Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
Pharmacogenetic Practice of Anticancer Drugs: Multiple Approaches for an Accurate and Comprehensive Genotyping
Authors Montrasio C ![]() , Cheli S
, Cheli S ![]() , Clementi E
, Clementi E
Received 30 March 2023
Accepted for publication 19 June 2023
Published 27 July 2023 Volume 2023:16 Pages 739—746
DOI https://doi.org/10.2147/PGPM.S412430
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Cristina Montrasio,1 Stefania Cheli,1 Emilio Clementi2,3
1Unit of Clinical Pharmacology, ASST Fatebenefratelli Sacco, L. Sacco University Hospital, Milan, Italy; 2Clinical Pharmacology Unit, Department of Biomedical and Clinical Sciences, L. Sacco University Hospital, Università degli Studi di Milano, Milan, Italy; 3Scientific Institute IRCCS Eugenio Medea, Bosisio Parini, Italy
Correspondence: Cristina Montrasio, Unit of Clinical Pharmacology, Department of Laboratory Medicine, ASST Fatebenefratelli-Sacco, Milan, Italy, Email [email protected]
Abstract: The application of pharmacogenetics in oncology is part of the routine clinical practice. In particular, genotyping of dihydropyrimidine dehydrogenase (DPYD) and UDP-glucuronosyltransferase (UGT1A1) is crucial to manage the treatment of patients taking fluoropyrimidines and irinotecan. The unique approach of our laboratory to the pharmacogenetic diagnostic service in oncology is to combine two real-time PCR methods, LightSNiP assay (TIB MOLBIOL), and more recently FRET (Fluorescent Resonance Energy Transfer) probes technology (Nuclear Laser Medicine), plus TaqMan assay (Thermo Fisher) for the confirmation of the presence of variant alleles on DNA from a second extraction. We found that both the FRET and LightSNiP assays, where detection occurs by melting curve analysis, offer an advantage over the competing TaqMan technology. Whereas unexpected genetic variants may be missed using a mutation-specific TaqMan assay, the information thus obtained can be useful to adjust the therapy in case of unexpected post-treatment toxicity. The combination of TaqMan and FRET assays helped us to achieve more accurate genotyping and a correct result for the patient. The added value of the DPYD FRET assay is the possibility of detecting, with the same amplification profile of the polymorphisms detailed in the guidelines, also the c.2194G>A (*6 rs1801160), cited in the recommendations as a variant to be investigated in case of severe toxicity. Regarding the UGT1A1 (TA)n promoter polymorphism (rs3064744), the distinctive and positive feature of the FRET assay is to allow clearly identifying all those potential variant alleles, including the (TA)5 and (TA)8 alleles, that are frequent in African Americans. Our clinical practice emphasizes the importance of not only rapid and easy-to-use assays, such as the new FRET ones, but also of accurate and comprehensive genotyping for good pharmacogenetic diagnostic activity.
Keywords: real-time PCR, melting curve analysis, DPYD, UGT1A1, pharmacogenetics
A Letter to the Editor has been published for this article.
A Response to Letter by Mrs López López-Cepero has been published for this article.
Introduction
Fluoropyrimidines (5-fluorouracil and its oral pro-drug capecitabine) and irinotecan are anticancer agents widely used to treat solid tumors, such as head and neck, gastrointestinal tract, breast and pancreatic cancers. Due to heterogeneity in drug response and tolerability, up to one-third of exposed patients are at risk of developing severe, life-threatening adverse events, that negatively impact patient safety and quality of life and may compromise drug efficacy.1,2 The increased risk of toxicity is due in part to germline variants in the dihydropyrimidine dehydrogenase (DPYD) and uridine diphosphate-glucuronosyltransferase isoform 1A1 (UGT1A1) genes, encoding the enzymes responsible for the metabolism of fluoropyrimidines and irinotecan, respectively.3,4 The application of pharmacogenetics in oncology, with genotyping of DPYD and UGT1A1, is thus crucial for managing the treatment of patients taking fluoropyrimidines and irinotecan and has become part of routine clinical practice, especially over the last few years, with the diffusion of recommendations on the application of genetic information to patient care for therapeutic optimization.5–8 Although not uniquely, the US Food and Drug Administration and the European Medicines Agency, as well as the major expert groups, ie the Clinical Pharmacogenetics Implementation Consortium (CPIC), the Dutch Pharmacogenetics Working Group (DPWG), the Italian Association of Medical Oncology jointly with the Italian Society of Pharmacology, provide therapeutic recommendations for these gene-drug associations.9–12 Additionally, in April 2020, the EMA recommended DPYD pre-treatment testing in a communication to healthcare professionals.13 Regarding UGT1A1, FDA and EMA do not mandate pre-treatment genotyping, but recommend starting dose reductions for patients homozygous for UGT1A1*28.14,15 Instead, pre-treatment UGT1A1 genotyping is proposed by the AIOM-SIF indications as an option.16
We describe here the unique approach of our laboratory to the pharmacogenetic diagnostic service in oncology, emphasizing the importance of using multiple methods for a more comprehensive genotyping.
Materials and Methods
Patients
This study is based on a retrospective analysis of routine requests for PGx analysis for fluoropyrimidines and irinotecan anticancer drugs carried out as day-by-day diagnostic service by the Unit of Clinical Pharmacology at the Luigi Sacco University Hospital, Milan, Italy, between 2017 and 2021.
The PGx testing panel, based on guidelines published by the CPIC, in collaboration with The Pharmacogenomics Knowledge Base (PharmGKB), as well as regulatory administrations such as FDA and EMA, includes the variants c.1905+1G>A (*2A rs3918290), c.1129–5923C>G (rs75017182), c.1679T>G (*13 rs55886062), c.2846A>T (rs67376798), c.2194G>A (*6 rs1801160), c.496A>G (rs2297595) for the DPYD gene and the UGT1A1*28 allele (rs3064744) for the UGT1A1 gene.
Ethic Statement
This retrospective research was conducted on data collected for clinical purposes. All data used in the study were previously anonymized, according to the requirements set by Italian Data Protection Code (leg. decree 196/2003) and by the General authorizations issued by the Data Protection Authority. Approval by Ethics Committee was unnecessary because, under Italian law, such an approval is required only in the hypothesis of prospective clinical trials on medical products for clinical use (art. 6 and art. 9, leg. decree 211/2003). The study complies with the Declaration of Helsinki; a written informed consent for genetic testing and publication of clinical data for research purposes was provided.
Genotyping
Genomic DNA was isolated from peripheral blood cells using an automatic DNA extraction system (EZ1 Advanced XL System, Qiagen) according to the manufacturer’s instructions. DNA concentration and purity were evaluated by absorbance methodology using NanoDrop 1000 Spectrophotometer V3.7 (Thermo Fisher Scientific). The detection of the polymorphisms was performed by Real-Time PCR methods. From 2021, for the fluoropyrimidines PGx analysis, five SNPs (single nucleotide polymorphisms) of the DPYD gene are genotyped with the DPYD Real-Time CE-IVD kit (Nuclear Laser Medicine, NLM), based on the FRET (Fluorescent Resonance Energy Transfer) probe technology; the DPYD c.496A>G (rs2297595) is genotyped with LightSNiP assay (TIB MOLBIOL). In the previous years, the four polymorphisms of the CPIC guidelines were detected by Multi-SNiP DPYD kit (TIB MOLBIOL) and the other two (c.2194G>A and c.496A>G) with LightSNiP assay (TIB MOLBIOL). The presence of variant alleles is confirmed on DNA from a second extraction with TaqMan Assays® (Thermo Fisher Scientific). From 2021 also UGT1A1*28 (rs3064744) genotyping for the irinotecan PGx analysis is performed with the UGT1A1 Real Time (FRET) CE-IVD kit (NLM) and the homozygosity for the variant allele is confirmed with LightSNiP assay (TIB MOLBIOL). Any discrepancies between the different methods were resolved with Sanger sequencing performed by an external service.
Genotyping performance was estimated through the use, in each analysis, of known-genotype internal quality controls and through participation, once a year, to an External Quality Assessment program, testing the genes and related variants included in our panel.17
Statistical Considerations
The data analyzer used was the management software linked to the Information System Laboratory (DNLab-NoemaLife) that collects, processes and archives the results.
All genotypes were tested for Hardy– Weinberg equilibrium using χ2 test.
Results
Between 2017 and 2021, we carried out 2413 fluoropyrimidines and 1076 irinotecan pharmacogenetic analyses, the increase being due to a growing consciousness of the clinical utility of these tests (Figure 1).
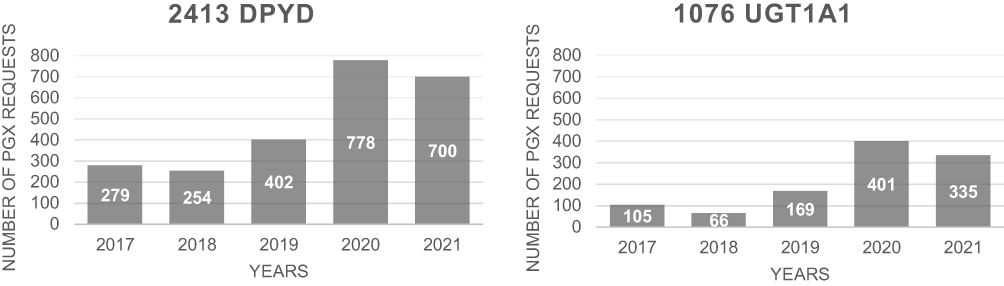 |
Figure 1 Number of requests in the period 2017–2021. |
Allele frequencies were similar to the ones reported for Caucasians on the NCBI SNP database (dbSNP) and the genotype distributions were in Hardy-Weinberg equilibrium (p > 0.05).
DPYD Analysis
Of the 2413 requests collected, 915 had at least one mutated allele (about 38%). Taking into account the four variants of the CPIC guidelines (c.1905+1G>A or *2A, c.1129–5923C>G, c.1679T>G or *13, c.2846A>T), 99 subjects were heterozygous for one of those (4.1%) and 1 was homozygous for the c.2846A>T (0.04%). So about 4% of our population had an intermediate phenotype, that is a partial DPD deficiency. Furthermore, regarding the c.496A>G and the c.2194G>A polymorphisms, we found 776 heterozygous subjects (32.1%) and 39 homozygous subjects (1.6%); in particular 19.6% were heterozygous and 1.1% were homozygous for the c.496, 12.5% were heterozygous and 0.5% were homozygous for the c.2194. About 2% of the subjects were compound heterozygous for the c.496 and the c.2194.
In the FRET assay, as in the LightSNiP one, the SNPs are detected by melting curve analysis, which allows a clear discrimination between normal allele and homozygous/heterozygous variants. Also unusual genotypes, ie with variants other than the ones known, were identified thanks to the analysis of specific melting profiles. We report here some cases. One example refers to a variation near to the c.1679T>G: as shown in Figure 2, the melting profile of the sample B.M. was apparently heterozygous for the variant c.1679T>G, but a TaqMan assay on second extraction did not confirm the presence of the searched polymorphism. Then, Sanger sequencing was performed to understand this discrepancy and the sequence showed a T > G substitution four bases before the position of c.1679. This variation, reported in dbSNP with rs1648179549, is the c.1676T>G and its clinical significance to date is unknown.18 Another example is related to a quite frequent variant close to the DPYD*2A: in this case the analysis of the known polymorphism showed an abnormal melting profile, as you can see in Figure 3, the peak is rounder than the wild-type one and had a melting temperature slightly anticipated. A TaqMan assay for the DPYD*2A did not detect the near variant. A LightSNiP assay confirmed the presence of the c.1896T>C (rs17376848) in heterozygosity.19 There was not a TaqMan assay for this SNP, due to its location. It is assigned a normal function by CPIC, but conflicting evidence has been reported.20 In the last case, c.496A>G genotyping showed the presence of a heterozygous melting profile different from that of the control (Figure 4). A TaqMan assay on second extraction gave a heterozygous profile, not completely overlapping that of the heterozygous control, as if the binding efficiency of the probe on the alternative allele were reduced in this sample. Sanger sequencing was necessary to define the right genotype and showed the presence of another polymorphism in addition to the c.496, one base after it, also in heterozygosity. This is a rare variant identified by the rs763061658 and it is a T>C substitution at position 497 (p.Met166Thr), its clinical significance to date is unknown.21
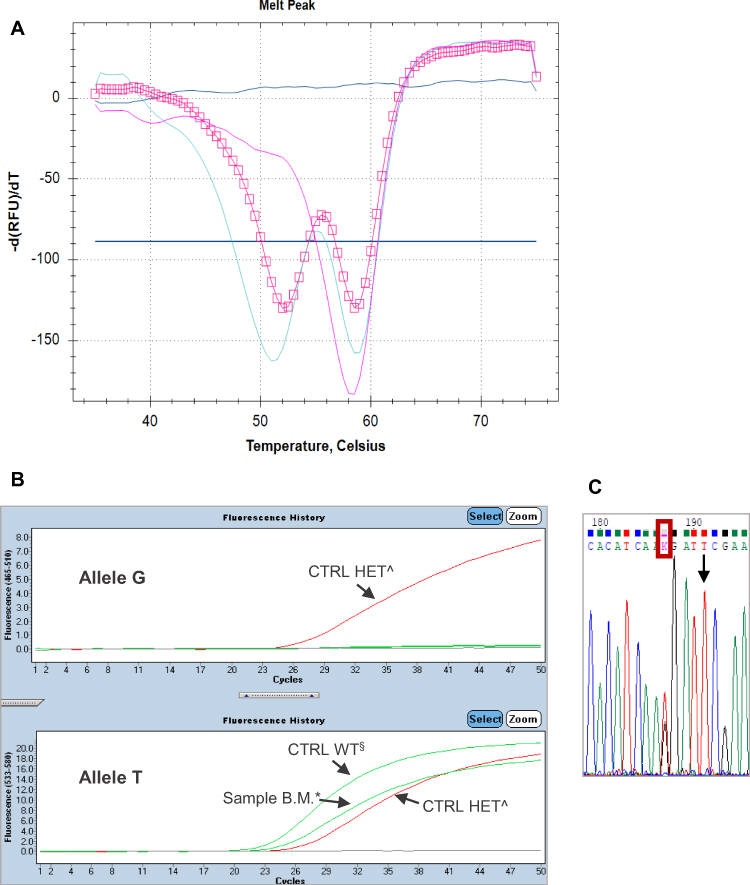 |
Figure 2 (A) The pink square line represents the melting profile of the sample B.M. (Tm allele 1: 52°C; Tm allele 2: 58.5°C). The melting profile of the heterozygous control is represented by the light-blue solid line (Tm allele T: 58.5°C; Tm allele G: 51°C) and the melting profile of the wild-type (WT) control is represented by the pink solid line (Tm allele T: 58.5°C). (B) TaqMan assay on second extraction detects only the reference allele T in the sample B.M.*, as in the control WT§; the heterozygous control^ has both alleles. (C) The sequence shows a T>G substitution (red rectangle) four bases before the position of c.1679, indicated by the row. |
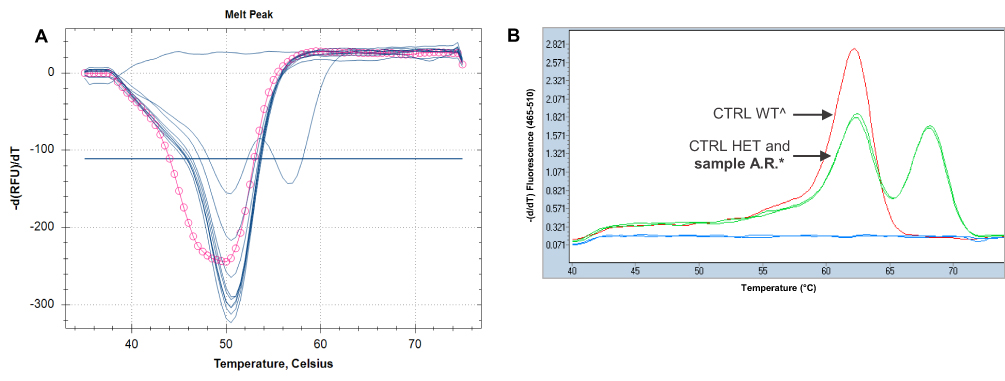 |
Figure 3 (A) DPYD*2A genotyping: the pink circle line represents an abnormal melting profile of that sample, compared to the others with GG genotype (wild-type samples, represented by single blue peaks). The double blue peak represents the heterozygous control (Tm allele G: 50.5°C; Tm allele A: 56.5°C). (B) The melting profile of the sample A.R.* confirms the presence of the variant close to the DPYD*2A, c.1896T>C in heterozygosity (the double green peaks, one of the sample A.R. and the other one of the heterozygous control, are completely overlapped; Tm allele T: 62.2°C and Tm allele C: 68.2°C). The wild-type control^ is represented by the single red peak (Tm allele T: 62.2°C). |
 |
Figure 4 (A) The star line represents the melting profile of the sample C.L. (Tm allele 1: 53.2°C; Tm allele 2: 62.8°C). Instead, the solid line represents the double peak of the c.496A>G heterozygous control (Tm allele G: 58°C; Tm allele A: 63.3°C). (B) TaqMan assay on second extraction shows an amplification plot (red dash line) in-between the wild-type (blue solid line) and heterozygous (red solid line) controls. The green solid line represents the mutated control. (C) The sequence shows a T>C substitution (red rectangle) one base after the position of c.496A>G, indicated by the row, also in heterozygosity. |
UGT1A1 Analysis
Of the 1076 requests collected, 649 had at least one mutated allele (about 60%). The heterozygosis of the UGT1A1*28 allele was found in 510 subjects (47.4%), whereas 139 subjects were homozygous (13%).
All variant alleles of the UGT1A1 (TA)n polymorphism (rs3064744), with 5 (UGT1A1*36), 6 (UGT1A1*1), 7 (UGT1A1*28) or 8 (UGT1A1*37) repeats were detected unambiguously with the UGT1A1 Real Time (FRET) kit, because each of them has a distinctive melting curve, as shown in the chromatogram (Figure 5).
 |
Figure 5 A distinctive melting profile is obtained for each different genotype of the UGT1A1 (TA)n polymorphism. On the right of the chromatogram, lines of different colours are listed, corresponding to the different genotypes. The melting temperatures of each allele are: 43.5°C±1 for the wild-type *1 allele; 47°C±1 for the *28 mutated allele; 39°C±1 for the *36 allele and 51.5°C±1 for the *37 allele. |
We found 6 subjects heterozygous for the *36 allele (0.6%; 5 of them were *28/*36) and 3 subjects heterozygous for the *37 allele (0.3%; all of them were *28/*37).
Discussion
The increase in the number of requests for pharmacogenetic testing, since the introduction in 2020 of a clear indication on when to perform them,13 confirms the usefulness of pharmacogenetics in the management of anticancer therapy. The choice of the most appropriate technology for accurate genotyping is crucial in clinical and scientific perspective.
We found that both FRET assay and LightSNiP assay, where the detection occurs by melting curve analysis, offer an advantage over competing TaqMan technology. Unexpected genetic variants may be missed using a mutation-specific TaqMan assay and any mismatch under the probe may reduce its binding efficiency, as shown by our results. It may be useful to know that a variant exists, even if it is not included in the guidelines or if it is a new variant of unknown significance; such an information would not be used to change therapy pre-emptively, rather to adjust it in case of unexpected post-treatment toxicity. The combination of the two methods we propose is helpful in that respect; in one of the cases we described a TaqMan assay helped us to understand that the variant detected by FRET assay was not the one searched, c.1679T>G, but another variant close to it, the characterization of which was only possible by direct sequencing. For small to medium-sized laboratories such as ours and for routine pharmacogenetic diagnostic activity looking for variants of well-known clinical significance, undoubtedly the best compromise is the use of a highly sensitive and rapid genotyping method such as real-time PCR with melting curve analysis, to be then combined with TaqMan assay and direct sequencing, if necessary, for more comprehensive genotyping and a correct result to the patient. Next-generation sequencing (NGS) technologies, which are emerging as a more comprehensive and time- and cost-effective approach in pharmacogenetics, represent an alternative strategy for optimizing treatment.22 Targeted sequencing panels of known pharmacogenes are being developed as pre-emptive profiling, in a new model of personalized medicine.23 A high-throughput approach is more suited to large-volume laboratories, with specific instruments and supportive bioinformatics tools. Furthermore, the real challenge of sequencing strategies, which do not allow a widespread implementation in the routine clinical practice, is the management of rare variants, variants of unknown clinical significance (VUS), and the insufficient coverage of some complex regions.22
The added value of the DPYD Real Time (FRET) CE-IVD kit is the possibility of detection, with the same amplification profile, also of the c.2194G>A, not yet included in the CPIC guidelines, but cited in recommendations as a variant to be investigated in case of severe toxicity, after the start of treatment. We also analyze the more frequent c.496A>G that, especially in cases of homozygosity for the alternative allele or in combination with other variants, seems to increase the risk of developing toxicity. Some studies in the literature state this24–27 and we also have evidence in support, as shown by the results on the patient with the double heterozygosity for the c.496 and the close c.497, an 80-year-old man diagnosed with rectal cancer, whose treatment with capecitabine concomitant to radiotherapy was stopped due to pancytopenia. Pharmacogenetic testing was performed one year later, at the onset of a second stomach cancer, given the previous toxicity, and indeed the result can at least partially account for this adverse event. This was not the only case we encountered in our clinical practice; another representative one was of a 64-year-old patient diagnosed with low-grade intestinal adenocarcinoma, who underwent the test because he developed an unexpected severe haematological toxicity (neutropenia and thrombocytopenia of grade 4) after the first cycle of adjuvant chemotherapy with capecitabine at 70% of the standard dose and he was GG homozygous for the 496 variant.
The positive distinctive feature of the UGT1A1 Real Time (FRET) CE-IVD kit is that it allows identification of all the potential variant alleles of the (TA)n polymorphism (rs3064744) in the gene promoter. This testing could have a double value, namely maximizing treatment efficacy by increasing the dose in patients with low-risk genotypes (UGT1A1*1/*1, *1/*28)28–30 and reducing the risk of irinotecan toxicity in patients with high-risk genotypes (UGT1A1*28/*28).12,14–16,31 There is a need for unambiguous results, particularly for this polymorphism, because several technologies are inappropriate for use in obtaining specific genotypes.32 It is true that the (TA)5 and (TA)8 alleles are more frequent in African Americans, but with the current racial admixture this should not be overlooked. Moreover, until now, therapeutic recommendations for other genotypes than UGT1A1*28/*28 were not established, but one of the reasons may be the lack of data on *36 and *37 alleles.
In conclusion, our clinical practice emphasizes the importance of not only rapid and easy-to-use assays, such as the new FRET ones, but also accurate and comprehensive genotyping for good pharmacogenetic diagnostic activity. Our combined approach has proved itself successful for this purpose. Based on our experience, we safely recommend the use of multiple approaches, in line with good genetic practice, to both confirm the presence of clinically relevant variants and resolve any unexpected results.
Acknowledgments
We thank TIB MOLBIOL and Nuclear Laser Medicine for the technical support provided in the sequencing of cases with uncertain genotypes.
Disclosure
This study was performed as part of our routine work and was not specifically funded.
The authors declare no conflict of interest.
References
1. Knikman JE, Gelderblom H, Beijnen JH, Cats A, Guchelaar HJ, Henricks LM. Individualized dosing of fluoropyrimidine-based chemotherapy to prevent severe fluoropyrimidine-related toxicity: what are the options? Clin Pharmacol Ther. 2021;109(3):591–604. doi:10.1002/cpt.2069
2. Dean L. Irinotecan therapy and UGT1A1 genotype. In: Pratt VM, Scott SA, Pirmohamed M, Esquivel B, Kattman BL, Malheiro AJ, editors. Medical Genetics Summaries [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2012.
3. Varughese LA, Lau-Min KS, Cambareri C, et al. DPYD and UGT1A1 pharmacogenetic testing in patients with gastrointestinal malignancies: an overview of the evidence and considerations for clinical implementation. Pharmacotherapy. 2020;40(11):1108–1129. doi:10.1002/phar.2463
4. Mhandire DZ, Goey AKL. The value of pharmacogenetics to reduce drug-related toxicity in cancer patients. Mol Diagn Ther. 2022;26(2):137–151. doi:10.1007/s40291-021-00575-x
5. Begré UBM, Jörger M, Aebi S, Amstutz U, Largiadèr CR. Clinical implementation of DPYD pharmacogenetic testing to prevent early-onset fluoropyrimidine-related toxicity in cancer patients in Switzerland. Front Pharmacol. 2022;13:885259. doi:10.3389/fphar.2022.885259
6. Wigle TJ, Povitz BL, Medwid S, et al. Impact of pretreatment dihydropyrimidine dehydrogenase genotype-guided fluoropyrimidine dosing on chemotherapy associated adverse events. Clin Transl Sci. 2021;14(4):1338–1348. doi:10.1111/cts.12981
7. Innocenti F, Mills SC, Sanoff H, Ciccolini J, Lenz HJ, Milano G. All you need to know about DPYD genetic testing for patients treated with fluorouracil and capecitabine: a practitioner-friendly guide. JCO Oncol Pract. 2020;16(12):793–798. doi:10.1200/OP.20.00553
8. Carrato A. Precision medicine: UGT1A1 genotyping to better manage irinotecan-induced toxicity. JCO Oncol Pract. 2022;18(4):278–280. doi:10.1200/OP.21.00858
9. Pharmacogenetic associations for which the data support therapeutic management recommendations. Available from: https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations.
10. Human medicine European public assessment report (EPAR). Available from: https://www.ema.europa.eu/en/medicines.
11. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing; 2017. Available from: https://cpicpgx.org/guidelines/guideline-for-fluoropyrimidines-and-dpyd/.
12. Dutch pharmacogenomics guidelines. Available from: https://upgx.eu/guidelines/.
13. EMA recommendations on DPD testing prior to treatment with fluorouracil, capecitabine, tegafur and flucytosine; 2020. Available from: https://www.ema.europa.eu/en/documents/press-release/ema-recommendations-dpd-testing-prior-treatment-fluorouracil-capecitabine-tegafur-flucytosine_en.pdf.
14. US Food and Drug Administration. Highlights of prescribing information: camptosar (irinotecan); 2014. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020571s048lbl.pdf.
15. European Medicines Agency. Summary of product characteristics onivyde (irinotecan hydrochloride trihydrate); 2021. Available from: https://www.ema.europa.eu/en/documents/product-information/onivyde-pegylated-liposomal-epar-product-information_en.pdf.
16. Recommendations for pharmacogenetic testing by AIOM-SIF working group. Available from: https://www.aiom.it/wp-content/uploads/2019/10/2019_Racc-analisi-farmacogenetiche_v26.3.2020.pdf.
17. EMQN’s EQA schemes: pharmacogenetics (panel testing). Available from: https://www.emqn.org/eqa-scheme-catalogue/.
18. rs1648179549 RefSNP report. Available from: https://www.ncbi.nlm.nih.gov/snp/.
19. Cheli S, Pietrantonio F, Clementi E, Falvella FS. LightSNiP assay is a good strategy for pharmacogenetics test. Front Pharmacol. 2015;6:114. doi:10.3389/fphar.2015.00114
20. rs17376848 RefSNP report. Available from: https://www.ncbi.nlm.nih.gov/snp/.
21. rs763061658 RefSNP report. Available from: https://www.ncbi.nlm.nih.gov/snp/.
22. Tafazoli A, Guchelaar HJ, Miltyk W, Kretowski AJ, Swen JJ. Applying next-generation sequencing platforms for pharmacogenomic testing in clinical practice. Front Pharmacol. 2021;12:693453. doi:10.3389/fphar.2021.693453
23. van der Wouden CH, van Rhenen MH, Jama WOM, et al. Development of the PGx-passport: a panel of actionable germline genetic variants for pre-emptive pharmacogenetic testing. Clin Pharmacol Ther. 2019;106(4):866–873. doi:10.1002/cpt.1489
24. Palmirotta R, Lovero D, Delacour H, Le Roy A, Cremades S, Silvestris F. Rare dihydropyrimidine dehydrogenase variants and toxicity by floropyrimidines: a case report. Front Oncol. 2019;9:139. doi:10.3389/fonc.2019.00139
25. Gentile G, Botticelli A, Lionetto L, et al. Genotype-phenotype correlations in 5-fluorouracil metabolism: a candidate DPYD haplotype to improve toxicity prediction. Pharmacogenomics J. 2016;16(4):320–325. doi:10.1038/tpj.2015.56
26. Ruzzo A, Graziano F, Galli F, et al. Dihydropyrimidine dehydrogenase pharmacogenetics for predicting fluoropyrimidine-related toxicity in the randomised, Phase III adjuvant TOSCA trial in high-risk colon cancer patients. Br J Cancer. 2017;117(9):1269–1277. doi:10.1038/bjc.2017.289
27. Gross E, Busse B, Riemenschneider M, et al. Strong association of a common dihydropyrimidine dehydrogenase gene polymorphism with fluoropyrimidine-related toxicity in cancer patients. PLoS One. 2008;3(12):e4003. doi:10.1371/journal.pone.0004003
28. Sharma MR, Joshi SS, Karrison TG, et al. A UGT1A1 genotype-guided dosing study of modified FOLFIRINOX in previously untreated patients with advanced gastrointestinal malignancies. Cancer. 2019;125(10):1629–1636. doi:10.1002/cncr.31938
29. Páez D, Tobeña M, Fernández-Plana J, et al. Pharmacogenetic clinical randomised Phase II trial to evaluate the efficacy and safety of FOLFIRI with high-dose irinotecan (HD-FOLFIRI) in metastatic colorectal cancer patients according to their UGT1A 1 genotype. Br J Cancer. 2019;120(2):190–195. doi:10.1038/s41416-018-0348-7
30. Karas S, Innocenti F. All you need to know about UGT1A1 genetic testing for patients treated with irinotecan: a practitioner-friendly guide. JCO Oncol Pract. 2022;18(4):270–277. doi:10.1200/OP.21.00624
31. Etienne-Grimaldi MC, Boyer JC, Thomas F, et al. Collective work by the Group of Clinical Onco-Pharmacology (GPCO-Unicancer); French National Network of Pharmacogenetics (RNPGx). UGT1A1 genotype and irinotecan therapy: general review and implementation in routine practice. Fundam Clin Pharmacol. 2015;29(3):219–237. doi:10.1111/fcp.12117
32. Sissung TM, Barbier RH, Price DK, et al. Comparison of eight technologies to determine genotype at the UGT1A1 (TA)n repeat polymorphism: potential clinical consequences of genotyping errors? Int J Mol Sci. 2020;21(3):896. doi:10.3390/ijms21030896
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.