Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 10
Pharmacogenetics in inflammatory bowel disease: understanding treatment response and personalizing therapeutic strategies
Authors Yamamoto-Furusho JK
Received 22 December 2016
Accepted for publication 6 April 2017
Published 26 May 2017 Volume 2017:10 Pages 197—204
DOI https://doi.org/10.2147/PGPM.S109648
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin Bluth
Jesús K Yamamoto-Furusho
Inflammatory Bowel Disease Clinic, Department of Gastroenterology, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Tlalpan, Mexico
Abstract: Inflammatory bowel disease (IBD) is a chronic and heterogeneous disorder characterized by remitting and relapsing periods of activity. Pharmacogenetics refers to the study of the effect of inheritance on individual variation in drug responses. Several drug-related markers in IBD patients have been identified in order to predict the response to medical treatment including biological therapy as well as the reduction of adverse events. In the future, the treatment of IBD should be personalized in its specific profile to provide the most efficacious treatment with lack of adverse events.
Keywords: individualized medicine, pharmacogenomics, genetic, serologic, profile, treatment, prognostic factors
Introduction
Inflammatory bowel disease (IBD) comprises Crohn’s disease (CD) and ulcerative colitis (UC) characterized by intestinal chronic inflammation of unknown etiology. It has been postulated that it is a multifactorial disease involving interplay among aberrant immune response, environmental factors, and multiple genes.1
The incidence of IBD is now rising in developing countries, and it is being increasingly considered an emerging global disease.2,3 Traditionally, developing nations have reported a lower prevalence of IBD, but the incidence is currently rising in many of these countries as they become more industrialized in Latin America and Asia.4–7
Knowledge development in IBD has permitted classification of the disease into different phenotypes according to the current clinical Montreal classification that considers clinical characteristics such as age at diagnosis, location of disease, behavior in CD, and extent of disease in UC.8 Clinical traits such as the extent of inflammation or disease location, extraintestinal manifestations, and disease behavior enabled predicting the future course of the disease and response to medical therapy to allow disease categorization which in turn guides present-day care recommendations.9
On the other hand, understanding the involvement of several molecular pathways by different technologies such as genomic, transcriptomic, epigenetic, and miRNAs studies has been focused on the identification of new genetic risk factors for specific disease and application to clinical practice such as prognostic factors involved in the clinical course and response to medical therapy.10,11
Finally, pharmacogenetics is the study of the association between variability in drug response, drug toxicity, and polymorphisms in genes in order to adapt drugs to a patient’s specific genetic background and therefore make them more efficacious and safe.12
The vast heterogeneity of IBD patients has motivated a comprehensive evidence-based search of novel biomarkers for appropriate patient stratification that accounts for the interindividual differences in severity, drug efficacy, side effects, or prognosis and would help guide clinicians in the management of patients with IBD and represents a major step toward personalized medicine.13
This study aimed to review the role of pharmacogenetics in the potential selection of personalized treatment in patients with IBD in the future.
Several drug-related markers classified according to pharmacological groups with clinical utility in patients with IBD are described below and summarized in Table 1.
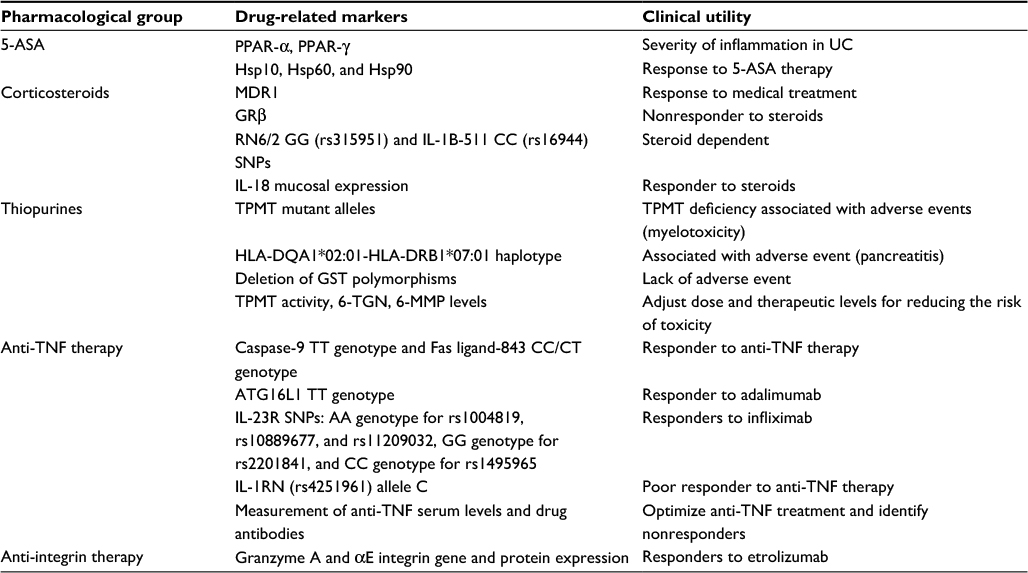 | Table 1 Drug-related markers in inflammatory bowel disease Abbreviations: SNPs, single nucleotide polymorphisms; PPAR, peroxisome proliferator-activated receptors; 5-ASA, 5-aminosalicylic acid; IL, interleukin; Hsp, heat shock protein; MDR, multidrug resistant; TNF, tumor necrosis factor; GRβ, glucocorticoid receptor β; TPMT, thiopurine methyltransferase; HLA, human leukocyte antigen; GST, glutathione-S-transferase; 6-TGN, 6-thioguanine nucleotide; 6-MMP, 6-methylmercaptopurine; ATG16L1, autophagy-related 16-like 1; UC, ulcerative colitis. |
Pharmacogenetics in IBD treatment
Mesalazine (5-aminosalicylic acid [5-ASA])
Mesalazine constitutes the first line of treatment for induction and maintenance of remission in UC. It appears to act locally on colonic mucosa and reduces inflammation through a variety of anti-inflammatory processes by the activation of γ-form peroxisome proliferator-activated receptors (PPAR-γ). PPAR-γ has a role in the regulation of intestinal inflammation and is highly expressed in the colon, where epithelial cells and macrophages are the main cellular sources of this nuclear receptor.14
A study showed decreased gene expression of PPAR-γ in colonic biopsies of patients with active UC and its expression was negatively correlated with severity of endoscopic disease activity.15
In another study, the PPAR alpha gene expression was significantly decreased in patients with active UC compared with remission UC group (P=0.001) and normal controls (P=0.001). Yamamoto-Furusho et al16 found that low gene expression of PPAR alpha in colonic mucosa is associated with high risk of UC activity (P≤0.0001, odds ratio [OR]=22.6). They observed an increase of PPAR alpha expression in patients with UC who were treated with 5-aminosalicylates compared with those who received any other combined therapy (P=0.03, OR=0.08). PPAR-γ gene expression was decreased in the active UC group compared with remission UC (P=0.001) and control group (P=0.001). An increased expression of PPAR-γ gene was associated with mild clinical course of the disease (P≤0.001, OR=0.05).16
Heat shock proteins (Hsp) are a family of molecules typically involved in folding, refolding, translocation, and degradation of intracellular proteins under normal and stress conditions.17 Hsp60 and Hsp10 (Hsp60 co-chaperonin) are increased in the affected intestinal mucosa in patients with CD and UC.18
A study demonstrated that mucosal Hsp60 levels in UC patients decrease after therapy with either mesalazine alone or mesalazine plus probiotics. They also demonstrated that Hsp90 levels are elevated in colonic mucosa from UC patients, both in epithelium and lamina propria.19 Treatment with 5-ASA plus probiotics reduced the Hsp90 levels in the lamina propria, while 5-ASA alone did not have any effect and a linear correlation was also reported between Hsp90 and CD4 levels in lamina propria in UC patients at both diagnosis and 6 months after 5-ASA only therapy.20
Corticosteroids
Corticosteroids constitute the second line of therapy in patients who fail to respond to the maximal dose of mesalazine or present moderate-to-severe disease activity of IBD. The mechanism of action of corticosteroids is based on the inhibition of T-cell activation and the production of pro-inflammatory cytokines.
A review has proposed different markers associated with steroid therapy outcomes in patients with IBD.21 The most-studied molecule is the multidrug-resistant (MDR1) gene code for a drug efflux pump P-glycoprotein-170 (permeability glycoprotein or Pgp), which is expressed on the apical surface of lymphocytes and intestinal epithelial cells. Its function comprises active transportation of toxins and drugs out of target cells.22 Pgp and MDR expression have been shown to be significantly higher in CD and UC patients requiring surgery due to failure of medical therapy.23 On the other hand, MDR1 expression on colonic biopsies decreased in patients with active UC compared to UC patients in remission and the normal control group (P=0.034 and 0.002, respectively). However, in this study, the relevant finding was that the medical treatment response and long-term remission were associated with high gene expression of MDR1 (P=0.009 and 0.002, respectively).24
Other studies have reported an increased expression of glucocorticoid receptor β in 83% of the patients with steroid-resistant UC compared to only 9% in steroid-responsive patients and 10% in healthy controls.25,26
Yamamoto-Furusho et al27 found a significant association between RN6/2 GG (rs315951) and interleukin (IL)-1B-511 CC (rs16944) genotypes and the presence of steroid dependence in UC patients (p corrected=0.0001, OR=15.6 and pC=0.008, OR=4.09, respectively).
A previous study identified several predictor gene panels containing genes involved in immune mechanisms (PTN, OLFM4, LILRA2, CD36), autophagy, or GC response (STS, MDM2) with potential value to predict GC response and need of surgery as well as with diagnostic value for IBD patients.28 Villeda-Ramirez et al29 showed that IL-18 mRNA expression was significantly increased in the mucosa of patients with active and remission UC compared to the healthy control group (P=0.006 and 0.007, respectively). The high gene expression of IL-18 was associated with the use of steroids (P=0.04).
Thiopurines
Immunomodulator drugs have become the mainstay of IBD with proven efficacy in reducing relapses, permitting steroid withdrawal, and closing fistulas.30
Thiopurines such as azathioprine (AZA) and 6-mercaptopurine (6-MP) are usually used in patients with corticosteroid dependence or resistance and combined with tumor necrosis factor (TNF) therapy. The gene encoding thiopurine methyltransferase (TPMT) is located on chromosome 6 (6p22.3) and contains 10 exons. Two wild-type alleles (TPMT*1 and *1S) and 20 mutant alleles (TPMT*2, *3A, *3B, *3C, *3D, *4, *5, *6, *7, *8, *9, *10, *11,*12, *13, *14, *15, *16, *17, *18) are responsible for TPMT deficiency.31,32 The distribution of TPMT mutant alleles differs significantly among ethnic populations. TPMT*3A (3.2%–5.7%) is the most occurring mutant allele in white populations, followed by TPMT*2 (0.2%–0.5%) and TPMT*3C (0.2%–0.8%), accounting for the vast majority (>95%) of mutant alleles.33–36 In Asian and African populations, however, TPMT*3C is the most frequent mutant allele.37,38
A genome-wide association study found a 2.59-fold risk of pancreatitis in IBD patients taking thiopurines who had the single nucleotide polymorphism rs2647087 within the human leukocyte antigen (HLA)-DQA1*02:01-HLA-DRB1*07:01 haplotype.39
Traditionally, AZA and 6-MP are initiated at a low dose and then gradually increased to a full therapeutic dose of 2.0–2.5 and 1.5 mg/kg/day, respectively; however, this strategy requires tight monitoring in order to detect adverse events such as myelotoxicity and hepatotoxicity. This strategy has been replaced by an approach based on the assessment of TPMT phenotype or activity as shown in Table 1. TPMT testing is recommended before initiating AZA or 6-MP therapy for IBD to decrease the risk of leukopenia. For patients who have absent or low TPMT, activity leading to elevated 6-thioguanine nucleotide (6-TGN) concentrations during thiopurine therapy is significantly associated with an increased risk of development of bone marrow suppression.40 In patients with very high TPMT, activity develops suboptimal 6-TGN concentrations, which have been associated with treatment failure.41 Population studies have shown that the distribution of TPMT activity is trimodal: 0.3% of the population have low-to-absent activity, 11% have intermediate activity, and 89% inherit normal-to-high enzyme activity.42–44
A review by Stocco et al45 has reported the role of enzyme glutathione-S-transferase (GST) genetic polymorphisms which may influence decreased sensitivity to AZA. Some studies have found that the frequency of GST-M1 deletion was significantly lower in patients who developed an adverse event in comparison to patients who tolerated AZA treatment with no adverse event.46–48
On the other hand, measurement of the thiopurine metabolites, 6-TGN and 6-methylmercaptopurine (6-MMP), is useful in clinical practice. Several studies have demonstrated that 6-TGN levels >230 pmol/8×108 red blood cells (RBCs) are associated with increased efficacy.49,50 However, supra-therapeutic levels, generally >400 pmol/8×108 RBCs, are associated with an increased risk of myelosuppression.51 6-MMP can be measured to predict the risk of hepatotoxicity; levels >5,700 pmol/8×108 RBCs carry a 3-fold risk of hepatotoxicity.52
Anti-TNF therapy
Anti-TNF-α drugs are indicated in patients with moderate-to-severe IBD who do not tolerate or respond to conventional therapies. The use of anti-TNF therapy has improved several outcomes in patients with IBD such as better quality of life, reduction of surgeries and hospitalizations, steroid free remission, mucosal healing, and others. However, one third of the patients do not respond to anti-TNF treatment.
Several studies have focused on studying genetic markers that may predict individual response to anti-TNF therapy.53 In luminal CD, the response rate to anti-TNF therapy was 74.7% in patients with Fas ligand (FASLG) -843 CC/CT genotype compared to a response rate of 38.1% in patients with the TT genotype (OR=0.11; 95% confidence interval [CI]=0.08–0.56, P<0.01). On the other hand, patients with caspase-9 TT genotype responded to anti-TNF therapy, in contrast to 66.7% of patients with the CC and CT genotypes (OR=1.50; 95% CI=1.34–1.68, P=0.04). Another variant in FASLG, rs763110, was able to predict the therapeutic response to infliximab in patients with fistulizing CD at week 10.54 A Japanese study reported that GG genotype of FCGR3A had a better response to anti-TNF therapy at week 8 in CD patients.55
Autophagy-related 16-like 1 (ATG16L1) is an autophagy-related gene that processes intracellular bacteria and is expressed in intestinal epithelial cell lines. ATG16L1 TT genotype for rs10210302 responded better to adalimumab after 12, 20, and 30 weeks of treatment compared to the CC genotype in CD patients.56
The cytokine IL-23 is involved in the pathogenesis of IBD. Several genetic variants in IL-23R have been associated with response to infliximab in patients with moderate-to-severe UC at week 14. For instance, AA genotype for rs1004819, rs10889677, and rs11209032, GG genotype for rs2201841, and CC genotype for rs1495965 in IL-23R gene increased the probability to respond to infliximab. However, GG genotype for rs7517847 and rs11465804, CC genotype for rs10489629, and AA genotype for rs1343151 in IL-23R decreased the probability to respond to this drug.57 Bank et al found that the TC/CC genotype for rs10499563 in IL-6 and the GA/AA genotype had a better response to anti-TNF but the effect of IL-1RN (rs4251961) allele C was associated with poorer response to anti-TNF therapy.58
In some patients who had a genetically increased MD-2 level (rs11465996) and TNFRSF1A (TNFR1) expression (rs4149570) and genetically determined decreased TNFAIP3 (A20) expression (rs6927172), IL-1β (rs3804099 and rs4848306), IL-6 (rs3804099 and rs10499563), IL-17 (rs2275913), and interferon-γ (rs2430561) levels were associated with beneficial response among patients with IBD. Fujino et al59 found mRNA expression and serum levels of IL-17 to be increased in patients with IBD and suggested that IL-17 might be associated with altered immune and inflammatory responses in the intestinal mucosa.
Therapeutic drug monitoring (TDM) and measurement of antidrug antibodies (ADAs) for anti-TNF agents have been useful in clinical practice to optimize the efficacy of biologics and minimize adverse events (Figure 1). TDM has been best studied for infliximab and adalimumab including the measurement of both drug and antibodies to infliximab (ATIs) or antibodies to adalimumab (ATAs). Several studies have reported concentrations predictive of response ranging from 1.4 to 12.0 μg/mL.60–63 For adalimumab, cutoffs predictive of mucosal healing range from 4.9 to 7.5 μg/mL.64,65 Low levels of anti-TNF agents are associated with developing ADAs and preceded the formation of ATIs and ATAs66,67 and these are implicated in the increased drug clearance of anti-TNF agents related with lower serum drug levels as well as active disease and loss of response.68,69
 | Figure 1 Optimization of anti-TNF agents according drug serum levels and anti-drug antibodies Abbreviation: TNF, tumor necrosis factor. |
Anti-integrin therapy
Etrolizumab is a monoclonal antibody against β7 integrin subunit that has shown efficacy in patients with moderate-to-severe UC. A study evaluated biomarkers in the colonic mucosa such as granzyme A and integrin αE measured by gene and protein expression levels; both markers were found to be associated with clinical response and mucosal healing in UC patients treated with etrolizumab.70
Microbiota
A potential link between genetics and the microbiome has been documented in patients with IBD. Some studies have shown the effect of NOD2 mutations associated with increased numbers of mucosa-adherent bacteria71 and decreased transcription of the anti-inflammatory cytokine IL-10.72
IBD patients with NOD2 and ATG16L1 have significant alterations in the structure of their gut microbiota, including decreased levels of Faecalibacterium and increases in Escherichia.73 Individuals homozygous for loss-of-function alleles for fucosyltransferase 2 are “nonsecretors,” who do not express ABO antigen on the gastrointestinal mucosa and bodily secretions. Nonsecretors are at increased risk for CD28 and exhibit substantial alterations in the mucosa-associated microbiota.74
Several studies have shown specific taxonomic shifts in IBD patients. Enterobacteriaceae are increased in IBD patients.75 Escherichia coli, particularly adherent-invasive E. coli strains, has been isolated from CD biopsies in culture-based studies76 and is enriched in UC patients.77 Another type of adherent and invasive bacteria is Fusobacteria. The genus Fusobacterium has been found in higher abundance in the colonic mucosa of patients with UC relative to control individuals.78,79
On the other hand, specific groups of gut bacteria may have protective effects against IBD. Bacteroides and Clostridium species have been shown to induce the expansion of Treg cells, reducing intestinal inflammation.80
Other bacterial species such as Bifidobacterium, Lactobacillus, and Faecalibacterium may protect the host from mucosal inflammation by several mechanisms, including downregulation of inflammatory cytokines81 or stimulation of IL-10.82 Faecalibacterium prausnitzii is a member of the microbiota with anti-inflammatory properties. F. prausnitzii has been found depleted in CD biopsy samples concomitant with an increase in E. coli abundance,83 and low levels of mucosa-associated F. prausnitzii are associated with higher risk of recurrent CD following surgery.82 Conversely, recovery of F. prausnitzii after relapse is associated with maintenance of clinical remission of UC.84
A recent study has shown a microbial signature for CD that identified eight groups of microorganisms including Faecalibacterium, Peptostreptococcaceae, Anaerostipes, Methanobrevibacter, Christensenellaceae, Collinsella, Fusobacterium, and Escherichia; the signature achieved an overall sensitivity of 80% and a specificity of 94% for the detection of CD versus healthy controls.85
Fecal microbiota transplantation (FMT) aims to restore gut microbiota diversity by transferring feces from a healthy donor to a sick patient. To date, FMT has been assessed as a novel therapeutic for UC. A randomized controlled trial reported 24% and 5% of those who received FMT and placebo, respectively, reached clinical remission after 7 weeks. This study identified that those patients who had been recently diagnosed of UC (within the year previous to FMT) had a high rate of clinical remission compared to UC patients with longer disease duration (75% vs 18% respectively).86
On the other hand, another study by Rossen et al87 showed no statistically significant difference in clinical remission between UC patients who received FMT sourced from healthy donors or autologous FMT (their own fecal microbiota), and only 41% of patients who received donor FMT achieved clinical and endoscopic remission. The findings of this study suggest that microbial ecosystems of patients who responded to FMT from a healthy donor increased in the numbers of bacterial species from Clostridium clusters.
A detailed assessment of the fecal microbiota taxonomic composition pre- and post-FMT need to be performed in order to identify the responders to FMT in patients with UC. A selective FMT of certain species such as Bifidobacterium, Lactobacillus, and F. prausnitzii could be effective as personalized treatment in patients with IBD.
In conclusion, the combination of genetic markers with clinical, biochemical, serological, and microbiome data for subgroups of IBD patients might permit individualized risk stratification and treatment selection to ensure high efficacy of medical treatment with lack of adverse events.
Disclosure
The author has received honoraria from AbbVie, Takeda, Janssen, UCB, Almirall, Pfizer, Novartis, and Danone as speaker, key opinion leader, and member of the advisory board at national and international levels. He is the President of the Pan American Crohn’s and Colitis Organization.
References
Yamamoto-Furusho JK, Podolsky DK. Innate immunity in inflammatory bowel disease. World J Gastroenterol. 2007;13:5577–5580. | ||
Bernstein CN, Shanahan F. Disorders of a modern lifestyle: reconciling the epidemiology of inflammatory bowel diseases. Gut. 2008; 57(9):1185–1191. | ||
Goh K, Xiao SD. Inflammatory bowel disease: a survey of the epidemiology in Asia. J Dig Dis. 2009;10:1–6. | ||
Torres EA, De Jesús R, Pérez CM, et al. Prevalence of inflammatory bowel disease in an insured population in Puerto Rico during 1996. P R Health Sci J. 2003;22:253–258. | ||
Yamamoto-Furusho JK. Clinical epidemiology of ulcerative colitis in Mexico: a single hospital-based study in a 20-year period (1987–2006). J Clin Gastroenterol. 2009;43:221–224. | ||
Ng SC, Tang W, Ching JY, et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-pacific Crohn’s and colitis epidemiology study. Gastroenterology. 2013;145:158–165. | ||
Parente JM, Coy CS, Campelo V, et al. Inflammatory bowel disease in an underdeveloped region of Northeastern Brazil. World J Gastroenterol. 2015;21:1197–1206. | ||
Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol. 2005;19(Suppl A):5A–36A. | ||
Gerich ME, McGovern DP. Towards personalized care in IBD. Nat Rev Gastroenterol Hepatol. 2014;11:287–299. | ||
Yamamoto-Furusho JK, Fonseca-Camarillo G. Genetic markers associated with clinical outcomes in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2015;21:2683–2695. | ||
Quetglas EG, Mujagic Z, Wigge S, et al. Update on pathogenesis and predictors of response of therapeutic strategies used in inflammatory bowel disease. World J Gastroenterol. 2015;21:12519–12543. | ||
Kirchheiner J, Fuhr U, Brockmöller J. Pharmacogenetics-based therapeutic recommendations – ready for clinical practice? Nat Rev Drug Discov. 2005;4:639–647. | ||
Katsanos KH, Papadakis KA. Pharmacogenetics of inflammatory bowel disease. Pharmacogenomics. 2014;15:2049–2062. | ||
Lawson MM, Thomas AG, Akobeng AK. Tumour necrosis factor alpha blocking agents for induction of remission in ulcerative colitis [review]. Cochrane Database Syst Rev. 2006;3:CD005112. | ||
Yamamoto-Furusho JK, Peñaloza-Coronel A, Sánchez-Muñoz F, Barreto-Zúñiga R, Domínguez-López A. Peroxisome proliferator-activated receptor-gamma (PPAR gamma) expression is down-regulated in patients with active ulcerative colitis. Inflamm Bowel Dis. 2011;17:680–681. | ||
Yamamoto-Furusho JK, Jacintez-Cazares M, Furuzawa-Carballeda J, Fonseca-Camarillo G. Peroxisome proliferator-activated receptors family is involved in the response to treatment and mild clinical course in patients with ulcerative colitis. Dis Markers. 2014;2014:932530. | ||
Prohászka Z, Füst G. Immunological aspects of heat-shock proteins – the optimum stress of life. Mol Immunol. 2004;41:29–44. | ||
Rodolico V, Tomasello G, Zerilli M, et al. Hsp60 and Hsp10 increase in colon mucosa of Crohn’s disease and ulcerative colitis. Cell Stress Chaperones. 2010;15:877–884. | ||
Tomasello G, Rodolico V, Zerilli M, et al. Changes in immunohistochemical levels and subcellular localization after therapy and correlation and colocalization with CD68 suggest a pathogenetic role of Hsp60 in ulcerative colitis. Appl Immunohistochem Mol Morphol. 2011; 19:552–561. | ||
Tomasello G, Sciumé C, Rappa F, et al. Hsp10, Hsp70, and Hsp90 immunohistochemical levels change in ulcerative colitis after therapy. Eur J Histochem. 2011;55:e38. | ||
Gabryel M, Skrzypczak-Zielinska M, Kucharski MA, Slomski R, Dobrowolska A. The impact of genetic factors on response to glucocorticoids therapy in IBD. Scand J Gastroenterol. 2016;51:654–665. | ||
Annese V, Valvano MR, Palmieri O, et al. Multidrug resistance 1 gene in inflammatory bowel disease: a meta-analysis. World J Gastroenterol. 2006;12:3636–3644. | ||
Farrell RJ, Murphy A, Long A, et al. High multidrug resistance (P-glycoprotein 170) expression in inflammatory bowel disease patients who fail medical therapy. Gastroenterology. 2000;118:279–288. | ||
Yamamoto-Furusho JK, Villeda-Ramírez MA, Fonseca-Camarillo G, et al. High gene expression of MDR1 (ABCB1) is associated with medical treatment response and long term remission in patients with ulcerative colitis. Inflamm Bowel Dis. 2010;16:541–719. | ||
Honda M, Orii F, Ayabe T, et al. Expression of glucocorticoid receptor beta in lymphocytes of patients with glucocorticoid-resistant ulcerative colitis. Gastroenterology. 2000;118:859–866. | ||
Fujishima S, Takeda H, Kawata S, Yamakawa M. The relationship between the expression of the glucocorticoid receptor in biopsied colonic mucosa and the glucocorticoid responsiveness of ulcerative colitis patients. Clin Immunol. 2009;133:208–217. | ||
Yamamoto-Furusho JK, Santiago-Hernández JJ, Pérez-Hernández N, Ramírez-Fuentes S, Fragoso JM, Vargas-Alarcón G. Interleukin 1β (IL-1B) and IL-1 antagonist receptor (IL-1RN) gene polymorphisms are associated with the genetic susceptibility and steroid dependence in patients with ulcerative colitis. J Clin Gastroenterol. 2011;45:531–535. | ||
Montero-Meléndez T, Llor X, García-Planella E, Perretti M, Suárez A. Identification of novel predictor classifiers for inflammatory bowel disease by gene expression profiling. PLoS One. 2013;8:e76235. | ||
Villeda Ramírez MA, Mendivil Rangel EJ, Domínguez López A, Yamamoto-Furusho JK. Interleukin-18 upregulation is associated with the use of steroids in patients with ulcerative colitis. Inflamm Bowel Dis. 2011;17:E50–E51. | ||
Dubinsky MC. Azathioprine, 6-mercaptopurine in inflammatory bowel disease: pharmacology, efficacy, and safety. Clin Gastroenterol Hepatol. 2004;2:731–743. | ||
Schaeffeler E, Lang T, Zanger UM, Eichelbaum M, Schwab M. High-throughput genotyping of thiopurine S-methyltransferase by denaturing HPLC. Clin Chem. 2001;47:548–555. | ||
Schaeffeler E, Fischer C, Brockmeier D, et al. Comprehensive analysis of thiopurine S-methyltransferase phenotype–genotype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics. 2004;14:407–417. | ||
Reuther LO, Sonne J, Larsen N, Dahlerup JF, Thomsen OO, Schmiegelow K. Thiopurine methyltransferase genotype distribution in patients with Crohn’s disease. Aliment Pharmacol Ther. 2003;17:65–68. | ||
Yates CR, Krynetski EY, Loennechen T, et al. Molecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med. 1997;126:608–614. | ||
Ansari A, Hassan C, Duley J, et al. Thiopurine methyltransferase activity and the use of azathioprine in inflammatory bowel disease. Aliment Pharmacol Ther. 2002;16:1743–1750. | ||
Kurzawski M, Gawronska-Szklarz B, Drozdzik M. Frequency distribution of thiopurine S-methyltransferase alleles in a Polish population. Ther Drug Monit. 2004;26:541–545. | ||
Kubota T, Chiba K. Frequencies of thiopurine S-methyltransferase mutant alleles (TPMT*2, *3A, *3B and *3C) in 151 healthy Japanese subjects and the inheritance of TPMT*3C in the family of a propositus. Br J Clin Pharmacol. 2001;51:475–477. | ||
Ameyaw MM, Collie-Duguid ES, Powrie RH, Ofori-Adjei D, McLeod HL. Thiopurine methyltransferase alleles in British and Ghanaian populations. Hum Mol Genet. 1999;8:367–370. | ||
Heap GA, Weedon MN, Bewshea CM, et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat Genet. 2014;46:1131–1134. | ||
Lennard L. TPMT in the treatment of Crohn’s disease with azathioprine. Gut. 2002;51:143–146. | ||
Dubinsky MC, Lamothe S, Yang HY, et al. Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology. 2000;118:705–713. | ||
Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32:651–662. | ||
Moon W, Loftus EV Jr. Review article: recent advances in pharmacogenetics and pharmacokinetics for safe and effective thiopurine therapy in inflammatory bowel disease. Aliment Pharmacol Ther. Epub 2016 Feb 14. | ||
Fong SC, Blaker PA, Arenas-Hernandez M, Marinaki AM, Sanderson JD. Getting the best out of thiopurine therapy: thiopurine S-methyltransferase and beyond. Biomark Med. 2015;9:51–65. | ||
Stocco G, Pelin M, Franca R, et al. Pharmacogenetics of azathioprine in inflammatory bowel disease: a role for glutathione-S-transferase? World J Gastroenterol. 2014;20:3534–3541. | ||
Stocco G, Martelossi S, Barabino A, et al. Glutathione-S-transferase genotypes and the adverse effects of azathioprine in young patients with inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:57–64. | ||
Dervieux T, Boulieu R. Simultaneous determination of 6-thioguanine and methyl 6-mercaptopurine nucleotides of azathioprine in red blood cells by HPLC. Clin Chem. 1998;44:551–555. | ||
Stocco G, Cuzzoni E, De Iudicibus S, et al. Deletion of glutathione-S-transferase M1 reduces azathioprine metabolite concentrations in young patients with inflammatory bowel disease. J Clin Gastroenterol. 2014;48:43–51. | ||
Dubinsky MC, Lamothe S, Yang HY, et al. Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology. 2000;118:705–713. | ||
Osterman MT, Kundu R, Lichtenstein GR, Lewis JD. Association of 6-thioguanine nucleotide levels and inflammatory bowel disease activity: a meta-analysis. Gastroenterology. 2006;130:1047–1053. | ||
Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2006;24:331–342. | ||
Yarur AJ, Abreu MT, Deshpande AR, Kerman DH, Sussman DA. Therapeutic drug monitoring in patients with inflammatory bowel disease. World J Gastroenterol. 2014;20:3475–3484. | ||
Prieto-Pérez R, Almoguera B, Cabaleiro T, Hakonarson H, Abad-Santos F. Association between genetic polymorphisms and response to anti-TNFs in patients with inflammatory bowel disease. Int J Mol Sci. 2016;17:225. | ||
Hlavaty T, Pierik M, Henckaerts L, et al. Polymorphisms in apoptosis genes predict response to infliximab therapy in luminal and fistulizing Crohn’s disease. Aliment Pharmacol Ther. 2005;22:613–626. | ||
Moroi R, Endo K, Kinouchi Y, et al. FCGR3A-158 polymorphism influences the biological response to infliximab in Crohn’s disease through affecting the ADCC activity. Immunogenetics. 2013;65:265–271. | ||
Koder S, Repnik K, Ferkolj I, et al. Genetic polymorphism in ATG16l1 gene influences the response to adalimumab in Crohn’s disease patients. Pharmacogenomics. 2015;16:191–204. | ||
Jürgens M, Laubender RP, Hartl F, et al. Disease activity, ANCA, and IL23R genotype status determine early response to infliximab in patients with ulcerative colitis. Am J Gastroenterol. 2010;105:1811–1819. | ||
Bank S, Andersen PS, Burisch J, et al. Associations between functional polymorphisms in the NFB signaling pathway and response to anti-TNF treatment in Danish patients with inflammatory bowel disease. Pharmacogenom J. 2014;14:526–534. | ||
Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2013;52:65–70. | ||
Afif W, Loftus EV Jr, Faubion WA, et al. Clinical utility of measuring infliximab and human anti-chimeric antibody concentrations in patients with inflammatory bowel disease. Am J Gastroenterol. 2010;105:1133–1139. | ||
Bortlik M, Duricova D, Malickova K, et al. Infliximab trough levels may predict sustained response to infliximab in patients with Crohn’s disease. J Crohns Colitis. 2013;7:736–743. | ||
Van Moerkercke W, Ackaert C, Compernolle G, et al. High infliximab trough levels are associated with mucosal healing in Crohn’s disease. Gastroenterology. 2010;138:S60. | ||
Baert F, Noman M, Vermeire S, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N Engl J Med. 2003;348:601–608. | ||
Roblin X, Marotte H, Rinaudo M, et al. Association between pharmacokinetics of adalimumab and mucosal healing in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2014;12:80–84. | ||
Yarur AJ, Jain A, Hauenstein SI, et al. Higher adalimumab levels are associated with histologic and endoscopic remission in patients with Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2016;22:409–415. | ||
Seow CH, Newman A, Irwin SP, Steinhart AH, Silverberg MS, Greenberg GR. Trough serum infliximab: a predictive factor of clinical outcome for infliximab treatment in acute ulcerative colitis. Gut. 2010;59:49–54. | ||
Karmiris K, Paintaud G, Noman M, et al. Influence of trough serum levels and immunogenicity on long-term outcome of adalimumab therapy in Crohn’s disease. Gastroenterology. 2009;137:1628–1640. | ||
Nanda KS, Cheifetz AS, Moss AC. Impact of antibodies to infliximab on clinical outcomes and serum infliximab levels in patients with inflammatory bowel disease (IBD): a meta-analysis. Am J Gastroenterol. 2013;108:40–47. | ||
Velayos FS, Sheibani S, Lockton S, et al. Prevalence of antibodies to adalimumab (ATA) and correlation between ATA and low serum drug concentration on CRP and clinical symptoms in a prospective sample of IBD patients. Gastroenterology. 2013;144:S91. | ||
Tew GW, Hackney JA, Gibbons D, et al. Association between response to etrolizumab and expression of integrin αE and granzyme A in colon biopsies of patients with ulcerative colitis. Gastroenterology. 2016;150:477–487. | ||
Swidsinski A, Ladhoff A, Pernthaler A, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology. 2002;122:44–54. | ||
Philpott DJ, Girardin SE. Crohn’s disease-associated Nod2 mutants reduce IL10 transcription. Nat Immunol. 2009;10:455–457. | ||
Frank DN, Robertson CE, Hamm CM, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:179–184. | ||
Rausch P, Rehman A, Kunzel S, et al. Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (secretor) genotype. Proc Natl Acad Sci U S A. 2011;108:19030–19035. | ||
Lupp C, Robertson ML, Wickham ME, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:119–129. | ||
Darfeuille-Michaud A, Boudeau J, Bulois P, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127:412–421. | ||
Sokol H, Lepage P, Seksik P, Dore J, Marteau P. Temperature gradient gel electrophoresis of fecal 16S rRNA reveals active Escherichia coli in the microbiota of patients with ulcerative colitis. J Clin Microbiol. 2006;44:3172–3177. | ||
Ohkusa T, Sato N, Ogihara T, Morita K, Ogawa M, Okayasu I. Fusobacterium varium localized in the colonic mucosa of patients with ulcerative colitis stimulates species-specific antibody. J Gastroenterol Hepatol. 2002;17:849–853. | ||
Ohkusa T, Yoshida T, Sato N, Watanabe S, Tajiri H, Okayasu I. Commensal bacteria can enter colonic epithelial cells and induce proinflammatory cytokine secretion: a possible pathogenic mechanism of ulcerative colitis. J Med Microbiol. 2009;58:535–545. | ||
Atarashi K, Tanoue T, Oshima K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. | ||
Llopis M, Antolin M, Carol M, et al. Lactobacillus casei downregulates commensals’ inflammatory signals in Crohn’s disease mucosa. Inflamm Bowel Dis. 2009;15:275–283. | ||
Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008; 105:16731–16736. | ||
Willing B, Halfvarson J, Dicksved J, et al. Twin studies reveal specific imbalances in the mucosa associated microbiota of patients with ileal Crohn’s disease. Inflamm Bowel Dis. 2009;15:653–660. | ||
Varela E, Manichanh C, Gallart M, et al. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment Pharmacol Ther. 2013;38:151–161. | ||
Pascal V, Pozuelo M, Borruel N, et al. A microbial signature for Crohn’s disease. Gut. 2017;66(5):813–822. | ||
Moayyedi P, Surette MG, Kim PT, et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology. 2015;1:102–109. | ||
Rossen NG, Fuentes S, van der Spek MJ, et al. Findings from a randomized controlled trial of fecal transplantation for patients with ulcerative colitis. Gastroenterology. 2015;1:110–118. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.