.49 The mechanism that explains these effects has not yet been clarified. It is possible that the ability of DUSP1 to address the p38 MAPK signaling pathway may be changed in carriers of the rs881152 SNP due to a corticosteroid-induced reduction of the DUSP1 expression.50
Histone deacetylase (HDAC) is critical in regulating inflammatory genes because it removes acetyl groups from histones.51 Increased airway inflammation and larger type 2 cytokine production were observed after deletion of HDAC1 in T-cells in an in vivo mouse model of allergic airway disease.52 Corticosteroids perform their anti-inflammatory effects by inducing acetylation of anti-inflammatory genes, but particularly by recruiting HDAC2 to activated inflammatory genes. Acetylated GRs are deacetylated by HDAC2 and subsequently they can suppress activated inflammatory genes.53 The rs1741981 polymorphism in the HDAC1 gene located on chromosome 1p35.2-p35.1 and containing 14 exons is significantly associated with a smaller improvement in FEV1 in response to corticosteroid therapy in asthmatics.54 However, the rs58677352 polymorphism in the HDAC2 gene, which maps to chromosome 6q21 and is composed of 14 exons, does not show any relationship with possible changes in pulmonary function induced by corticosteroids.54
Genetic Variation in the Pharmacokinetics of Corticosteroids
The safety and efficacy profile of any ICS is influenced by its pharmacokinetic properties, and genetic polymorphisms can modify not only its pharmacodynamic profile, but also the pharmacokinetic behavior producing variability in response to ICSs.55
Corticosteroid metabolism occurs primarily in the liver. Generally, ICSs are metabolized to inactive metabolites through enzymatic transformations. They are substrates for CYP3A4, which is also present in the intestine and lung.56
The presence of the rs35599367 (CYP3A4*22) allele in patients receiving fluticasone propionate induced a significant improvement in asthma control scores.57 In another study, the rs776746 (CYP3A5*3) SNP was related to asthma control scores in subjects under inhaled beclomethasone treatment.58 CYP3A4*22 and CYP3A5*3, which are members of the CYP3A family of genes located on chromosome 7, abolish the CYP3A4 and CYP3A5 expression and activity, respectively, and thereby extend the presence of the active ICS in the airways, thus prolonging its anti-inflammatory effects.
Also the association between 11β-hydroxysteroid dehydrogenase and glutathione S-transferases enzyme family gene variation and corticosteroid treatment has been reported but all the available information is focused on patients suffering from acute lymphoblastic leukemia.16
Although not strictly connected with the classical parameters of pharmacokinetics, there are two aspects of the movement and intracellular distribution of corticosteroids that have prompted pharmacogenetic considerations.
Gene mutations in corticosteroid transporters should be more important for asthmatic patients. P-glycoprotein 170 (P-gp) pumps corticosteroids out of cells and, therefore, it might also be involved in corticosteroid activity/resistance.59 When its expression is increased, the intracellular drug concentrations decrease and, consequently, also the response to ICSs may decrease The multidrug-resistance gene MDR1 (ABCB1) that maps on chromosomal region 7q21 encodes P-gp.16,59 Three common MDR1 SNPs, C1236T (rs1128503), G2677T/A (rs2032582), and C3435T (rs1045642) located on the coding region have an impact in corticosteroid treatment. The rs1128503 polymorphism is associated with an increased risk of steroid resistance at least in children with idiopathic nephrotic syndrome.60
Long-term exposure to ICSs may increase P-gp expression and thus reduce the effectiveness of other known P-gp substrate medications, including systemic corticosteroids, when they are administered using standard doses. This could be a issue when a systemic corticosteroid is needed to treat severe asthma.61 However, it is generally believed that the increased expression of P-gp is not an important mechanism of induction of corticosteroid resistance in asthma16 and, therefore, it would not compromise the ICS effectiveness.61
GR is shuttling between the cytoplasm and the nucleus and its subcellular localization depends on the rate of nuclear import and export. Nuclear import of GR is mediated through its nuclear localization signals (NLSs) and interaction with importins (IPOs) or karyopherins, with IPOα selectively binding to NLS1 and IPO7 and 8 binding to both NLS1 and NLS2.23 IPOs are known to direct the ligand-activated GR to gated channels of the nuclear membrane, the nuclear pore complex, and cause the translocation of the GR to the nucleus.62 Small interfering RNAs that inhibited IPO13 synthesis, which also shows export activity,63 inhibit nuclear translocation of GR.55 Silencing of IPO13, which is a corticosteroid-inducible gene located on chromosome 1p34.1, also abolishes the inhibitory effect of cortisol on TNF-α stimulation of the inflammatory cytokine IL-8 expression.64 These findings suggest that the anti-inflammatory effects of corticosteroids or, conversely, the induction of corticosteroid resistance, are influenced by variation in cellular levels of IPO13 and intracellular IPO13 shuttling rates. There is documentation that genetic variation of IPO13 is associated with reduction in airway responsiveness in children with mild-to-moderate asthma not treated with long-term ICSs.65 Since this improvement was similar to the improvements noted among patients on long-term ICSs, it was suggested that that IPO13 variation could increase endogenous corticosteroid bioavailability in the cell nucleus. It is obvious to wonder whether the presence of such a variation could lead us to avoid the use of ICSs or to recommend their use at lower doses. Regrettably, no other studies have been conducted on this topic, so the question is still unanswered.
Other Genetic Variations That Can Modify the Corticosteroid Activity
Many other SNPs that are even outside the corticosteroid pathway, but have the potential to influence the corticosteroid activity have been described.
Thus, the presence of a deletion polymorphism (rs3842689) of the pregnane X receptor gene (nuclear receptor subfamily 1, group I, member 2, NR1I2), which is located on chromosome 13q12-q13.3, has been associated with unresponsiveness to corticosteroids. In effect, a reduced expression of pregnane X receptor leads to an underexpression of GRs.66
Improvement in bronchial hyperresponsiveness or better bronchoprotection during ICS treatment have been associated with another SNP, rs2240017, which is encoded by the T-box expressed in T-cell transcription that regulates naïve T-lymphocyte development (TBX21) gene which maps on chromosome 17q21.32.67
However, in these last years genome-wide association studies (GWASs), which allow analyzing hundreds of thousands of SNPs with the aim of associating them to a better or worse drug response, have been performed.
A GWAS revealed that there is a significant association between the genetic polymorphism rs37972 in the promoter region of the glucocorticoids-induced transcript 1 (GLCCI1) gene, which is located on chromosome 7p21.3 and encodes a protein of unknown function, and a decreased response to ICS therapy in asthmatic patients.68 This SNP is in complete linkage disequilibrium in the same gene with the rs37973 SNP, which can influence GLCCI1 gene expression. However, another study did not confirm that GLCCI1 rs37973 is able to predict the response to ICSs.69 Little is known about the real function of GLCCI1 in humans. It was originally described as a thymocyte-specific transcript that is quickly upregulated in response to dexamethasone treatment.70 GLCCI1 deficiency in asthmatic mice does not allow the activation of GR and mitogen-activated protein kinase phosphatase 1. Furthermore, it causes an increased phosphorylation of p38 MAPK, leading to a decremented response to corticosteroids.71
The intronic rs10044254 polymorphism has been identified in two independent pediatric cohorts (average age 8.9 and 10.4 years, respectively), but failed to be replicated in an adult asthmatic population (33–34 years old on average).72 It is associated with both decreased expression of the F-Box and Leucine Rich Repeat Protein 7 (FBXL7) gene, which maps on chromosome 5p15.1, and improved symptomatic response to ICSs. This finding suggests that there might be a specific genetic mechanism able to regulate symptomatic response to ICSs only in children. However, recently FBXL7 has been identified as a male specific gene in epithelial cells of asthmatic subjects although, unfortunately, it was not specified whether they were adults or children.73
Two variants of the orosomucoid like-3 (ORM1-like protein 3; ORMDL3) gene, which is located in chromosome 17q21 and encodes for a protein that is an important mediator in diminishing the synthesis of sphingolipids, rs2872507, that is intergenic,74 and rs72821893, which was located in the same chromosomal area,75 are associated with ICSs treatment response in atopic subjects. Sphingolipids have a central role in synthesizing many inflammatory proteins.76
The polymorphism rs2146323 of the vascular endothelial growth factor A (VEGFA) gene, mapped to chromosome 6p12, was associated with response to ICS therapy in asthma patients.77 In an animal model, VEGF A increases mucus production, collagen deposition, as well as smooth muscle hyperplasia.78 However, the functional implications of rs2146323 are uncertain because this SNP is located in a non-coding exon area.79
Another GWAS found a significant association between the SNP rs2392165 near the Bardet-Biedl syndrome 9 (BBS9) gene mapped on chromosome 7p14.3 and ICS use when treating wheezing/coughing, with better ICS response.80 BBS9 is implicated in lung development and ciliogenesis.
The Fc fragment of IgE receptor II (FCER2) gene, which is an 11‐exon gene located at chromosome 19p13.2 and encodes for a low affinity IgE receptor, offers the most consistent results in candidate gene studies. The rs28364072 (T2206C) polymorphism in FCER2 gene located 7bp 3ʹ of exon 9 can predict low-response to ICS in childhood asthma.81,82 Mutations in the FCER2 gene might alter the ability of a subject to downregulate the IgE pathway that is inherently relatively resistant to corticosteroid-induced downregulation, with resulting poorer long-term outcomes in both lung function and exacerbations.2
However, other GWASs on ICS response in asthmatic population did not find any significant finding.83 A fundamental GWAS on the pharmacogenetics of ICS response in 2672 patients with asthma investigated more than 9.8 million genetic variants (minor allele frequency ≥1%).84 Common genetic variants or rare genetic variants with large effect sizes did not influence the variability in ICS treatment response in these patients and did not predict an individual’s response.
Differences in ICS Response by Genetic Ancestry
There is often a link between the frequency of a SNP and race/ethnicity.85 Actually, SNPs cluster within groups and are more likely to share a common region of origin or ancestral lineage. Data from the NHLBI Severe Asthma Research Program suggest that several genetic polymorphisms, such as rs17142727 in GLCCI1 and rs1429032 and rs6867762 in FBXL7 in non-Hispanic Whites, and rs1476823 in GLCCI1, rs7801671 and rs7801671 in CYP3A4, rs4701641 and rs12152734 in FBXL7, and rs17689966 in CRHR1 in African-Americans could be associated with changed ICS responsiveness.85 Apparently, the polymorphism rs37972 of GLCCI1 gene rs37972 is less common in populations of African descent compared with a European population.68 A GWAS in asthmatic Hispanic/Latino and African-American children and young adults was unable to find any consistent association with ICS response of 22 SNPs previously associated with this response in another GWAS.86 However, it discovered that the A allele of rs5995653, located 5.8 kb from the 3ʹUTR of the apolipoprotein B mRNA-editing catalytic polypeptide 3 (APOBEC3) gene, was associated with improvement in FEV1 in patients treated with ICS.
In any case, the Study of Asthma Phenotypes and Pharmacogenomic Interactions by Race-ethnicity (SAPPHIRE) documented that the self-declared race ethnicity and genetic ancestry do not predict ICS response, in contrast to the baseline measures of lung function and self-reported asthma control.87 This finding strongly supports the opinion that ICSs are useful in treating asthma, regardless of racial background.87
Conclusion
Theoretically, responses to ICS in asthma are influenced by different genetic variants. However, the replication of pharmacogenetic findings is scarce and we are still far from using them in our clinical asthma practice. This is arguably explained by the fact that response to corticosteroids is too complex to be mainly conditioned by a few genetic variants.13 Nevertheless, it is a real limitation because in an era of precision medicine that links the right patient to the right treatment,88 knowing and dealing with the genetic factors that influence the therapeutic ICS response is fundamental for making precision medicine socially and scientifically accurate.
Although there has been considerable progress, currently known genetic loci only account for a fraction of variability in drug response. In fact, too many different proteins are involved in the response to asthma drugs,89 in particular ICSs. We strongly believe that in examining the role of single SNPs, the epigenetic changes must always be considered because they can modify genetic effects in time-, environment-, and tissue-specific manners.13 ICS use has been shown to affect epigenetic patterns. Active and passive smoking is a typical example because it impairs HDAC2 activity, which could contribute to corticosteroid-insensitive inflammation in patients with severe asthma.90 Furthermore, since genes interact epistatically (ie, together) in networks, this gene-gene interaction must always be considered being critical for expanding the amount of phenotypic varieties that can be attributed to genetics.91
In order to overcome this big issue the possibility of integrating multiple omics layers seems an interesting new approach.92 Connecting genetics with two or more layers will likely maximize the information obtained from different layers. It will allow better understanding of the underlying biological pathways of the multifactorial disease process of asthma. If we will be able to analyze and integrate this information, we likely will generate a systems medicine view of the molecular processes underlying the development and progression of asthma. This will allow us to prescribe the right dose of ICS to the right patient at the right time.
Author Contributions
All authors made substantial contributions to conception of the article; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
Mario Cazzola has participated as a faculty member, and advisor in scientific meetings and courses under the sponsorship of Almirall, AstraZeneca, Biofutura, Boehringer Ingelheim, Chiesi Farmaceutici, GlaxoSmithKline, Menarini Group, Lallemand, Mundipharma, Novartis, Pfizer, Recipharm, Verona Pharma, and Zambon, and is or has been a consultant to ABC Farmaceutici, AstraZeneca, Chiesi Farmaceutici, Recipharm, Lallemand, Novartis, Ockham Biotech, Verona Pharma, and Zambon. His department was funded by Almirall, Boehringer Ingelheim, Novartis, and Zambon.
Paola Rogliani participated as a lecturer and advisor in scientific meetings and courses under the sponsorship of Almirall, AstraZeneca, Biofutura, Boehringer Ingelheim, Chiesi Farmaceutici, GlaxoSmithKline, Menarini Group, Mundipharma, Novartis, and Recipharm. Her department was funded by Almirall, Boehringer Ingelheim, Chiesi Farmaceutici, Novartis, and Zambon.
Luigino Calzetta has participated as an advisor in scientific meetings under the sponsorship of Boehringer Ingelheim and Novartis, received non-financial support from AstraZeneca, a research grant partially funded by Chiesi Farmaceutici, Boehringer Ingelheim, Novartis, and Almirall, and is or has been a consultant to ABC Farmaceutici, Recipharm, Zambon, Verona Pharma, and Ockham Biotech.
Maria Gabriella Matera has participated as a lecturer and advisor in scientific meetings and courses under the sponsorship of Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi Farmaceutici, GlaxoSmithKline, and Novartis, and has been a consultant to ABC Farmaceutici, and Chiesi Farmaceutici. Her department was funded by Novartis.
The authors have no other relevant affiliations or financial involvement with any organization or entity with financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
1. Pelaia G, Vatrella A, Gallelli L, Cazzola M, Maselli R, Marsico SA. Potential genetic influences on the response to asthma treatment. Pulm Pharmacol Ther. 2004;17(5):253–261. doi:10.1016/j.pupt.2004.04.005
2. Rogers AJ, Tantisira KG, Fuhlbrigge AL, et al. Predictors of poor response during asthma therapy differ with definition of outcome. Pharmacogenomics. 2009;10(8):1231–1242. doi:10.2217/pgs.09.86
3. Shastry BS. Genetic diversity and new therapeutic concepts. J Hum Genet. 2005;50(7):321–328. doi:10.1007/s10038-005-0264-6
4. Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol. 2017;17(4):233–247. doi:10.1038/nri.2017.1
5. Strehl C, Buttgereit F. Optimized glucocorticoid therapy: teaching old drugs new tricks. Mol Cell Endocrinol. 2013;380(1–2):32–40. doi:10.1016/j.mce.2013.01.026
6. Parente L, Solito E. Annexin 1: more than an anti-phospholipase protein. Inflamm Res. 2004;53(4):125–132. doi:10.1007/s00011-003-1235-z
7. De Bosscher K, Haegeman G. Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol. 2009;23(3):281–291. doi:10.1210/me.2008-0283
8. Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9(1):62–70. doi:10.1038/nri2470
9. Girol AP, Mimura KK, Drewes CC, et al. Anti-inflammatory mechanisms of the annexin A1 protein and its mimetic peptide Ac2-26 in models of ocular inflammation in vivo and in vitro. J Immunol. 2013;190(11):5689–5701. doi:10.4049/jimmunol.1202030
10. Barnes PJ. Mechanisms and resistance in glucocorticoid control of inflammation. J Steroid Biochem Mol Biol. 2010;120(2–3):76–85. doi:10.1016/j.jsbmb.2010.02.018
11. Cazzola M, Coppola A, Rogliani P, Matera MG. Novel glucocorticoid receptor agonists in the treatment of asthma. Expert Opin Investig Drugs. 2015;24(11):1473–1482. doi:10.1517/13543784.2015.1078310
12. Keskin O, Farzan N, Birben E, et al. Genetic associations of the response to inhaled corticosteroids in asthma: a systematic review. Clin Transl Allergy. 2019;9:2. doi:10.1186/s13601-018-0239-2
13. Matera MG, Rinaldi B, Calzetta L, Cazzola M. Pharmacogenetic and pharmacogenomic considerations of asthma treatment. Expert Opin Drug Metab Toxicol. 2017;13(11):1159–1167. doi:10.1080/17425255.2017.1391215
14. Ortega VE, Bleecker ER. The pharmacogenetics of asthma and the road to personalized medicine. Pulmão RJ. 2012;21(2):41–52.
15. Duong-Thi-Ly H, Nguyen-Thi-Thu H, Nguyen-Hoang L, Nguyen-Thi-Bich H, Craig TJ, Duong-Quy S. Effects of genetic factors to inhaled corticosteroid response in children with asthma: a literature review. J Int Med Res. 2017;45(6):1818–1830. doi:10.1177/0300060516683877
16. Song QQ, Xie WY, Tang YJ, Zhang J, Liu J. Genetic variation in the glucocorticoid pathway involved in interindividual differences in the glucocorticoid treatment. Pharmacogenomics. 2017;18(3):293–316. doi:10.2217/pgs-2016-0151
17. Tantisira KG, Lake S, Silverman ES, et al. Corticosteroid pharmacogenetics: association of sequence variants in CRHR1 with improved lung function in asthmatics treated with inhaled corticosteroids. Hum Mol Genet. 2004;13(13):1353–1359. doi:10.1093/hmg/ddh149
18. Kim WJ, Sheen SS, Kim TH, et al. Association between CRHR1 polymorphism and improved lung function in response to inhaled corticosteroid in patients with COPD. Respirology. 2009;14(2):260–263. doi:10.1111/j.1440-1843.2008.01425.x
19. Awasthi S, Gupta S, Agarwal S, Sharma N. CRHR1 Gene SNPs and response to systemic corticosteroids in Indian asthmatic children during acute exacerbation. Indian J Pediatr. 2015;82(9):781–786. doi:10.1007/s12098-015-1702-x
20. Mougey EB, Chen C, Tantisira KG, et al. Pharmacogenetics of asthma controller treatment. Pharmacogenomics J. 2013;13(3):242–250. doi:10.1038/tpj.2012.5
21. Dijkstra A, Koppelman GH, Vonk JM, Bruinenberg M, Schouten JP, Postma DS. Pharmacogenomics and outcome of asthma: no clinical application for long-term steroid effects by CRHR1 polymorphisms. J Allergy Clin Immunol. 2008;121(6):1510–1513. doi:10.1016/j.jaci.2008.04.015
22. Keskin O, Uluca Ü, Birben E, et al. Genetic associations of the response to inhaled corticosteroids in children during an asthma exacerbation. Pediatr Allergy Immunol. 2016;27(5):507–513. doi:10.1111/pai.12566
23. Ito K, Chung KF, Adcock IM. Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 2006;117(3):522–543. doi:10.1016/j.jaci.2006.01.032
24. Rivers C, Levy A, Hancock J, Lightman S, Norman M. Insertion of an amino acid in the DNA-binding domain of the glucocorticoid receptor as a result of alternative splicing. J Clin Endocrinol Metab. 1999;84(11):4283–4286. doi:10.1210/jcem.84.11.6235
25. Lu NZ, Cidlowski JA. Glucocorticoid receptor isoforms generate transcription specificity. Trends Cell Biol. 2006;16(6):301–307. doi:10.1016/j.tcb.2006.04.005
26. Panek M, Pietras T, Fabijan A, et al. Effect of glucocorticoid receptor gene polymorphisms on asthma phenotypes. Exp Ther Med. 2013;5(2):572–580. doi:10.3892/etm.2012.809
27. Leventhal SM, Lim D, Green TL, Cantrell AE, Cho K, Greenhalgh DG. Uncovering a multitude of human glucocorticoid receptor variants: an expansive survey of a single gene. BMC Genet. 2019;20(1):16. doi:10.1186/s12863-019-0718-z
28. Maltese P, Canestrari E, Palma L, et al. High resolution melting (HRM) analysis for the detection of ER22/23EK, BclI, and N363S polymorphisms of the glucocorticoid receptor gene. J Steroid Biochem Mol Biol. 2009;113(3–5):269–274. doi:10.1016/j.jsbmb.2009.01.012
29. Reimondo G, Chiodini I, Puglisi S, et al. Analysis of BCLI, N363S and ER22/23EK polymorphisms of the glucocorticoid receptor gene in adrenal incidentalomas. PLoS One. 2016;11(9):e0162437. doi:10.1371/journal.pone.0162437
30. Russcher H, van Rossum EF, de Jong FH, Brinkmann AO, Lamberts SW, Koper JW. Increased expression of the glucocorticoid receptor-A translational isoform as a result of the ER22/23EK polymorphism. Mol Endocrinol. 2005;19(7):1687–1696. doi:10.1210/me.2004-0467
31. Cuzzoni E, De Iudicibus S, Bartoli F, Ventura A, Decorti G. Association between BclI polymorphism in the NR3C1 gene and in vitro individual variations in lymphocyte responses to methylprednisolone. Br J Clin Pharmacol. 2012;73(4):651–655. doi:10.1111/j.1365-2125.2011.04130.x
32. Pietras T, Panek M, Tworek D, et al. The Bcl I single nucleotide polymorphism of the human glucocorticoid receptor gene h-GR/NR3C1 promoter in patients with bronchial asthma: pilot study. Mol Biol Rep. 2011;38(6):3953–3958. doi:10.1007/s11033-010-0512-5
33. Huizenga NA, Koper JW, De Lange P, et al. A polymorphism in the glucocorticoid receptor gene may be associated with and increased sensitivity to glucocorticoids in vivo. J Clin Endocrinol Metab. 1998;83(1):144–151. doi:10.1210/jcem.83.1.4490
34. Jewell CM, Cidlowski JA. Molecular evidence for a link between the N363S glucocorticoid receptor polymorphism and altered gene expression. J Clin Endocrinol Metab. 2007;92(8):3268–3277. doi:10.1210/jc.2007-0642
35. Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids. 2010;75(1):1–12. doi:10.1016/j.steroids.2009.09.002
36. Panek M, Pietras T, Antczak A, Górski P, Kuna P, Szemraj J. The role of functional single nucleotide polymorphisms of the human glucocorticoid receptor gene NR3C1 in Polish patients with bronchial asthma. Mol Biol Rep. 2012;39(4):4749–4757. doi:10.1007/s11033-011-1267-3
37. Szczepankiewicz A, Breborowicz A, Sobkowiak P, Popiel A. No association of glucocorticoid receptor polymorphisms with asthma and response to glucocorticoids. Adv Med Sci. 2008;53(2):245–250. doi:10.2478/v10039-008-0042-8
38. Koper JW, Stolk RP, de Lange P, et al. Lack of association between five polymorphisms in the human glucocorticoid receptor gene and glucocorticoid resistance. Hum Genet. 1997;99(5):663–668. doi:10.1007/s004390050425
39. Huizenga NA, de Lange P, Koper JW, et al. Five patients with biochemical and/or clinical generalized glucocorticoid resistance without alterations in the glucocorticoid receptor gene. J Clin Endocrinol Metab. 2000;85(5):2076–2081. doi:10.1210/jcem.85.5.6542
40. Green TL, Tung K, Lim D, Leventhal SM, Cho K, Greenhalgh DG. A novel human glucocorticoid receptor SNP results in increased transactivation potential. Biochem Biophys Rep. 2016;9:140–145. doi:10.1016/j.bbrep.2016.12.003
41. Hawkins GA, Lazarus R, Smith RS, et al. The glucocorticoid receptor heterocomplex gene STIP1 is associated with improved lung function in asthmatic subjects treated with inhaled corticosteroids. J Allergy Clin Immunol. 2009;123(6):1376–1383. doi:10.1016/j.jaci.2009.01.049
42. Ouyang J, Chen P, Jiang T, Chen Y, Li J. Nuclear HSP90 regulates the glucocorticoid responsiveness of PBMCs in patients with idiopathic nephrotic syndrome. Int Immunopharmacol. 2012;14(3):334–340. doi:10.1016/j.intimp.2012.08.012
43. Maltese P, Palma L, Sfara C, et al. Glucocorticoid resistance in Crohn’s disease and ulcerative colitis: an association study investigating GR and FKBP5 gene polymorphisms. Pharmacogenomics J. 2012;12(5):432–438. doi:10.1038/tpj.2011.26
44. Koper JW, van Rossum EF, van den Akker EL. Glucocorticoid receptor polymorphisms and haplotypes and their expression in health and disease. Steroids. 2014;92:62–73. doi:10.1016/j.steroids.2014.07.015
45. Kobayashi Y, Mercado N, Barnes PJ, Ito K, Hartl D. Defects of protein phosphatase 2A causes corticosteroid insensitivity in severe asthma. PLoS One. 2011;6(12):e27627. doi:10.1371/journal.pone.0027627
46. Kobayashi Y, Ito K, Kanda A, Tomoda K, Mercado N, Barnes PJ. Impaired dual-specificity protein phosphatase DUSP4 reduces corticosteroid sensitivity. Mol Pharmacol. 2017;91(5):475–481. doi:10.1124/mol.116.107656
47. Pelaia G, Cuda G, Vatrella A, et al. Mitogen-activated protein kinases and asthma. J Cell Physiol. 2005;202(3):642–653. doi:10.1002/jcp.20169
48. Wang X, Nelin LD, Kuhlman JR, Meng X, Welty SE, Liu Y. The role of MAP kinase phosphatase-1 in the protective mechanism of dexamethasone against endotoxemia. Life Sci. 2008;83(19–20):671–680. doi:10.1016/j.lfs.2008.09.003
49. Jin Y, Hu D, Peterson EL, et al. Dual-specificity phosphatase 1 as a pharmacogenetic modifier of inhaled steroid response among asthmatic patients. J Allergy Clin Immunol. 2010;126(3):618–625. doi:10.1016/j.jaci.2010.06.007
50. Shaw D, Portelli MA, Sayers I. Asthma. In: Padmanabhan S, editor. Handbook of Pharmacogenomics and Stratified Medicine. London: Academic Press; 2014.
51. Bhavsar P, Ahmad T, Adcock IM. The role of histone deacetylases in asthma and allergic diseases. J Allergy Clin Immunol. 2008;121(3):580–584. doi:10.1016/j.jaci.2007.12.1156
52. Grausenburger R, Bilic I, Boucheron N, et al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J Immunol. 2010;185(6):3489–3497. doi:10.4049/jimmunol.0903610
53. Barnes PJ. Therapy of airway disease: epigenetic potential. In: Tollefsbol T, editor. Epigenetics in Human Disease. Amsterdam: Elsevier; 2012:387–393.
54. Kim MH, Kim SH, Kim YK, et al. A polymorphism in the histone deacetylase 1 gene is associated with the response to corticosteroids in asthmatics. Korean J Intern Med. 2013;28(6):708–714. doi:10.3904/kjim.2013.28.6.708
55. Matera MG, Rinaldi B, Calzetta L, Rogliani P, Cazzola M. Pharmacokinetics and pharmacodynamics of inhaled corticosteroids for asthma treatment. Pulm Pharmacol Ther. 2019;58:101828. doi:10.1016/j.pupt.2019.101828
56. Kelly HW. Comparison of inhaled corticosteroids: an update. Ann Pharmacother. 2009;43(3):519–527. doi:10.1345/aph.1L546
57. Stockmann C, Fassl B, Gaedigk R, et al. Fluticasone propionate pharmacogenetics: CYP3A4*22 polymorphism and pediatric asthma control. J Pediatr. 2013;162(6):1222–1227. doi:10.1016/j.jpeds.2012.11.031
58. Stockmann C, Reilly CA, Fassl B, et al. Effect of CYP3A5*3 on asthma control among children treated with inhaled beclomethasone. J Allergy Clin Immunol. 2015;136(2):505–507. doi:10.1016/j.jaci.2015.02.009
59. Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet. 2009;373(9678):1905–1917. doi:10.1016/S0140-6736(09)60326-3
60. Han SS, Xu YQ, Lu Y, Gu XC, Wang Y. A PRISM: a-compliant meta-analysis of MDR1 polymorphisms and idiopathic nephrotic syndrome: susceptibility and steroid responsiveness. Medicine (Baltimore). 2017;96(24):e7191. doi:10.1097/MD.0000000000007191
61. Crowe A, Tan AM. Oral and inhaled corticosteroids: differences in P-glycoprotein (ABCB1) mediated efflux. Toxicol Appl Pharmacol. 2012;260(3):294–302. doi:10.1016/j.taap.2012.03.008
62. Vandevyver S, Dejager L, Libert C. On the trail of the glucocorticoid receptor: into the nucleus and back. Traffic. 2012;13(3):364–374. doi:10.1111/j.1600-0854.2011.01288.x
63. Mingot JM, Kostka S, Kraft R, Hartmann E, Görlich D. Importin 13: a novel mediator of nuclear import and export. EMBO J. 2001;20(14):3685–3694. doi:10.1093/emboj/20.14.3685
64. Tao T, Lan J, Lukacs GL, Haché RJ, Kaplan F. Importin 13 regulates nuclear import of the glucocorticoid receptor in airway epithelial cells. Am J Respir Cell Mol Biol. 2006;35(6):668–680. doi:10.1165/rcmb.2006-0073OC
65. Raby BA, Van Steen K, Lasky-Su J, Tantisira K, Kaplan F, Weiss ST. Importin-13 genetic variation is associated with improved airway responsiveness in childhood asthma. Respir Res. 2009;10(1):67. doi:10.1186/1465-9921-10-67
66. Turolo S, Edefonti A, Lepore M, et al. SXR rs3842689: a prognostic factor for steroid sensitivity or resistance in pediatric idiopathic nephrotic syndrome. Pharmacogenomics. 2016;17(11):1227–1233. doi:10.2217/pgs-2016-0029
67. Tantisira KG, Hwang ES, Raby BA, et al. TBX21: a functional variant predicts improvement in asthma with the use of inhaled corticosteroids. Proc Natl Acad Sci USA. 2004;101(52):18099–18104. doi:10.1073/pnas.0408532102
68. Tantisira KG, Lasky-Su J, Harada M, et al. Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N Engl J Med. 2011;365(13):1173–1183. doi:10.1056/NEJMoa0911353
69. Chapman MS, Qu N, Pascoe S, et al. Isolation of differentially expressed sequence tags from steroid-responsive cells using mRNA differential display. Mol Cell Endocrinol. 1995;108(1–2):R1–R7. doi:10.1016/0303-7207(95)03481-L
70. Hosking L, Bleecker E, Ghosh S, et al. GLCCI1 rs37973 does not influence treatment response to inhaled corticosteroids in white subjects with asthma. J Allergy Clin Immunol. 2014;133(2):587–589. doi:10.1016/j.jaci.2013.08.024
71. Hu CP, Xun QF, Li XZ, et al. Effects of glucocorticoid-induced transcript 1 gene deficiency on glucocorticoid activation in asthmatic mice. Chin Med J (Engl). 2018;131(23):2817–2826. doi:10.4103/0366-6999.246061
72. Park HW, Dahlin A, Tse S, et al. Genetic predictors associated with improvement of asthma symptoms in response to inhaled corticosteroids. J Allergy Clin Immunol. 2014;133(3):664–669. doi:10.1016/j.jaci.2013.12.1042
73. Gautam Y, Afanador Y, Abebe T, López JE, Mersha TB. Genome-wide analysis revealed sex-specific gene expression in asthmatics. Hum Mol Genet. 2019;28(15):2600–2614. doi:10.1093/hmg/ddz074
74. Berce V, Kozmus CE, Potočnik U. Association among ORMDL3 gene expression, 17q21 polymorphism and response to treatment with inhaled corticosteroids in children with asthma. Pharmacogenomics J. 2013;13(6):523–529. doi:10.1038/tpj.2012.36
75. Leusink M, Vijverberg SJ, Koenderman L, et al. Genetic variation in uncontrolled childhood asthma despite ICS treatment. Pharmacogenomics J. 2016;16(2):158–163. doi:10.1038/tpj.2015.36
76. Ryan JJ, Spiegel S. The role of sphingosine-1-phosphate and its receptors in asthma. Drug News Perspect. 2008;21(2):89–96. doi:10.1358/dnp.2008.21.2.1188195
77. Balantic M, Rijavec M, Skerbinjek Kavalar M, et al. Asthma treatment outcome in children is associated with vascular endothelial growth factor A (VEGFA) polymorphisms. Mol Diagn Ther. 2012;16(3):173–180. doi:10.1007/BF03262206
78. Lee KS, Kim SR, Park HS, Jin GY, Lee YC. Cysteinyl leukotriene receptor antagonist regulates vascular permeability by reducing vascular endothelial growth factor expression. J Allergy Clin Immunol. 2004;114(5):1093–1099. doi:10.1016/j.jaci.2004.07.039
79. García-Menaya JM, Cordobés-Durán C, García-Martín E, Agúndez JAG. Pharmacogenetic factors affecting asthma treatment response. Potential implications for drug therapy. Front Pharmacol. 2019;10:520. doi:10.3389/fphar.2019.00520
80. Wang RS, Croteau-Chonka DC, Silverman EK, Loscalzo J, Weiss ST, Hall KT. Pharmacogenomics and placebo response in a randomized clinical trial in asthma. Clin Pharmacol Ther. 2019;106(6):1261–1267. doi:10.1002/cpt.1646
81. Tantisira KG, Silverman ES, Mariani TJ, et al. FCER2: a pharmacogenetic basis for severe exacerbations in children with asthma. J Allergy Clin Immunol. 2007;120(6):1285–1291. doi:10.1016/j.jaci.2007.09.005
82. Koster ES, Maitland-van der Zee A-H, Tavendale R, Maitland-van der Zee AH, Tavendale R, et al. FCER2 T2206C variant associated with chronic symptoms and exacerbations in steroid-treated asthmatic children. Allergy. 2011;66(12):1546–1552. doi:10.1111/j.1398-9995.2011.02701.x
83. Farzan N, Vijverberg SJ, Arets HG, Raaijmakers JA, Maitland-van der Zee AH. Pharmacogenomics of inhaled corticosteroids and leukotriene modifiers: a systematic review. Clin Exp Allergy. 2017;47(2):271–293. doi:10.1111/cea.12844
84. Mosteller M, Hosking L, Murphy K, et al. No evidence of large genetic effects on steroid response in asthma patients. J Allergy Clin Immunol. 2017;139(3):797–803. doi:10.1016/j.jaci.2016.05.032
85. Cazzola M, Calzetta L, Matera MG, Hanania NA, Rogliani P. How does race/ethnicity influence pharmacological response to asthma therapies? Expert Opin Drug Metab Toxicol. 2018;14(4):435–446. doi:10.1080/17425255.2018.1449833
86. Hernandez-Pacheco N, Farzan N, Francis B, et al. Genome-wide association study of inhaled corticosteroid response in admixed children with asthma. Clin Exp Allergy. 2019;49(6):789–798. doi:10.1111/cea.13354
87. Wells KE, Cajigal S, Peterson EL, et al. Assessing differences in inhaled corticosteroid response by self-reported race-ethnicity and genetic ancestry among asthmatic subjects. J Allergy Clin Immunol. 2016;137(5):1364–1369. doi:10.1016/j.jaci.2015.12.1334
88. Ulrik CS, Vijverberg S, Hanania NA, Diamant Z. Precision medicine and treatable traits in chronic airway diseases - where do we stand? Curr Opin Pulm Med. 2020;26(1):33–39. doi:10.1097/MCP.0000000000000639
89. Kersten ET, Koppelman GH. Pharmacogenetics of asthma: toward precision medicine. Curr Opin Pulm Med. 2017;23(1):12–20. doi:10.1097/MCP.0000000000000335
90. Kobayashi Y, Bossley C, Gupta A, et al. Passive smoking impairs histone deacetylase-2 in children with severe asthma. Chest. 2014;145(2):305–312. doi:10.1378/chest.13-0835
91. Park HW, Tantisira KG, Weiss ST. Pharmacogenomics in asthma therapy: where are we and where do we go? Annu Rev Pharmacol Toxicol. 2015;55:129–147. doi:10.1146/annurev-pharmtox-010814-124543
92. Abdel-Aziz MI, Neerincx AH, Vijverberg SJ, Kraneveld AD, Maitland-van der Zee AH. Omics for the future in asthma. Semin Immunopathol. 2020;42(1):111–126. doi:10.1007/s00281-019-00776-x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.