Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 10
Pharmacogenetic testing revisited: 5′nuclease real-time polymerase chain reaction test panels for genotyping CYP2D6 and CYP2C19
Authors Larsen JB, Rasmussen JB
Received 4 January 2017
Accepted for publication 2 February 2017
Published 18 April 2017 Volume 2017:10 Pages 115—128
DOI https://doi.org/10.2147/PGPM.S131580
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin Bluth
Jens Borggaard Larsen, Jan Borg Rasmussen
Laboratory Unit, Danish Epilepsy Centre, Filadelfia, Dianalund, Denmark
Abstract: Due to their involvement in the metabolization of commonly prescribed psychopharmaceutical drugs, the cytochrome oxidase genes CYP2D6 and CYP2C19 are extensive targets for pharmacogenetic testing. The existence of common allelic variants allows the prediction of a metabolic phenotype based on a genotype result, hereby supplying a clinical tool for optimizing prescription and minimizing adverse effects. In this study, we present the development of two 5′ nuclease real-time polymerase chain reaction (PCR) test panels, capable of detecting eight of the most clinically relevant alleles of the CYP2D6 gene (*2, *3, *4, *6, *9, *10, 17, *41) and the three most common nonfunctional alleles of CYP2C19 (*2, *3, *4). The assays have been thoroughly validated using a large collection of reference samples, by parallel testing and by DNA sequencing. The reanalysis of reference samples provided the calculation of the frequency of the CYP2D6*4K allele in a population, not previously reported. Furthermore, original test results from CYP2D6*41, generated based on the presence of the 2850T and the lack of the −1584G single-nucleotide polymorphism (SNP), were compared with genotyping based on the current acknowledged founder SNP 2988G of this allele. These results indicate that up to 17.7% of the patients originally tested as carriers of the CYP2D6*41 allele may have had an incorrect phenotypic result assigned. The two 5′ nuclease real-time PCR test panels have subsequently been optimized for use in the clinical laboratory, using a standard real-time PCR instrument and software.
Keywords: genotyping, 5′ nuclease assay, drug metabolization, CYP2C19, CYP2D6, pharmacogenetics
Introduction
Interindividual variation in drug response is a common problem in the therapy of psychiatric patients, leading to nonoptimal treatments and potentially serious adverse effects.1,2 Together with noncompliance, this has a significant impact on the clinical success and hereby also carries social–economic importance.3–5 This discrepancy is primarily caused by biological variations in uptake, metabolization, and excretion rates, which leads to differences between patients despite having the same or comparable dosages administrated.6 Traditionally, in vivo phenotyping of a patient’s drug metabolization capability has been accomplished by administration of selective enzyme substrates, followed by quantification of their metabolic ratio in a matrix like plasma or urine.6,7 Although such methods hold the benefit of taking all the biological factors into account, hereby giving a direct quantitative measure as the phenotypic result, suitable and specific substrates are not available for all enzyme/protein systems of interest.6 Also, these methods demand a specifically dedicated clinical setting for their application.
In the last 25 years, the realization that a patient’s phenotype for metabolizing a drug can be linked to the genotype of specific enzymes or transporter proteins has led to the development of a range of molecular tests.8–12 The hypothesis behind these tests is the prediction of a metabolic phenotype based on the genotype, with the overall goal leading to personalized medicine – medicine prescribed based on the genetic information of an individual person.13,14 The genes that have received the most attention in this context are those of the cytochrome P450 oxidase family (CYP genes).15,16 These genes primarily code for liver enzymes involved in the metabolization and excretion of a wide range of commonly prescribed drugs. Genetic mutations causing decreased or impaired activity of the mature enzyme have been identified and correlated with increased plasma levels of the substrate and adverse effects in patients.13,17–20 Genetic variations have also been identified causing increased production of an enzyme, and hereby increased metabolism and higher therapeutic levels of prescribed drugs.21,22
The most extensively tested cytochrome oxidase genes for clinical purposes are CYP2D6 and CYP2C19.4,23,24 Both of these genes have nonfunctional variants at a relatively high frequency, causing abolished or decreased metabolization of drugs by the mature enzyme.25,26 However, while less than a handful of alleles account for almost all poor metabolizers of the CYP2C19 enzyme, genotyping of CYP2D6 is much more complex. More than 100 alleles have been identified for this gene.27 Besides a high number of nonfunctional alleles, an intermediate metabolizer (IM) phenotype also exists having decreased activity compared to the wildtype *1.26 Genotyping of CYP2D6 is further complicated by the existence of pseudogenes (highly homolog genes at the DNA sequence level), the existence of allelic variants generated by hybrid crossovers between CYP2D6 and CYP2D7, and by duplication of the gene in some individuals (copy number variations).21,28 Because of this, aside from cloning and sequencing that are too expensive for routine use, there is no single molecular method capable of genotyping all identified alleles of the CYP2D6 and CYP2C19 genes.26 Current genotyping techniques therefore focus on the most common clinically relevant alleles. For CYP2C19, these are the nonfunctional alleles *2, *3, and *4, while for CYP2D6, these include the nonfunctional alleles *3, *4, *5 (gene deletion), and *6, as well as alleles *9, *10, *17, and *41 with decreased activity.29,30 Due to the presence of pseudogenes, the genotyping techniques commonly rely on one of the two approaches: direct detection where the primers in a single polymerase chain reaction (PCR) assay ensure the specificity to the intended target, or the secondary approach where a primary PCR selectively amplifies the gene/region of interest prior to the actual genotyping assay.31 The latter method is frequently applied in commercial DNA chip/bead chip-based methods and allows discrimination between higher numbers of alleles.12,32,33 It ensures less bias generated due to low-quality primers but comes at a higher cost of reagents and specialized equipment, as well as workload in the laboratory.31 In contrast, direct detection techniques are generally faster, omitting the secondary PCR as well as potential, purification and hybridization steps.
A large range of chemistries have been developed for genotyping both CYP2C19 and CYP2D6 using the direct genotyping approach.34,35 These include probe-based methods (SimpleProbes, HybProbes, TaqMan 5′ nuclease9,36,37), enzymatic digestion (restriction fragment length polymorphism, RFLP), and high-resolution melting (HRM).38,39 The quality of all these assays relies on the design of PCR primers specific to the intended target.26 Given the restriction inferred by the homology to pseudogenes, both primers developed in-house and commercially available ones must be presumed to target the same nucleotide sequences. Therefore, although specific patented techniques or chemistries might apply, the underlying basic design of the PCR primers still governs the quality of the genotyping assay and is the same for published as well as commercial tests. A consequence of this is that with correct validation and optimization, noncommercial assays can significantly decrease the total cost of reagents and hereby the price of genetic tests.38
Here, we present two 5′ nuclease real-time PCR test panels, capable of identifying eight of the most common CYP2D6 alleles and three of the most common alleles of the CYP2C19 gene. The panels have been thoroughly validated and optimized for routine testing. Furthermore, the applied dyes should be compatible with most real-time PCR platforms currently available on the market.
Method
Background
Since 2002, genotyping of CYP2D6 and CYP2C19 has been provided as a service by the Laboratory Unit of the Danish Epilepsy Centre. Besides pharmacogenetic tests, the laboratory is also a regional center of Therapeutic Drug Monitoring (TDM). The annual volume of ~300 genetic samples may however still be considered small due to the demographic size of the country. The laboratory participates in a proficiency scheme for CYP2D6 and CYP2C19 (https://www.rfb.bio), and in addition maintains a program with Diakonhjemmet Sykehus, Oslo, Norway, where every 6 months, the laboratory exchanges 2×2 samples for confirmation of each other’s test performances. Over the years, the applied molecular genotyping techniques used at the laboratory have progressed from primarily RFLP methods using gel electrophoresis to real-time PCR technologies. The original RFLP tests included CYP2D6 alleles *2, *3, *4, *5, *6, *7, *8, *9, *10, *11, *12, *17, and *41, as well as the deletion of the alleles *5, *13, *16 and duplication of the alleles *1, *2, *4, and *41.37 The CYP2C19 test consisted of alleles *2 and *3. Due to the labor involved in running the initial CYP2D6 PCR-RFLP genotyping setup, the analysis was performed by selectively excluding individual alleles in a branch-like fashion, based on the presence of common single-nucleotide polymorphisms (SNPs) found in a group of alleles.24,36 More recently, testing has been done using real-time PCR methods, based on the LightCycler hybridization probes and simple probe technology.9,35,36
Reference material
As reference DNA for validation of the 5′ nuclease assays, original test material submitted to the laboratory was used. Upon retrieval, the samples were anonymized and given a laboratory identification number. DNA extraction was performed using the MagnaPure system (Roche Diagnostics, Basel, Switzerland) applying 200 µL of full blood and eluting with 100 µL buffer, resulting in an average yield of 31.1 ng/µL (±9.8 standard deviation) of genomic DNA. Following testing by the original method, extracted DNA was stored at −20°C for up to 5 years prior to its use in the validation of the 5′ nuclease assays.
Assay design
Two alignments of consensus sequences, one of the CYP2D6 and its pseudogenes CYP2D7 and 2D8, and one of the CYP2C19 with the homolog sequence of CYP2C9, were generated (Supplementary materials). The alignments were used to design specific PCR primers and 5′ nuclease probes targeting eight characteristic SNPs/mutations of the CYP2D6 gene (alleles *2, *3, *4, *6, *9, *10, *17, *41; Table 1) and three of CYP2C19 (alleles *2, *3, *4). Primers were placed into the alignment by visual inspection and located so that they covered heterolog bases between target and pseudogenes (Supplementary materials). Standard primer and probe design rules were used to ensure their quality.40 These included a theoretical annealing temperature of 57°C–63°C, 18–25 nt size, and 3′ destabilization by minimizing the number of C′ and G′ in the last five bases. If possible, bases heterolog to the pseudogene sequences were placed in the 3′ end of the primers, and the amplicon size was minimized.
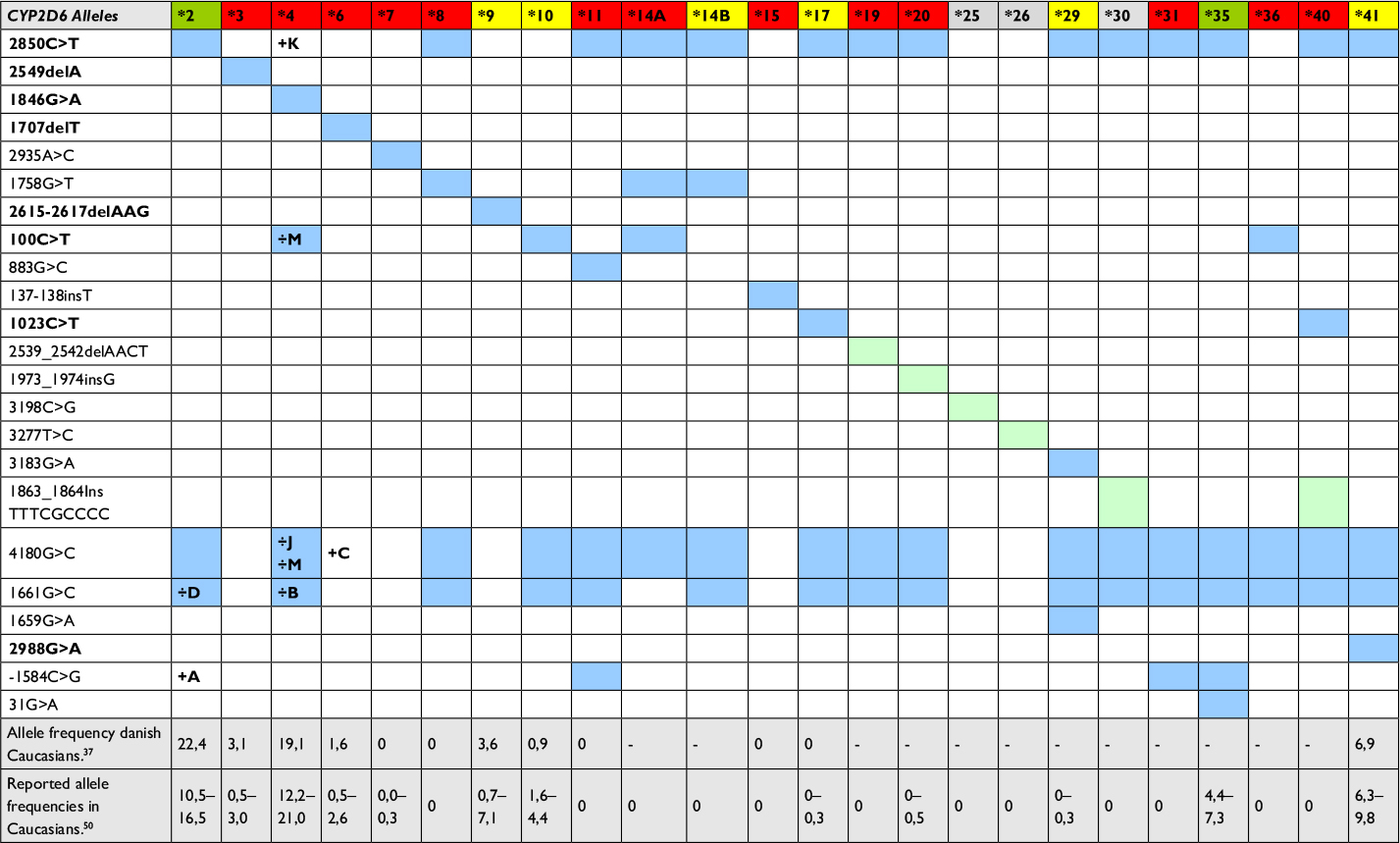 | Table 1 CYP2D6 alleles identified by the 5’nuclease assays compared to those of the Luminex XTag CYP2D6 v.3 and Roche AmpliChip P450 kit Notes: Comparison between SNPs/mutations detected by the 5′ nuclease assays, the AmpliChip P450, and Luminex XTag CYP2D6 kit. Top row shows the list of CYP2D6 alleles identified by the Roche AmpliChip Cytochrome P450 kit (excluding duplications), with their enzymatic activity indicated by color codes: green – normal, yellow – decreased, red – none, and gray – activity unknown. Proposed SNPs/mutations used for the identification of all of the alleles identified by the AmpliChip method are listed on the left side of the table, with the ones included in the 5′ nuclease assay highlighted in bold. The presence of each SNP/mutation in alleles is marked by colored squares. Light green indicate mutations only identified by the AmpliChip kit, while blue indicate mutations detected by both the AmpliChip and the Luminex XTag kit. Subtype of an allele that does not contain a given mutation is marked with a “÷” and their letter (e.g., the CYP2D6*2D is lacking the 1661G>C SNP reported in other CYP2D6*2 alleles). (http://www.cypalleles.ki.se),59 Rasmussen et al,37 and Dodgen et al.50 Abbreviation: SNP, single-nucleotide polymorphism |
Probes were designed with a theoretical annealing temperature of 8–12°C higher than that of the primers, and with the SNP/mutation located in the center region. A G′ immediately adjacent to the 5′ labeling was avoided, and the strand containing the most C′ in the probe sequence was preferred. Wildtype and mutant probes were further designed, so the maximum difference in the theoretical annealing temperature between them was <1°C.
Before ordering, each primer and probe sequence was validated in silico using the two online software tools Netprimer (Premier Biosoft; http://www.premierbiosoft.com/netprimer/) and FastPCR (primerdigital.com).40 The specificity of primers and probes to their intended targets was confirmed by Primer-Blast search (National Center for Biotechnology Information [NCBI]; www.ncbi.nlm.nih.gov/tools/primer-blast/). Probes specific for the wildtype sequence were ordered labeled 5′ with 6-carboxyfluorescein (FAM) as dye and 3′ with black hole quencher-1 (BHQ-1) as quencher, while the mutant probe was labeled 5′ with Cal Fluor 540 (a tetrachlorofluorescein [TET] analog; Biosearch, Novato, CA, USA).
Assay optimization
Primer and probe concentrations for all allele-discriminating assays were optimized using a standard PCR setup on a Bio-Rad CFX connect real-time PCR instrument (Bio-Rad Laboratories, Hercules, CA, USA). The program consisted of 3 minutes of polymerase activation at 95°C, followed by 45 cycles of a collective annealing and elongation step at 60°C for 1 minute (30 seconds for the CYP2C19 assays), and denaturization at 95°C for 15 seconds. Reactions were performed in a 12 µL volume, consisting of 6 µL Bio-Rad ITaq 2× Universal Probe master mix and 2 µL genomic DNA. For optimization of the primer concentration, a titration series of each pair was prepared going from 200 to 600 nM, with 150 nM of each of the two probes added, and using a heterozygotic sample as template DNA. Optimal concentrations of primers were selected based on the efficiency of the real-time PCR amplification. Presence of unspecific amplification product and primer dimers was investigated by melt curve analysis, using Bio-Rad’s High Precision Melting supermix at the same PCR conditions and primer concentrations but without probes added to the reaction mix (Supplementary materials).
Next, the concentration ratio between the wildtype and mutant probe was optimized for each of the assays. Depending on the observed efficiency at the initial PCR setup with 150 nM of each of the probes, the ratio between the two was varied to minimize differences in the fluorescence signal. This was done by lowering the concentration of the more efficient of the two, while increasing or maintaining the concentration of the inferior probe. For each ratio tested, optimization was performed on template DNA from a wildtype homozygote, a heterozygote, and when available a mutant homozygote sample. The probe concentration ratio with the best discrimination capability between each of the tree sample types was selected (Supplementary materials).
Validation of the assays
Validation of each assay was done separately on reference material previously tested in the laboratory using the validated RFLP or real-time PCR methods.9,36,37,41 For each of the 11 assays, 48 genotyped samples were retested: 16 wildtype, 16 heterozygotes, and 16 homozygotes for the mutant allele. For rare alleles, replicates of the same heterozygote and/or mutant homozygote sample were used. This was the case for CYP2D6 alleles *3, *6, *9, and *17 and CYP2C19 alleles *3 and *4. Also, since insufficient samples with a heterozygote or homozygote genotype for CYP2D6*10 were available, samples previously tested as carriers of allele *4 were used for validation of the assay targeting the 100C>T transition (Table 2).27
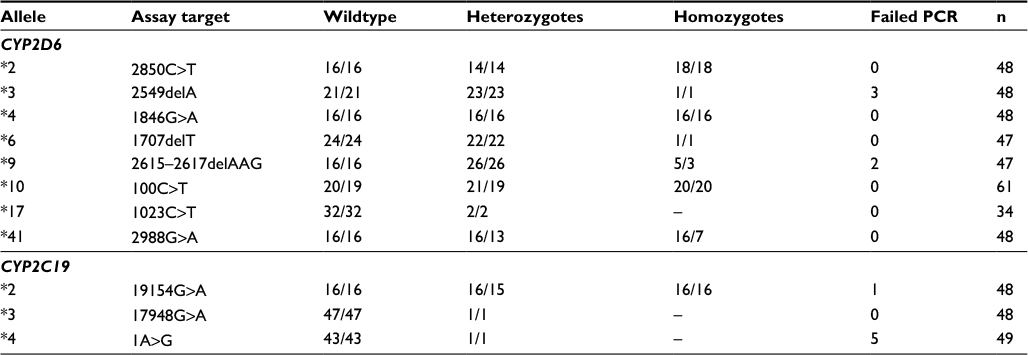 | Table 2 Validation of 5′ nuclease test panels Notes: This table presents the number of samples tested and the result of the individual validation of each of the 11 5′ nuclease assays. Figures to the left are the number of samples used for validating the assay, and right are the number of samples in agreement between the original test and the 5′ nuclease assay. Abbreviation: PCR, polymerase chain reaction. |
Due to the recent finding of the 2988G>A transition in CYP2D6*41 and its classification as the founder SNP of this allele,42,43 all archival samples previously designated as carriers of the *41 allele (n=97) were retested using the 5′ nuclease assay. Tests differing from that of the original RFLP assay were validated by parallel testing with Diakonhjemmet Sykehus, Oslo, Norway. Additionally, sequencing of ambiguous samples was performed at Roskilde University Hospital using a standard Sanger sequencing technique (BigDye 3.1; Thermo Fisher Scientific, Waltham, MA, USA).
Results
The high sequence homology between the two genes targeted for genotyping in this paper, and their respective pseudogenes (CYP2D7/CYP2D8 in the case of CYP2D6 and CYP2C9 in the case of CYP2C19), makes the design of specific PCR primers difficult.26,28,44 This is especially true for CYP2D6 where the high number of polymorphisms further puts restrictions on their location in the sequence.26 For the validation of the 11 assays, a total of 541 retests were performed. Only one sample did not comply with the outcome of the original test result or could not be verified by parallel testing, while another 11 samples failed to amplify, most likely due to degradation of DNA during storage.
CYP2C19 assays
Although it encodes a protein similar in size, the gene of CYP2C19 is >80,000 nt longer than that of CYP2D6. Also, while CYP2D6 has two pseudogenes, only CYP2C9 shows strong sequence resemblance to CYP2C19.45 This, together with significantly fewer clinically relevant alleles, makes the design of primers and probes generally easier for this gene, compared to CYP2D6.27
The CYP2C19 allele *2 is the most common nonfunctional variant found in Caucasians.25 The founder SNP is the 19154G>A transition located outside exon 5, which causes a splicing defect of the transcript and hereby abolishment of enzyme activity in the protein. A second SNP, 12122G>A, is however also conserved between all nine subtypes currently listed.27 The primers designed for genotyping CYP2C19*2 span a 211 bp region (Table 3). Specificity is achieved by a two-base difference to the CYP2C9 sequence in both the forward and reverse primer, of which one base is located within the last 5 nt in the 3′ end. In addition, the probes targeting the 19154G>A SNP have a three-base heterogeneity to CYP2C9.
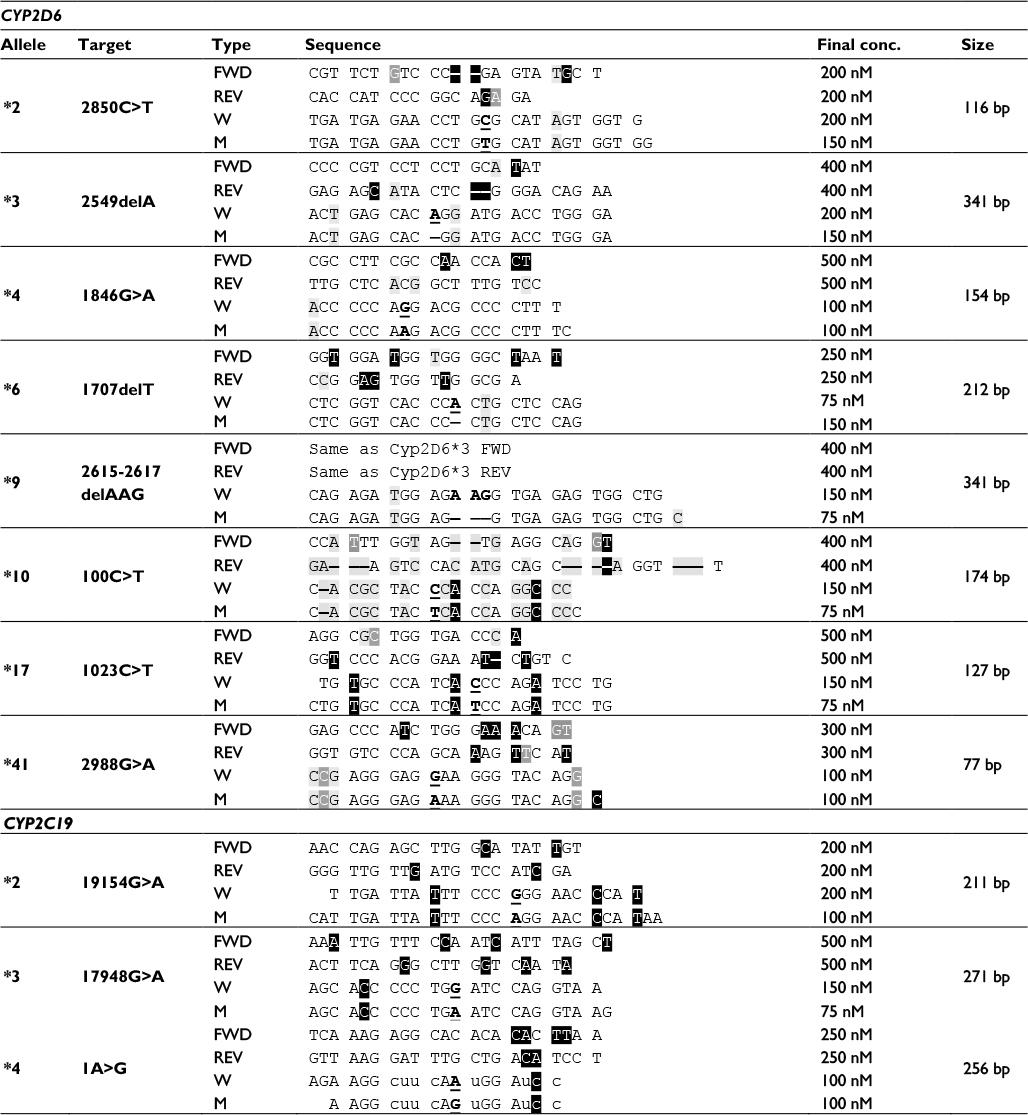 | Table 3 Specificity of primer and probe sequences with optimized reaction concentrations Notes: Specificity of sequences and optimized concentrations of primers and probes for each of the 11 assays presented in this paper. Wildtype probes were labeled 5′ with FAM and 3′ with BHQ-1, while mutant probes were labeled 5′ with CalFlour 540 (a TET analog), and 3′ with BHQ-1. The probes used for CYP2C19*4 were synthesized using phosphorothioate bases (u, c). Amplicon size generated by the primers is shown on the right. Sequence color codes: light gray – CYP2D6 nucleotides deviating from CYP2D8; medium gray – CYP2D6 nucleotides deviating from CYP2D7; reverse black – bases deviating from both CYP2D7 and CYP2D8, including inserted bases (CYP2C9 for the CYP2C19 assays). Underlined bold indicates target SNPs/mutations in either the wildtype or the mutant probe. - indicates one-base insert present in pseudogene or mutant target. Abbreviations: FAM, carboxyfluorescein; BHQ-1; black hole quencher-1; TET, tetrachlorofluorescein; SNP, single-nucleotide polymorphism. |
The CYP2C19*2 5′ nuclease assay was validated on 48 archival samples. These consisted of 16 wildtype homozygotes, 16 heterozygotes, and 16 homozygotes for the mutant SNP 19154A (Table 2). One heterozygote sample failed to amplify, while the result of the remaining 47 samples all complied with that of the original test.
Allele *3 is the second most common nonfunctional allele of CYP2C19. The founder SNP is the 17948G>A that causes premature stop of translation.27 The primers of the 5′ nuclease assay amplify a 271 bp fragment, and each contains four bases different to the corresponding sequence of CYP2C9 (Table 3).
Since it is present at a considerably lower frequency than CYP2C19*2 in Caucasians, only one heterozygote and no homozygote sample were available for validating the allele *3 5′ nuclease assay (Table 2). For this reason, the single heterozygote sample was tested in triplicate during the validation, together with 45 wildtype homozygote samples. All results of the assay conformed to that of the original test.
The nonfunctionality of CYP2C19 allele *4 is due to an A>G transition in the start codon of the gene (1A>G).27 The allele is rare in Caucasians, where it has been reported at a frequency of 0.003 and is neglectable in Africans and Hispanics.25 The PCR designed to flank the SNP generates a 256 bp amplicon. Each of the primers ensures specificity to the CYP2C19 gene based on a two-base heterogeneity, while the forward primer in addition also contains a two-base “-CA-” insert not present in CYP2C9 (Table 3). For the validation of the assay, six replicate tests of a single available heterozygote sample were used, together with 43 wildtype homozygotes (Table 2). Five of the wildtype samples failed to amplify, while all the other acquired results comformed to that of the original tests.
CYP2D6 assays
Identification of CYP2D6*2 was previously performed at the Danish Epilepsy Centre, using a SimpleProbe assay targeting the 2850C>T transition, and further exclusion of alleles *4 and *41 was based on the result of other tests.36,37 This SNP is located in exon 6 of the gene, in a region low on other polymorphisms.27,44 The same region is however well conserved between CYP2D6 and its two pseudogenes CYP2D7 and CYP2D8. For this reason, the forward primer of the 5′ nuclease assay CYP2D6*2 is located immediately upstream of the exon.44 Selective amplification of CYP2D6 by this primer is achieved by spanning a “-CT-” insert that is only present in the sequence of the two pseudogenes (Table 3). Further specificity is obtained by the reverse primer containing one to two bases that are heterolog to the sequence of the CYP2D7 and CYP2D8 gene. These bases are located within the last five bases of its 3′ end. Together, the two primers generate a 116 bp amplicon (Table 3). The immediate area surrounding the 2850C>T SNP also contains a low degree of polymorphism, and no other SNP of frequently occurring alleles of the CYP2D6 gene is found here. The 5′ nuclease assay was validated on previous test material including 16 wildtype homozygote samples, 16 heterozygotes, and 16 homozygotes for the *2 allele (Table 2). Two samples previously tested as heterozygotes were in the new assay tested as homozygote carriers of 2850T. This is in concordance with the original result with the genotype *2/*41, as both alleles *2 and *41 contain this polymorphism (Table 1).
The exon 5 of CYP2D6, where the allele *3 deletion 2549delA resides, is highly homolog to the sequence of its pseudogene CYP2D7.44 Because of this, locations for placing specific primers are significantly restricted. The forward primer for the 5′ nuclease CYP2D6*3 assay is located 67 bp upstream of the probe, providing specificity to the CYP2D6 gene sequence by a T>C difference to CYP2D7 within the last five bases of the 3′ end (Table 3). The reverse primer is located outside of exon 5, covering a sequence where CYP2D7 and CYP2D8 have a “-CT-” insert not present in the wildtype CYP2D6 gene. Together, the primers generate a 341 bp amplicon.
Although allele *3 is one of the most frequently found nonfunctional alleles of the CYP2D6 gene, homozygote carriers are rare.37,46 Only a single sample containing *3 in combination with a deleted CYP2D6 gene (allele *5) was therefore available for the validation of the 5′ nuclease assay (Table 2). The *3/*5 genotype results in a similar signal as a homozygote *3/*3 in the allele discrimination plot. For validating the 5′ nuclease assay, the single sample was therefore used as a homozygote mutant, together with 24 wildtype homozygotes and 23 heterozygotes for *3 allele. Of the total 48 samples used for validating the CYP2D6*3 assay, three failed to amplify, while the result of the remaining 45 conformed to the original test results (Table 2).
The 1846A SNP used for identifying the CYP2D6 allele *4 is located in exon 4 of the gene.44 The region has a high homology to the gene sequence of CYP2D7 and to a less extent to CYP2D8. Because of this, and to minimize the amplicon length, the specificity of the 5′ nuclease to the CYP2D6 gene sequence is achieved solely based on a three-base heterology between the forward primer and the homolog sequence of the CYP2D7 pseudogene (Table 3). Two of these bases are located in the immediate 3′ end of the primer, ensuring the specific amplification of the 154 bp product.
The 5′ nuclease assay for CYP2D6 allele *4 was validated on 16 samples homozygote for the 1846G (wildtype), 16 heterozygotes, and 16 samples homozygote for the mutant 1846A. All test results conformed to those of the original reports, and no samples failed to amplify in this assay (Table 2).
The immediate area (~100 bp upstream and ~40 bp downstream) surrounding the CYP2D6 allele *6 founder mutation 1707delT in exon 3 is highly conserved between the CYP2D6 and CYP2D7 gene.44 For this reason, the forward primer of the 5′ nuclease assay is located 131 bp upstream from the probe site, while the reverse primer is located 25 bases downstream. The latter primer covers the same sequence as that used for the forward primer of the allele CYP2D6*4 assay and also includes the *8 1758G>T in the 5′ end of its sequence.27 Similarly, the probe sequence of the assay includes the 1716G>A of alleles *45 and *46.27 The product generated using the two primers is 212 bp long (Table 3).
Although, allele *6 is found at a frequency of ~0.6 in Caucasians, homozygote carriers are rare.25,37,47 The only such sample available for validation of the 5′ nuclease assay was a *5/*6x2. This sample generates a signal that is similar to that of a *6/*6 genotype. When validating the assay, the *5/*6x2 sample was included in replicates together with 22 heterozygote samples and 24 previously tested as homozygote of the wildtype sequence. All results of the 5′ nuclease assay were consistent with that of the original tests (Table 2).
Due to the high homology between the sequence of CYP2D6 and that of CYP2D7, the 5′ nuclease assay for alleles *3 and *9 shares the same set of PCR primers (Table 3).44 For the verification of the 5′ nuclease assay of allele *9, five archival samples previously tested as either *5/*9 or *9/*9 were available. These samples were used (one in replicate), together with 26 heterozygotes and 16 wildtype homozygotes. Of the five homozygote samples, two failed to amplify. The results of all the remaining samples, including both heterozygotes and wildtype homozygotes, were consistent with that of the original test (Table 2).
The 100C>T used to identify CYP2D6*10 is located in exon 1 of the gene. This region also has a high sequence homology to that of CYP2D7 but low compared to CYP2D8.44 Several other mutations defining alleles of the CYP2D6 gene have been identified in this exon. These include the 31G>A of *35, 77G>A of alleles *43 and *46, the 124G>A of allele *12, and the T insertion at 137–138 found in allele *15.27 To avoid coamplification of CYP2D7, the forward primer of the 5′ nuclease assay is located, so it has three bases heterolog to this gene sequence, two of which are located at the 3′ end of the primer (Table 3). The reverse primer is located 134 bases downstream. It further increases the specificity for the CYP2D6 gene, by spanning a sequence containing a “T” insert in the CYP2D7 gene. In addition, the probe sequence also contains two-nucleotide differences to the sequence of this pseudogene (Table 3).
For validation of the CYP2D6*10 assay, 61 archival samples were retested. These consisted of 20 samples homozygote for the 100C, 21 heterozygotes, and additional 20 samples homozygote for the variant 100T. In the latter two groups, samples containing allele *4 were included to increase the total number of tests harboring this SNP.27 One sample originally genotyped as *2/*10 gave a wildtype signal for the 100C>T in the 5′ nuclease assay, while a second came out as heterozygote compared to the initial result that was as a homozygote wildtype (*1/*2). In both cases, the accuracy of the 5′ nuclease assay was confirmed by direct sequencing of the PCR product (data not shown).
The sequence of exon 2 harboring the CYP2D6*17 1023C>T is somewhat less conserved between CYP2D6 and CYP2D7. This, like in the case of allele *2, makes it easier to design a specific set of primers for the assay. The region contains a high number of other SNPs, including the 974C>A found in some forms of allele *4.27 The primers designed for the CYP2D6 allele *17 5′ nuclease assay amplify a 127 bp fragment (Table 3). The forward primer is located 27 bases upstream from the probe and covers the location of the 974C>A transversion. The reverse primer resides 43 bases downstream from the probe. Although the forward primer has a two- to three-base difference to that of the sequence of the two pseudogenes, most of the specificity is achieved by the reverse primer. This primer has a deletion of a C that is present in the sequence of CYP2D7 and CYP2D8, together with three additional heterolog base positions.
As the CYP2D6 allele *17 is rare in Caucasians, only two heterozygote samples and no homozygote mutants were available for the validation of the 5′ nuclease assay.37,47 Because of this, eight replicates of each of the two heterozygotes, together with 32 wildtype homozygote samples for the 1023C>T SNP, were used (Table 2). All retest results performed using the 5′ nuclease assay correlated with that of the previous findings.
The recently discovered 2988G>A SNP of allele *41 is located in the intron, 38 nucleotides upstream from exon 6.42,44 This region has a relatively high degree of sequence heterology between the CYP2D6 and the two pseudogenes, making it a more ideal target for designing a 5′ nuclease assay. As such, the forward and reverse primer is located in close proximity to the probe, generating an amplicon only 77 bp long (Table 3). Validation of the assay was performed using archival material consisting of 16 homozygote *1/*1 samples, 16 samples previously genotyped as heterozygote carriers of the *41 allele, and 16 samples tested as homozygotes. The *1/*1 samples were selected since these neither contain the −1584C>G nor the 2850C>T SNP.27 As such, the 5′ nuclease assay was expected to correlate with the original test result, although the RFLP assay did not target the same SNP as the 5′ nuclease assay does. Genotyping of the samples confirmed this prediction, while three samples of the heterozygote and seven of the homozygote for the 2988G>A SNP did not agree with the original test result of the RFLP (Table 2). The ten samples that differed were further validated by parallel testing at Diakonhjemmet Sykehus, Oslo, Norway, using a commercial TaqMan assay (Thermo Fischer Scientific). This test confirmed the results of the 5′ nuclease assay for all but one. The single sample that did not comply was originally genotyped as *1/*41, while it was typed as a *41/*41 homozygote by the 5′ nuclease assay (Figure 1A). In the allele-discriminating plot of the real-time assay, its signal was located between the other homozygote *41/*41 samples and those of the heterozygotes (Figure 1A). The heterozygosity for the 2988G>A was established by direct sequencing of the PCR product (Figure 1B). However, this method also identified a second SNP. This second SNP at 2980 nt is a heterozygote C>T transition in the sample, positioned at the second base in the probe sequence.
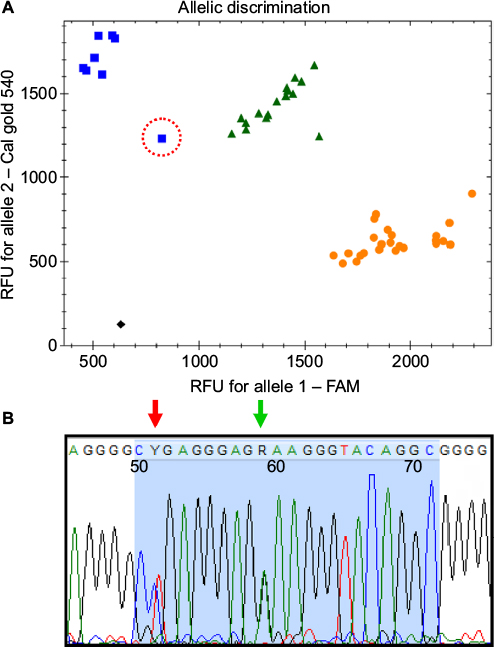 | Figure 1 (A) Allelic discrimination plot of the CYP2D*41 assay, showing the 48 samples used for validating the test; X-axis: FAM fluorescence – wildtype probe; Y-axis: CalFluor gold 540 – mutant probe. Ambiguous sample is marked with a red circle, while black square represents “no template” control (NTC). (B) Sequence of the probe region (blue area) of the ambiguous sample. Green arrow marks the 2988G>A SNP, while the red arrow shows the location of the 2980C>T SNP. Abbreviations: FAM, carboxyfluorescein; SNP, single-nucleotide polymorphism. |
To estimate the numbers of patients incorrectly genotyped with the *41 based on the RFLP assay, all available archival samples containing this allele were retested using the 5′ nuclease assay (Table 4). The result of the 5′ nuclease assay was in agreement with the original test results for 78 of these. As both 2850T and −1584C are also present in allele *4K,47 the remaining 19 unresolved samples that did not comply and which originally had been genotyped as *5/*41, *41/*41, 2×*41/*41, or *17/*41 were also tested using the CYP2D6*4 assay. This was done since the original RFLP setup for genotyping CYP2D6 branched off based on the result of the 2850C>T assay, and therefore, the allele *4K would have been wrongly identified as allele *41.36 Retesting these homozygotes of 2850C>T led to identification of four samples as heterozygote carriers of the 1846G>A SNP and hereby of the CYP2D6*4K allele.
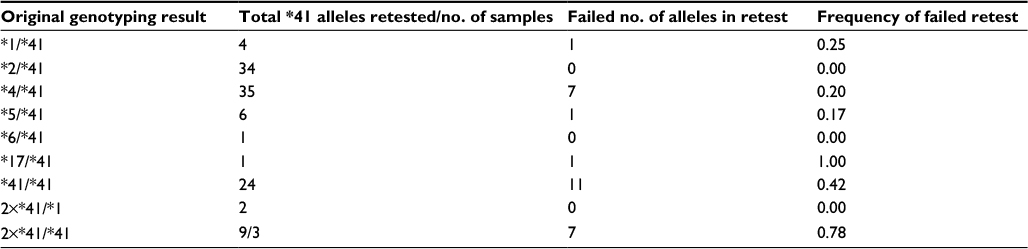 | Table 4 Retest of CYP2D6*41 alleles based on the presence of the 2988G>A SNP Notes: Retesting of samples originally tested as either heterozygote or homozygote carriers of CYP2D6 allele *41, based on the presence of the 2850C>T and lack of the −1584C>G SNP. Failed number of alleles are alleles not found to harbor the 2988G>A SNP in the retest. For samples with copy variations, the number of alleles has been set as either 2 (2×*41/*1) or 3 (2×*41/*41). Abbreviation: SNP, single-nucleotide polymorphism. |
Optimization of PCR setup for routine testing
The CYP2C19 and CYP2D6 test panels have been set up for routine testing in our laboratory. The CYP2D6 panel is used together with the copy number variation test previously published by Schaeffeler et al.41 This test provides identification of the *5 deletion together with copy number variations of the gene. The panels are optimized for a 96-well PCR-plate setup, where each of the CYP2C19/CYP2D6 assays is tested in a dedicated row. Using a primer/probe multimix and reaction mix, a master mix for each assay is prepared in the last cell in the row using 10 µL per reactions. This master mix is then distributed into each well in the same row. The setup includes three controls. A universal *1/*1 wildtype used for all assays (W), a homozygote mutant (M when available, else a heterozygote T), and a no template control. Their locations are fixed in the first three columns. Using repeated dispensing, 2 µL template DNA (~30–100 ng total) is added as a drop onto the sides of each well hereby minimizing the change of tips. The plates are then sealed and briefly centrifuged to mix DNA and mastermix. The whole procedure takes less than 1 hour for a full 96-well plate.
Discussion
Although there is strong scientific evidence linking the genotype of CYP2D6 and CYP2C19 to the metabolic status of their substrate drugs, the clinical application and routine use of these tests are still under debate. The ongoing discussion has been further intensified by the social–economic aspect of having expensive molecular assays, which do not give an unequivocal clinical answer.4 New genotyping tests should therefore preferably increase the precision by including the clinically most relevant alleles of each gene, while lowering the total cost by reducing price of reagents as well as labor.4,48
Following the decision to modernize the genotyping assays at the laboratory of the Danish Epilepsy Centre, several alternative techniques were evaluated before selecting the 5′ nuclease real-time PCR as the platform. Decisions were based upon the speed of setting up assays, total cost of reagents, hands-on time during routine testing, as well as number of recognizable alleles. Several commercial available platforms exist that are capable of discriminating between a high number of CYP2D6 alleles using DNA chip (GenoChip CYP2D6, http://www.pharmgenomics.de; AmpliChip Cyp450 kit, https://molecular.roche.com) or bead chip (Luminex, http://www.luminexcorp.com) technology.32,33,49 These together with HRM and the commercial TaqMan assays (Life Technologies) were considered before the decision was made to design the two test panels presented in this study. Table 1 shows a comparison between the 5′ nuclease panel for CYP2D6, the Luminex XTag CYP2D6 v.3 kit, and the Roche AmpliChip P450 kit.32,49 The two commercial kits are capable of recognizing 16 and 24 different signature SNPs/mutations of CYP2D6 (excluding duplications), compared to the eight identified by the 5′ nuclease assays. However, of the additional 16 alleles that the AmpliChip P450 kit can identify, allele *35 encodes normal enzyme activity, while the activity of *25, *26, and *30 is not known (Table 1). Also, of the nonfunctional alleles identified by this method, *8, *11, *14, *19, *31, and *40 are extremely rare in Caucasians (Table 1),37,46,47,50 while the remaining alleles *7, *15, *20, *29, and *36 have reported frequencies between 0.0 and 0.6, which are also too low to be categorized as polymorphisms according to Cavalli-Sforza and Bodmer.51 Therefore, although both the AmpliChip P450 and the Luminex XTag CYP2D6 kit unarguably deliver a better prediction of the actual genotype by their higher number of detectable SNPs/mutations, the difference is only marginal, if measurable, when predicting the clinically relevant phenotypic level.52
The main advantages of the direct approach for genotyping CYP2D6 are less hands-on time during setup, and that the PCR is performed in a closed system, hereby minimizing the risk of contamination. In contrast, the drawback is the restriction put on the design of the PCR primers, due to homology between the target and pseudogene.44 As a second direct genotyping method, HRM was considered, since several assays for genotyping CYP2D6 and CYP2C19 using this approach have already been published.38,39,53 This method has the benefit of reducing cost and maintenance of assays, since it does not rely on the use of specific fluorescence-labeled oligoprobes.38 While validating the primers designed for the two 5′ nuclease test panels, their suitability for use in HRM assays was also evaluated (data not shown). Most of the primer sets generated robust and consistent amplicons, facilitating genotyping using this method. However, the high degree of polymorphism in CYP2D6, and the tendency of these to cluster around certain regions of the gene, makes the method less suited for routine testing. A good example of this is the primer used for identifying CYP2D6 allele *6, which spans a 212 bp region at exon 3.44 This region also includes the common 1661G>C SNP as well as a number of more rare polymorphisms (e.g., 1704C>G, 1724C>T, 1757C>T, 1758G>A).27 These additional SNPs generate alternating melting curves, which would have to be characterized, and demand additional PCR controls, before robust genotype results can be obtained.53 The only way of reducing these alternating forms would be to minimize the size of the amplicon, centering it in on the 1707delT. This, however, is not possible due to the high homology in this region between CYP2D6 and CYP2D7. Therefore, HRM in our opinion would not in its current stage of development be the method of choice for routine testing of CYP2D6. However, for less polymorph genes, the technique may still be applied. One example where it can prove useful is for the detection of CYP2C19 allele *17. The C>T transition of this allele has an immediate area containing a high number of adenine and thymine bases (data not shown), which would result in a very long probe for the 5′ nuclease assay in order to obtain an optimal annealing temperature. During the current study, we tried without any success to design a 5′ nuclease assay capable of selecting against this SNP and are now pursuing the use of HRM instead.
During their time of storage, some of the reference samples were used as controls for the genotyping tests in the laboratory. In this process, they underwent cycles of repeated thawing and freezing that causes both degradation and fragmentation of DNA. For the validation of the assays, however, samples were not selected based on their DNA concentration nor was this normalized between them. Given that a variation in amplification efficiency hereby is expected, the overall success rate of both amplification and correct calls of genotype might be taken as an indicator of the robustness of the assays. Of the 381 samples used for validating the eight CYP2D6 assays, only five failed to amplify during the initial PCR (Table 2). These were all samples used for validating the CYP2D6*3 and *9 assays. Due to a particular high homology in this region between the gene sequences of CYP2D6 and 2D7, these two tests share a common set of primers that amplify a 341 bp fragment (Supplementary materials). This is a length commonly regarded as suboptimal for 5′ nuclease assays due to both an increased chance of abolishment of elongation by the polymerase and a higher chance of breaks in the DNA template to occur as the length increases. Efforts to optimize the two assays, by reducing the size of the amplified fragment, did not increase the strength of the signal (data not shown). Therefore, although the primed region is not an ideal length for 5′ nuclease genotyping, the proposed design for the two assays seems most optimal in order to avoid a nested PCR.
During the validation of the 11 5′ nuclease assays, only three out of 541 results did not comply with that of the original test. Two of these were samples that were used for validating the CYP2D6*10 assay (Table 2). In both cases, the result of the 5′ nuclease test was confirmed by sequencing the PCR product. In the case of the third sample, this was originally identified as a *1/*41 heterozygote using RFLP, while it was genotyped as a homozygote carrier of the 2988G>A SNP by the 5′ nuclease. Subsequent parallel testing at a second laboratory applying the commercial TaqMan test confirmed the original result of the RFLP assay. The allele-discriminating plot for this sample showed a decreased signal compared to both other homozygotes of the 2988G>A (CalFlour540 signal) and heterozygote samples (FAM signal) (Figure 1A). Sequencing of the amplicon identified a second SNP 2980C>T, located at the 5′ end of the probe. Given that the FAM signal is decreased for this sample (Figure 1B), this indicates that the SNP must interfere with the binding of the wildtype probe on the *1 allele in the sample. Since TaqMan MGB probes bind with higher affinity to their target, they are generally shorter than regular 5′ nuclease probes. This is the most likely reason why the parallel test performed at Diakonhjemmet Sykehus had an increased signal for the wildtype allele of this sample, compared to the 5′ nuclease assay, hereby assigning the correct genotype. To further investigate the occurrence of this novel SNP, a Blast search was performed using the 2980T sequence (https://blast.ncbi.nlm.nih.gov/Blast.cgi). This search did not return any direct hits in the GenBank nonredundant database. Also, this SNP is not included in any currently described alleles listed in the Human Cytochrome P450 allele database.27
In the first characterization of the CYP2D6 allele *41, this variant was recognized based on the presence of the 2850C>T SNP, and the lack of −1584C>G, distinguishing it from allele *2A.54,55 Later studies by Raimundo et al42 and Toscano et al43 have proven that the aberrant splicing of this variant is due to a single G>A transition located at 2988 nt in the gene. The paradigm shift leading to the changed definition of CYP2D6 allele *41 causes previous test results lacking the 2988A but containing the 2850T and −1584G to be reclassified as CYP2D6*2B-M.27,42,43 Therefore, in order to verify the results of the original RFLP assay, all achieved samples previously tested as homozygote or heterozygote carriers of the *41 allele were retested for the presence of the 2988G>A SNP. Of the 97 samples, 39 had in the original result been phenotyped as extensive metabolizers (EMs) having a normal functioning allele (*1 or *2) together with allele *41 (Table 4). The remaining 58 samples were IMs, with the RFLP test showing either a homozygote carrier of allele *41 or in combination with a decreased/nonfunctional allele (*3, *4, *5, *6, or *17). Of the EM group, only one sample did not confer to the original test result, hereby having a failed retest frequency per allele of 1.3% (1/76 alleles). The same calculated frequency for IM heterozygotes of allele *41 was 20.9% (9/43 alleles) and for homozygotes 41.6% (10/24). In 17 cases, retesting for the 2988G>A led to a change in phenotype from IM to EM. In one case, a sample previously tested as a 2×*41/*41 duplicate was by the 5′ nuclease assay genotyped as a 2×*2/*2, resulting in a change of phenotype from IM to ultrafast metabolizer. Given that the AmpliChip Cyp450 test was designed before the assignment of 2988G>A as the founder SNP of allele *41, a similar error rate when predicting the phenotype result is expected for patients genotyped using this system.33
The original RFLP CYP2D6 test used at the Danish Epilepsy Centre divided the alleles into two groups based on the presence of the 2850C>T SNP.36,37 Using this approach, any sample identified as either of the two types of homozygotes in the assay would subsequently only be tested in one of two subcategories. The group consisting of carriers of 2850C would be assayed for the alleles *3, *4, *6, *9, *10, and *13/*16, while the second group homozygote with the mutant 2850T would only be tested for the presence of alleles *2, *8, *11, *12, *17, and *41.24,37 While this setup was designed to facilitate fast clinical results reducing labor and overall cost of a test, one CYP2D6 subtype does not comply with this design. The allele *4K is characterized by harboring the 1846A founder SNP of CYP2D6 allele *4 as well as the 2850T used for deselecting samples from being tested in the CYP2D6*4 assay.47 The CYP2D6*4K was originally described by Sachse et al47 denoted as allele *4E in the paper; however, to our knowledge, the frequency of this particular allele is not known. As part of the retesting of CYP2D6*41, all original tests reported as *41/*41 homozygotes, which could not be confirmed as carriers of the 2988G>A SNP, were also retested using the CYP2D6 allele *4 assay. This identified four samples as *4K, leading to an estimated frequency of 0.4% in 1046 submitted samples. In all four samples, this caused a change of phenotype from IM to EM, as neither of these contained the 2988G>A transition.
Following validation of the CYP2D6*17 assay and setup of the test panel in the routine laboratory, a decreased signal for this assay was observed for samples tested as homozygotes for the *4 allele. Investigations showed this to be caused by the presence of the 974A on both alleles, which destabilizes the forward primer leading to reduced signal.27 For one rare sample identified as *4/*17, this caused a contradictive result as a homozygote (although with decreased signal) of the 1023C>T SNP, while correctly being genotyped as heterozygote for the 1846G>A. A similar ambiguous result was recently reported for the commercial TaqMan CYP2D6 assay (Life Technologies), although here, the mechanism behind could not be elucidated.56 Together with the report on ambiguous samples of CYP2D6*41 when comparing the Genochip and AmpliChip (in addition to other reports of failed test using commercial platforms), this highlights the complexities involved in genotyping the CYP2D6 gene and the need for expert knowledge on the performance of the assay in use.33,48,57,58 In our experience, this is best obtained using an open-source system, as it allows ambiguous or “no call” results to be resolved based on known primer and/or probe sequences. In the case of the CYP2D6*17 forward primer, its performance on a *4 background may be increased by using a degenerate base or including inosine at this position in the primer.
In conclusion, we have designed and thoroughly validated two test panels for genotyping CYP2D6 and CYP2C19. The panels have been optimized for routine use in the laboratory, providing a cost- and time-efficient method that we believe is comparable or superior to most commercial kits.
Acknowledgments
The authors thank method specialist Linda Hårstad Uthus from Centre of Psycofarmacology, Diakonhjemmet Sykehus, Oslo, Norway, for providing additional sample material and performing parallel testing. They would also like to thank Peter Bøhm and Lars Ødum MD at Zealand University Hospital, Roskilde for helping out with sequencing of samples. In addition, they are indebted to Dr. James Kennedy from the Icelandic Marine Research Institute, who helped during the preparation of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Koski A, Ojanperä I, Sistonen J, Vuori E, Sajantila A. A fatal doxepin poisoning associated with a defective CYP2D6 genotype. Am J Forensic Med Pathol. 2007;28(3):259–261. | ||
Ogu CC, Maxa JL. Drug interactions due to cytochrome P450. Proc (Bayl Univ Med Cent). 2000;13(4):421–423. | ||
Pouget JG, Shams TA, Tiwari AK, Müller DJ. Pharmacogenetics and outcome with antipsychotic drugs. Dialogues Clin Neurosci. 2014;16(4):555–566. | ||
Jürgens G, Jacobsen CB, Rasmussen HB, Werge T, Nordentoft M, Andersen SE. Utility and adoption of CYP2D6 and CYP2C19 genotyping and its translation into psychiatric clinical practice. Acta Psychiatr Scand. 2012;125(3):228–237. | ||
Ravyn D, Ravyn V, Lowney R, Nasrallah HA. CYP450 pharmacogenetic treatment strategies for antipsychotics: a review of the evidence. Schizophr Res. 2013;149(1–3):1–14. | ||
Streetman DS, Bertino JS Jr, Nafziger AN. Phenotyping of drug-metabolizing enzymes in adults: a review of in-vivo cytochrome P450 phenotyping probes. Pharmacogenetics. 2000;10(3):187–216. | ||
Jurica J, Bartecek R, Zourkova A, et al. Serum dextromethorphan/dextrorphan metabolic ratio for CYP2D6 phenotyping in clinical practice. J Clin Pharm Ther. 2012;37(4):486–490. | ||
Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76(3):391–396. | ||
Arneth B, Shams M, Hiemke C, Härtter S. Rapid and reliable genotyping procedure for detection of alleles with mutations, deletion, or/and duplication of the CYP2D6 gene. Clin Biochem. 2009;42(12):1282–1290. | ||
Sakuyama K, Sasaki T, Ujiie S, et al. Functional characterization of 17 CYP2D6 allelic variants (CYP2D6.2, 10, 14A–B, 18, 27, 36, 39, 47–51, 53–55, and 57). Drug Metab Dispos. 2008;36(12):2460–2467. | ||
Hosono N, Kato M, Kiyotani K, et al. CYP2D6 genotyping for functional-gene dosage analysis by allele copy number detection. Clin Chem. 2009;55(8):1546–1554. | ||
Ben S, Cooper-DeHoff RM, Flaten HK, et al. Multiplex SNaPshot-a new simple and efficient CYP2D6 and ADRB1 genotyping method. Hum Genomics. 2016;10:11. | ||
McElroy S, Sachse C, Brockmoller J, et al. CYP2D6 genotyping as an alternative to phenotyping for determination of metabolic status in a clinical trial setting. AAPS PharmSci. 2000;2(4):E33. | ||
Caudle KE, Klein TE, Hoffman JM, et al. Incorporation of pharmacogenomics into routine clinical practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr Drug Metab. 2014;15(2):209–217. | ||
Spina E, de Leon J. Clinical applications of CYP genotyping in psychiatry. J Neural Transm (Vienna). 2015;122(1):5–28. | ||
Zhou SF, Liu JP, Chowbay B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab Rev. 2009;41(2):89–295. | ||
Klein K, Tatzel S, Raimundo S, et al. A natural variant of the heme-binding signature (R441C) resulting in complete loss of function of CYP2D6. Drug Metab Dispos. 2007;35(8):1247–1250. | ||
Sachse C, Brockmöller J, Hildebrand M, Müller K, Roots I. Correctness of prediction of the CYP2D6 phenotype confirmed by genotyping 47 intermediate and poor metabolizers of debrisoquine. Pharmacogenetics. 1998;8(2):181–185. | ||
Shams ME, Arneth B, Hiemke C, et al. CYP2D6 polymorphism and clinical effect of the antidepressant venlafaxine. J Clin Pharm Ther. 2006;31(5):493–502. | ||
Steimer W, Zöpf K, von Amelunxen S, et al. Amitriptyline or not, that is the question: pharmacogenetic testing of CYP2D6 and CYP2C19 identifies patients with low or high risk for side effects in amitriptyline therapy. Clin Chem. 2005;51(2):376–385. | ||
Johansson I, Lundqvist E, Bertilsson L, Dahl ML, Sjöqvist F, Ingelman-Sundberg M. Inherited amplification of an active gene in the cytochrome P450 CYP2D locus as a cause of ultrarapid metabolism of debrisoquine. Proc Natl Acad Sci U S A. 1993;90(24):11825–11829. | ||
Ghasemi Z, Hashemi M, Ejabati M, et al. Development of a high-resolution melting analysis method for CYP2C19*17 genotyping in healthy volunteers. Avicenna J Med Biotechnol. 2016;8(4):193–199. | ||
Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005;5(1):6–13. | ||
Dorado P, Cáceres MC, Pozo-Guisado E, Wong ML, Licinio J, Llerena A. Development of a PCR-based strategy for CYP2D6 genotyping including gene multiplication of worldwide potential use. Biotechniques. 2005;39(4):571–574. | ||
Sistonen J, Fuselli S, Palo JU, Chauhan N, Padh H, Sajantila A. Pharmacogenetic variation at CYP2C9, CYP2C19, and CYP2D6 at global and microgeographic scales. Pharmacogenet Genomics. 2009;19(2):170–179. | ||
Gaedigk A. Complexities of CYP2D6 gene analysis and interpretation. Int Rev Psychiatry. 2013;25(5):534–553. | ||
Sim SC, Ingelman-Sundberg M. The Human Cytochrome P450 (CYP) Allele Nomenclature website: a peer-reviewed database of CYP variants and their associated effects. Hum Genomics. 2010;4(4):278–281. | ||
Black JL 3rd, Walker DL, O’Kane DJ, Harmandayan M. Frequency of undetected CYP2D6 hybrid genes in clinical samples: impact on phenotype prediction. Drug Metab Dispos. 2012;40(1):111–119. | ||
Strom CM, Goos D, Crossley B, et al. Testing for variants in CYP2C19: population frequencies and testing experience in a clinical laboratory. Genet Med. 2012;14(1):95–100. | ||
Hicks JK, Swen JJ, Thorn CF, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin Pharmacol Ther. 2013;93(5):402–408. | ||
Wiita A, Schrijver I. Clinical application of high throughput molecular screening techniques for pharmacogenomics. Pharmgenomics Pers Med. 2011;4:109–121. | ||
Rebsamen MC, Desmeules J, Daali Y, et al. The AmpliChip CYP450 test: cytochrome P450 2D6 genotype assessment and phenotype prediction. Pharmacogenomics J. 2009;9(1):34–41. | ||
Bank PC, Swen JJ, Guchelaar HJ, van der Straaten T. GenoChip CYP2D6 macroarray as a method to genotype for CYP2D6 variants: results of a validation study in a Caucasian population. Pharmacogenomics. 2015;16(7):681–687. | ||
Hiratsuka M, Agatsuma Y, Omori F, et al. High throughput detection of drug metabolizing enzyme polymorphisms by allele specific fluorogenic 5′ nuclease chain reaction assay. Biol Pharm Bull. 2000;23(10):1131–1135. | ||
Roijers JF, Jansen-Houtepen L, Jakobs BS, van Wijk EM. Detection of the CYP2D6*6 allele by LightCycler real-time PCR. Ned Tijdschr Klin Chem Labgeneesk. 2007;32(4):270–272. | ||
Nielsen KA, Hansen EL, Gille S. Genotyping of the cytochrome P450 2D6 4469 C>T polymorphism using SimpleProbesÔ. Scand J Clin Lab Invest. 2007;67(3):280–290. | ||
Rasmussen JO, Christensen M, Svendsen JM, Skausig O, Hansen EL, Nielsen KA. CYP2D6 gene test in psychiatric patients and healthy volunteers. Scand J Clin Lab Invest. 2006;66(2):129–136. | ||
Pindurová E, Zourková A, Zrůstová J, Juřica J, Pavelka A. Alternative reliable method for cytochrome P450 2D6 poor metabolizers genotyping. Mol Biotechnol. 2013;53(1):29–40. | ||
Chang YS, Lee CC, Liu TY, Chen YC, Lu HC, Chang JG. Direct assessment of cytochrome P450 2D6 genotypes by high-resolution melting analysis and DNA sequencing. Environ Toxicol Pharmacol. 2014;38(3):821–828. | ||
Kalendar R, Lee D, Schulman AH. FastPCR software for PCR primer and probe design and repeat search. Genes Genomes Genomics. 2009;3(1):1–14. | ||
Schaeffeler E, Schwab M, Eichelbaum M, Zanger UM. CYP2D6 genotyping strategy based on gene copy number determination by TaqMan real-time PCR. Hum Mutat. 2003;22(6):476–485. | ||
Raimundo S, Toscano C, Klein K, et al. A novel intronic mutation, 2988G>A, with high predictivity for impaired function of cytochrome P450 2D6 in white subjects. Clin Pharmacol Ther. 2004;76(2):128–138. | ||
Toscano C, Klein K, Blievernicht J, et al. Impaired expression of CYP2D6 in intermediate metabolizers carrying the *41 allele caused by the intronic SNP 2988G>A: evidence for modulation of splicing events. Pharmacogenet Genomics. 2006;16(10):755–766. | ||
Kimura S, Umeno M, Skoda RC, Meyer UA, Gonzalez FJ. The human debrisoquine 4-hydroxylase (CYP2D) locus: sequence and identification of the polymorphic CYP2D6 gene, a related gene, and a pseudogene. Am J Hum Genet. 1989;45(6):889–904. | ||
Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics. 2004;14(1):1–18. | ||
Sistonen J, Sajantila A, Lao O, Corander J, Barbujani G, Fuselli S. CYP2D6 worldwide genetic variation shows high frequency of altered activity variants and no continental structure. Pharmacogenet Genomics. 2007;17(2):93–101. | ||
Sachse C, Brockmöller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet. 1997;60(2):284–295. | ||
Gaedigk A, Garcia-Ribera C, Jeong HE, Shin JG, Hernandez-Sanchez J. Resolution of a clinical AmpliChip CYP450 TestÔ no call: discovery and characterization of novel CYP2D6*1 haplotypes. Pharmacogenomics. 2014;15(9):1175–1184. | ||
Melis R, Lyon E, McMillin GA. Determination of CYP2D6, CYP2C9 and CYP2C19 genotypes with Tag-It mutation detection assays. Expert Rev Mol Diagn. 2006;6(6):811–820. | ||
Dodgen TM, Hochfeld WE, Fickl H, et al. Introduction of the AmpliChip CYP450 Test to a South African cohort: a platform comparative prospective cohort study. BMC Med Genet. 2013;14:20. | ||
Cavalli-Sforza LL, Bodmer WF. The genetics of human populations. In: Livingstone FB, editor. American Journal of Physical Anthropology. Vol 39. San Francisco: W. H. Freeman; 1971:965. | ||
Gaedigk A, Sangkuhl K, Whirl-Carrillo M, Klein T, Leeder JS. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 2017;19(1):69–76. | ||
Lu HC, Chang YS, Chang CC, Lin CH, Chang JG. Developing and evaluating the HRM technique for identifying cytochrome P450 2D6 polymorphisms. J Clin Lab Anal. 2015;29(3):220–225. | ||
Panserat S, Mura C, Gérard N, et al. DNA haplotype-dependent differences in the amino acid sequence of debrisoquine 4-hydroxylase (CYP2D6): evidence for two major allozymes in extensive metabolisers. Hum Genet. 1994;94(4):401–406. | ||
Raimundo S, Fischer J, Eichelbaum M, Griese EU, Schwab M, Zanger UM. Elucidation of the genetic basis of the common “intermediate metabolizer” phenotype for drug oxidation by CYP2D6. Pharmacogenetics. 2000;10(7):577–581. | ||
Gaedigk A, Freeman N, Hartshorne T, et al. SNP genotyping using TaqMan technology: the CYP2D6*17 assay conundrum. Sci Rep. 2015;5:9257. | ||
Gaedigk A, Riffel AK, Berrocal BG, Solaesa VG, Dávila I, Isidoro-García M. Characterization of a complex CYP2D6 genotype that caused an AmpliChip CYP450 Test no-call in the clinical setting. Clin Chem Lab Med. 2014;52(6):799–807. | ||
Pratt VM, Zehnbauer B, Wilson JA, et al. Characterization of 107 genomic DNA reference materials for CYP2D6, CYP2C19, CYP2C9, VKORC1, and UGT1A1: a GeT-RM and Association for Molecular Pathology collaborative project. J Mol Diagn. 2010;12(6):835–846. | ||
The Human Cytochrome P450 (CYP) Allele Nomenclature Database. Available from: http://www.cypalleles.ki.se/cyp2d6.htm. Accessed March 13, 2017. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.