")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
pH-Responsive therapeutic solid lipid nanoparticles for reducing P-glycoprotein-mediated drug efflux of multidrug resistant cancer cells
Authors Chen H, Huang W, Chiang W, Liu T, Shen M, Hsu Y, Lin S, Chiu H
Received 3 April 2015
Accepted for publication 26 May 2015
Published 5 August 2015 Volume 2015:10(1) Pages 5035—5048
DOI https://doi.org/10.2147/IJN.S86053
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Hsin-Hung Chen,1 Wen-Chia Huang,2 Wen-Hsuan Chiang,2 Te-I Liu,2 Ming-Yin Shen,2,3 Yuan-Hung Hsu,4 Sung-Chyr Lin,1 Hsin-Cheng Chiu2
1Department of Chemical Engineering, National Chung Hsing University, Taichung, 2Department of Biomedical Engineering and Environmental Sciences, National Tsing Hua University, 3Department of Surgery, National Taiwan University Hospital-Hsinchu Branch, 4Pharmaceutical Optimization Technology Division, Biomedical Technology and Device Research Laboratory, Industrial Technology Research Institute, Hsinchu, Taiwan
Abstract: In this study, a novel pH-responsive cholesterol-PEG adduct-coated solid lipid nanoparticles (C-PEG-SLNs) carrying doxorubicin (DOX) capable of overcoming multidrug resistance (MDR) breast cancer cells is presented. The DOX-loaded SLNs have a mean hydrodynamic diameter of ~100 nm and a low polydispersity index (under 0.20) with a high drug-loading efficiency ranging from 80.8% to 90.6%. The in vitro drug release profiles show that the DOX-loaded SLNs exhibit a pH-controlled drug release behavior with the maximum and minimum unloading percentages of 63.4% at pH 4.7 and 25.2% at pH 7.4, respectively. The DOX-loaded C-PEG-SLNs displayed a superior ability in inhibiting the proliferation of MCF-7/MDR cells. At a DOX concentration of 80 µM, the cell viabilities treated with C-PEG-SLNs were approximately one-third of the group treated with free DOX. The inhibition activity of C-PEG-SLNs could be attributed to the transport of C-PEG to cell membrane, leading to the change of the composition of the cell membrane and thus the inhibition of permeability glycoprotein activity. This hypothesis is supported by the confocal images showing the accumulation of DOX in the nuclei of cancer cells and the localization of C-PEG on the cell membranes. The results of in vivo study further demonstrated that the DOX delivered by the SLNs accumulates predominantly in tumor via enhanced permeability and retention effect, the enhanced passive tumor accumulation due to the loose intercellular junctions of endothelial cells lining inside blood vessels at tumor site, and the lack of lymphatic drainage. The growth of MCF-7/MDR xenografted tumor on Balb/c nude mice was inhibited to ~400 mm3 in volume as compared with the free DOX treatment group, 1,140 mm3, and the group treated with 1,2 distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)] solid lipid nanoparticles, 820 mm3. Analysis of the body weight of nude mice and the histology of organs and tumor after the administration of DOX-loaded SLNs show that the SLNs have no observable side effects. These results indicate that the C-PEG-SLN is a promising platform for the delivery of therapeutic agents for MDR cancer chemotherapy.
Keywords: pH-responsive, solid lipid nanoparticles, multidrug resistance, permeability glycoprotein
Introduction
Nanotechnology has recently been used for the development of drug delivery systems for cancer therapy.1 Various biocompatible and biodegradable materials have been used to prepare drug-loaded nanocarriers that can be effectively delivered to tumor sites via the enhanced permeability and retention (EPR) effects.2–4 However, multidrug resistance (MDR) is frequently encountered in patients undergoing long-term chemotherapy, leading to the failure of chemotherapy. One of the well-known mechanisms of MDR is drug efflux mediated by permeability glycoprotein (P-gp), a membrane-bound translocase of the adenosine triphosphate (ATP)-binding cassette transporter superfamily encoded by MDR1 gene, overexpressed on the cancer cell membrane. Enhanced active transport of chemotherapeutic agents, such as doxorubicin (DOX), cisplatin, topotecan, and paclitaxel, out of cancer cells by P-gp prevents cellular drug accumulation, and thus reduces the efficacy of chemotherapy.5,6
To this end, several P-gp inhibitors, such as verapamil and disulfiram,7,8 have been co-encapsulated in nanocarriers to reduce the MDR of cancer cells. However, the inherent toxicity of these P-gp inhibitors, partly resulting from the inhibition of Ca2+ channels in smooth muscle that line blood vessels and heart, often leads to hypotension and bradycardia in cancer patients.9,10 Furthermore, such P-gp inhibitors may exhibit unfavorable pharmacokinetic interactions with chemotherapy drugs, and thus further enhance their toxicity to normal organs and tissues.11 Alternatively, relief of MDR by reducing the expression level of P-gp with small interfering RNA (siRNA) has also been proposed.12 To encapsulate the negatively charged siRNA, however, it is generally necessary to use positively charged materials, such as polyethylenimine, dioleoylphosphatidylethanolamine, and 1,2-dioleoyl-3-trimethylammonium-propane, for the preparation of nanoscale delivery systems.13–15 These cationic components are usually highly cytotoxic due to their extensive charge interactions with cell membrane. Furthermore, enhanced uptake of siRNA could lead to the inhibition of the translocase activity of P-gp of normal tissues.
Alternatively, reduction of the MDR effect of cancer cells on intracellular drug delivery by means of incorporating nonionic surfactants has been receiving increasing attentions. Several potential candidates, such as Tweens, Pluronic, D-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), and Brij,11,16–18 have emerged and been examined for their actions on reducing cellular drug efflux from MDR cancer cells by disrupting membrane structure of cells or mitochondria to inhibit the ATPase activity or ATP production. In addition to their relatively low cytotoxicity,19–21 these nonionic surfactants are also known for their ability in enhancing the stability and dispersity of nanocarriers in aqueous phases and thus their co-delivery to tumor sites with the anticancer drugs in the nanocarriers via EPR effect.22–24
Cholesterol, the most common steroid in animals, is well known for its vital role as the principal component of cell plasma membrane actively involved in the regulation of membrane fluidity and formation of lipid rafts with which the P-gp devices are intimately associated.25,26 It is thus rational to exploit new amphiphilic conjugates of cholesterol with poly(ethylene glycol) (C-PEGs) to elaborate nanoparticulate drug vehicles for the effective treatment of MDR cancer cells overexpressing P-gp by disturbing cell membrane structure, and thus the P-gp activity presumably through allosteric ATPase inhibition,27 in addition to the colloidal stabilization by PEG chain segments in aqueous phases. In this work, the effects of C-PEGs coated on the surfaces of the trilaurin-based solid lipid nanoparticles (SLNs) as the DOX carriers on reducing P-gp activity of human breast cancer cells (MCF-7/MDR) were evaluated and compared with that of 1,2 distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)] (DSPE-PEG) serving as the control. Neutralization of charges with laurate allows the incorporation of DOX/lauric acid complexes into the trilaurin-rich core of SLNs. C-PEG and DSPE-PEG were anchored on the surfaces of the SLNs via the hydrophobic moieties of the two nonionic surfactants, exposing the relatively hydrophilic PEG chain segments on the surface. It was demonstrated that the pH-responsive DOX-loaded SLNs elaborated by C-PEGs could significantly improve the therapeutic efficacy of chemotherapy to overcome MDR. The structure of the pH-responsive SLN and its delivery strategy to tumor cell of this study are shown in Figure 1.
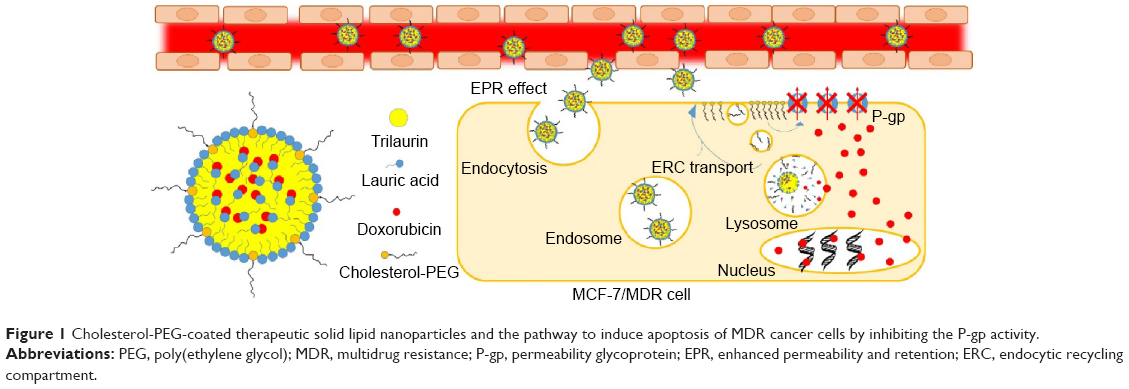 | Figure 1 Cholesterol-PEG-coated therapeutic solid lipid nanoparticles and the pathway to induce apoptosis of MDR cancer cells by inhibiting the P-gp activity. |
Materials and methods
Chemicals, drugs, and cell lines
Trilaurin, sodium laurate, monomethoxy-polyethylene glycols (mPEG), cholesteryl chloroformate, N-hydroxysuccinimide, N,N′-dicyclohexylcarbodiimide (DCC), cysteamine, verapamil, β-estradiol, 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO), Hoechst 33258, and rhodamine B were obtained from Sigma-Aldrich (St Louis, MO, USA, and Seelze, Germany). DOX and ω-maleimide-polyethylene glycol-amine were purchased from Seedchem (Melbourne, Australia) and Jenkem (Beijing, People’s Republic of China), respectively. DSPE-PEG was obtained from Avanti Polar Lipids (Alabaster, AL, USA). Chloroform, dimethyl sulfoxide (DMSO), and paraformaldehyde (PFA) were obtained from TEDIA (Fairfield, OH, USA). All organic reagents were of analytical grade. For cell culture studies, Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), and 0.25% trypsin–EDTA and penicillin–streptomycin solutions were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Alamar Blue, a cell viability reagent, was purchased from Thermo Fisher Scientific. MCF-7 and MCF-7/MDR were obtained from Food Industry Research and Development Institute (Hsinchu City, Taiwan) and courtesy of Dr San-Yuan Chen of Department of Materials Science and Engineering at National Chiao Tung University, Taiwan, respectively. Balb/c C57B nude mice were purchased from the National Laboratory Animal Center, Taiwan.
Synthesis and characterization of cholesterol-PEG derivatives
Cholesterol-PEG (C-PEG): monomethoxy-polyethylene glycols (Mw 2,000 and 5,000 g/mol) and cholesteryl chloroformate in a molar ratio of 1:1.1 were mixed in dichloromethane in the presence of 4-(dimethylamino) pyridine as the catalyst.28 Acylation of PEG was allowed to proceed at room temperature for 3 days. The C-PEG, denoted as C-PEG2k and C-PEG5k, thus obtained was precipitated from ethyl ether.29 The yield of C-PEG was determined by 1H-NMR (Bruker Avance-850NMR Spectrometer) in CDCl3 (Cambridge Isotope Laboratories, Cambridge, MA, USA).
Cholesterol-PEG-rhodamine (C-PEG-R): C-PEG-maleimide was obtained by the acylation of ω-maleimide-polyethylene glycol-amine (2,000/5,000) with cholesteryl chloroformate, as described earlier. C-PEG-maleimide was then further conjugated with cysteamine in dehydrated DMSO at room temperature for 24 hours. After precipitation from ethyl ether, C-PEG-NH2 obtained was collected by filtration. C-PEG-NH2, N-hydroxysuccinimide, N,N′-dicyclohexylcarbodiimide, and rhodamine B in the molar ratio of 1.0:1.1:1.1:1.1 were then mixed in dehydrated DMSO at room temperature for 24 hours. Finally, C-PEG-R thus obtained was purified by dialysis with Cellu-Sep T4 membrane (molecular weight cut-off 1,000, Membrane Filtration Products, Seguin, USA) against deionized water for 1 week. The chemical structure of C-PEG-R was confirmed by 1H-NMR in CDCl3 and the conversion yield was determined by Beer’s law based on the absorbance at 520 nm.
Preparation and characterization of DOX-loaded solid lipid nanoparticles
Trilaurin (24 mg) and lipid/PEG derivatives (C-PEG or DSPE-PEG, 4.8 mg) were dissolved in chloroform (0.7 mL) as the organic phase. DOX and lauric acid, in molar ratios from 1:1 to 1:5, were dissolved in deionized water (0.3 mL) as the aqueous phase. The aqueous drug solution was added dropwise into the organic phase. The mixture was then extensively homogenized by ultrasonication for 10 minutes in ice bath. Upon the completion of the first homogenization cycle, phosphate buffer (pH 7.4, ionic strength 10 mM, 2.5 mL) was added, and the mixture was further ultrasonicated for another 10 minutes in ice bath. Chloroform in the mixture was subsequently removed by rotary evaporator for 30 minutes. The unencapsulated DOX was removed from the DOX-loaded SLNs by dialysis against pH 7.4 phosphate buffer (ionic strength 10 mM) with Cellu-Sep T4 membrane (molecular weight cut-off 12,000–14,000) for 3 days.
The encapsulation efficiency was determined by the measurement of fluorescence intensity using a microplate reader (FLUOstar, OPTIMA, BGM Labtech, Ortenberg, Germany) with an excitation wavelength of 480 nm and an emission wavelength of 560 nm. The encapsulation efficiency herein was defined as the percentage of DOX remained in SLNs upon dialysis.30 The particle size, polydispersity index, and zeta potential of the SLNs were characterized by dynamic light scattering (DLS, Malvern ZetaSizer Nano Series instrument, Westborough, MA, USA) with He-Ne laser 4 mW, λ=633 nm. The morphology of the SLNs was examined by transmission electronic microscopy conducted on a JEOL JEM-1200 CXII microscope operating at an accelerating voltage of 120 kV.
Drug release of SLNs
The drug release profiles of DOX-loaded SLNs in buffers (ionic strength 0.15 M) of pH 4.7, 6.0 (succinate), and 7.4 (phosphate) were studied at 37°C, respectively. SLN solution (0.2 mL) was loaded into a dialysis bag (Cellu sep molecular weight cut-off 12,000–14,000) against 40 mL of the aforementioned outer buffer. At the prescribed time intervals, 1 mL outer buffer was withdrawn to quantify the amount of DOX released by fluorescence spectroscopy. After each sampling, 1 mL fresh buffer was added to the outer aqueous phase. The cumulative drug release was determined as the ratio of the amount of DOX released into the outer buffer to the original amount of DOX encapsulated in the SLNs.
Cytotoxicity analysis of SLNs
MCF-7 and MCF-7/MDR cells were seeded into 96-well plates at a concentration of 8×103 cells/well in 0.2 mL DMEM, containing 10% FBS and 1% penicillin, and incubated at 37°C overnight in an atmosphere of 5% CO2. The spent medium was then replaced with working suspensions/solutions containing DOX at concentrations ranging from 2.4 nM to 80 μM, prepared by twofold serial dilution of free DOX, DOX-loaded cholesterol-PEG adduct-coated solid lipid nanoparticles (C-PEG-SLNs), and DOX-loaded DSPE-PEG-SLNs stock solutions with DMEM, respectively. The plates were further incubated at 37°C for 6 hours. After being washed twice with phosphate buffer saline (PBS), the 96-well plates were then filled with fresh DMEM (0.2 mL/well) and re-incubated for additional 24 hours. To determine the cell viability of cancer cells, Alamar Blue (10% v/v) in RPMI 1640 medium was added into the 96-well plate (100 mL/well), followed by re-incubation at 37°C for 2 hours. The absorbance of each well at 570 and 600 nm was determined by the microplate reader.
Cellular uptake study
For intracellular DOX level in MCF-7/MDR cells study, MCF-7 and MCF-7/MDR cells were seeded in a six-well plate at 5×105 cells/well and incubated at 37°C in an atmosphere of 5% CO2 overnight. The cells were treated with free DOX, DOX-loaded C-PEG-SLNs, DOX-loaded DSPE-PEG-SLNs, or free DOX + verapamil (50 μM) at a DOX concentration of 20 μM for 6 hours and then washed with PBS twice. To study the effect of lipid/PEG derivative on the intracellular DOX level, the cells were treated simultaneously with free DOX (20 μM) and lipid/PEG adducts (C-PEG2k, C-PEG5k, DSPE-PEG2k, and DSPE-PEG5k) at concentrations ranging from 1.2 to 18.5 μg/mL for 6 hours and then washed with PBS twice. DMSO (1 mL) was then added into each well for the lysis of the cancer cells. The amount of DOX released from the cancer cells were quantitatively determined by the microplate reader. For laser scanning confocal microscopy imaging, cancer cells were seeded on 22 mm round glass coverslips placed in a six-well plate at 5×105 cells/well and incubated at 37°C overnight. The cells were treated with free DOX, DOX-loaded C-PEG-SLNs (DOX-loaded C-PEG-R-SLNs), DOX-loaded DSPE-PEG-SLNs at a DOX concentration of 20 μM for 6 hours, and then washed with PBS twice. Cancer cells on the glass coverslips were fixed with 4% PFA and then washed with PBS twice. Nuclei of cancer cells were stained with Hoechst 33258 at 37°C for 5 minutes and then washed with PBS twice. To examine the cellular distribution of C-PEG-R, the cell membrane was additionally stained with DiO. Finally, the cancer cells were then fixed with mounting gel, and the coverslips were covered with microslides. The distribution of DOX within cancer cells were examined by laser scanning confocal microscopy (ZEISS LSM-780) with an excitation wavelength of 488 nm and emission wavelength of 560 nm. Rhodamine was monitored with an excitation wavelength of 560 nm and emission wavelength of 630 nm. DiO was monitored with an excitation wavelength of 488 nm and emission wavelength of 510 nm.
Biodistribution of SLNs
The animal use protocol in this work has been approved by the Institutional Animal Care and Use Committee of National Tsing Hua University, Taiwan (IACUC approval number: 10244). All of the animal experiments were executed following the “Guide for the Care and Use of Laboratory Animals” published by the National Academy Press, revised in 1996.
MCF-7/MDR cells at a concentration of 5×106 cells/mL were suspended in PBS containing 50% matrigel. Seven-week-old Balb/c nude mice were subcutaneously injected with 0.1 mL cancer cells/PBS suspension on the right thigh and fed with β-estradiol at 2 mg/L in daily drinking water to induce the growth of the hormone-dependent MCF-7/MDR cells. The feeding of β-estradiol was terminated 1 week before the onset of chemotherapy treatment. The nude mice bearing tumor of the size ~100 mm3 were first injected with PBS via the tail vein and then followed by DOX-loaded C-PEG-SLNs and DSPE-PEG-SLNs at a dose of 20 mg/kg. One day later, the nude mice were sacrificed under CO2. Tumor and organs, including heart, liver, spleen, lung, and kidney, were collected for ex vivo DOX distribution examination with an in vivo imaging system (Caliper IVIS Spectrum) with excitation and emission wavelengths at 465 and 560 nm, respectively. The size of the tumor herein was evaluated as 0.52× length × width × height.31
Tumor inhibition
The MDR tumor model of nude mice bearing tumor of size 200 mm3 was established as described earlier. The nude mice were separated into six groups with three mice per group and intravenously injected with PBS, free DOX, DOX-loaded C-PEG-SLNs, or DOX-loaded DSPE-PEG-SLNs at a DOX dosage of 10 mg per kg of body weight every other day for three doses. Tumor volume, calculated as described earlier, was monitored for 45 days to estimate the extent of tumor inhibition.
Hematoxylin and eosin stain
Hematoxylin and eosin staining was performed according to the procedure reported in the literature32 and was briefly outlined. Major organs (heart, liver, spleen, lung, and kidney) and tumors were collected on day 45 after treatment, then cut into appropriate size, and stained with hematoxylin and eosin. The histology of stained organs and tumors were observed using microscopy (Olymplus IX70).
Statistical analysis
Unless specified otherwise, all experiments were performed in triplets, and data were reported as mean ± standard deviation. Statistical significance was determined using Student’s t-test. Significant differences were defined as (*) P<0.05 and (**) P<0.01. Not significant differences were displayed as NS (P>0.05).
Results and discussion
Synthesis and characterization of cholesterol/PEG adducts
To evaluate the inhibition efficiency of C-PEG-coated nanoparticles in overcoming the MDR of MCF-7, cholesterol-PEG2k/5k adducts were synthesized in this study. The 1H-NMR spectra of C-PEG2k/5k are shown in Figure S1 (supplementary data: https://www.dropbox.com/s/q82v4exsxwbh1p2/Supplementary_Data_05_17.docx?dl=0). The conversion yields of C-PEG2k and C-PEG5k, calculated with the characteristic resonances of PEG (5.37 ppm) and cholesterol (3.30 ppm), were 90.1% and 91.5%, respectively. For biodistribution study in tumor cells, C-PEG2k/5k additionally labeled with rhodamine B (C-PEG2k/5k-R) were synthesized. The presence of the characteristic resonances of PEG (3.56 ppm), cholesterol (5.37 ppm), and rhodamine B (7.03 ppm and 7.51 ppm) in the 1H NMR spectra of C-PEG2k/5k-R indicated that the conjugated compounds were successfully synthesized as shown in Figure S2. The conversion yields of C-PEG2k/5k-R, determined by Beer’s law at 520 nm, were 18.9% and 25.3%, respectively (data not shown).
Preparation and characterization of SLNs
The effect of the amount of lauric acid, which could associate with DOX via electrostatic attraction, on the drug loading efficiency of the SLNs was investigated. The DOX loading efficiency of the SLNs was found to increase with the amount of lauric acid. The loading efficiency reached a plateau of 80.2%±3.6% at a molar ratio of DOX to lauric acid at 1:5, beyond which the loading efficiency remained essentially constant (Figure S3). The effect of the amount of lipid/PEG derivatives (DSPE-PEG and C-PEG), which could prevent SLNs from aggregation with one another by virtue of the PEG chain segments of the derivatives, on the stability and size dispersity of the SLNs in aqueous phase was also studied. As expected, extensive aggregation induced by hydrophobic interactions in aqueous phase occurred for SLNs with insufficient lipid/PEG derivatives. At a concentration of C-PEG2k corresponding to 20% (w/w) of the trilaurin-rich hydrophobic domain, a clear transparent SLNs aqueous suspension was obtained, indicating that the SLNs were well dispersed in the medium. The SLNs thus prepared had a mean hydrodynamic diameter (Dh) of 108.7±3.2 nm and a fair polydispersity index of 0.128. A further increase in derivative concentration, however, led to an increase in particle size that could possibly limit the EPR effects for in vivo applications. Based on these results, SLNs with a DOX to lauric acid molar ratio of 1:5 containing 20% lipid/PEG derivatives were used for all the subsequent experiments, unless specified otherwise.
The mean particle sizes, particle size distributions, and zeta potentials of C-PEG2k/5k-SLNs and DSPE-PEG2k/5k-SLNs in pH 7.4 aqueous solution determined by DLS are summarized in Table 1. Note that, for efficient targeting of tumor, the drug-loaded nanocarriers must be of suitable particle sizes to facilitate the passive accumulation of nanoparticles in tumor.33–35 As shown in Table 1, the C-PEG-SLNs and DSPE-PEG-SLNs exhibit a mono-modal particle size distribution with the Dh values in the range 100–110 nm, pertinent for passive targeting to tumor by the EPR effect (≤200 nm)3,33 and for avoiding elimination by kidneys (≥10 nm).36 The transmission electronic microscopy images illustrate that the SLNs were solid spherical particles without aggregation (Figure 2). The SLNs exhibited neutral zeta potentials of 0.7–3.3 mV, due to the presence of PEG-derived coating as the stealth system on the surfaces of SLNs, which could further enhance storage stability and reduce albumin-associated aggregation in blood circulation,36,37 as the prerequisites for practical applications. This is supported by the results of DLS analysis showing that the SLNs maintained their original particle sizes after long-term storage (~5 weeks) in high ionic strength (0.15 M) PBS buffer (Figure S4A). Furthermore, no albumin-associated aggregation was observed for SLNs dispersed in DMEM containing 10% FBS over a time period of 5 days (Figure S4B). The SLNs in both PBS and DMEM also had mono-modal polydispersity indexes, indicating that the SLNs had excellent stability against self-aggregation for in vitro storage and in vivo applications. High loading efficiencies (>80%) and drug loading content (~11%) were achieved for all SLNs prepared.
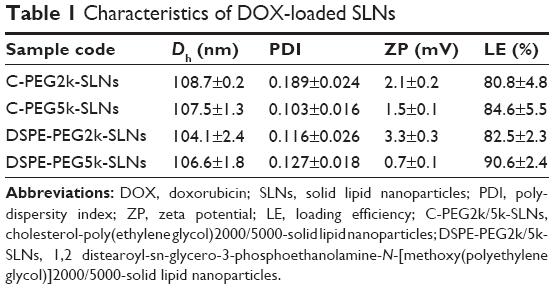 | Table 1 Characteristics of DOX-loaded SLNs |
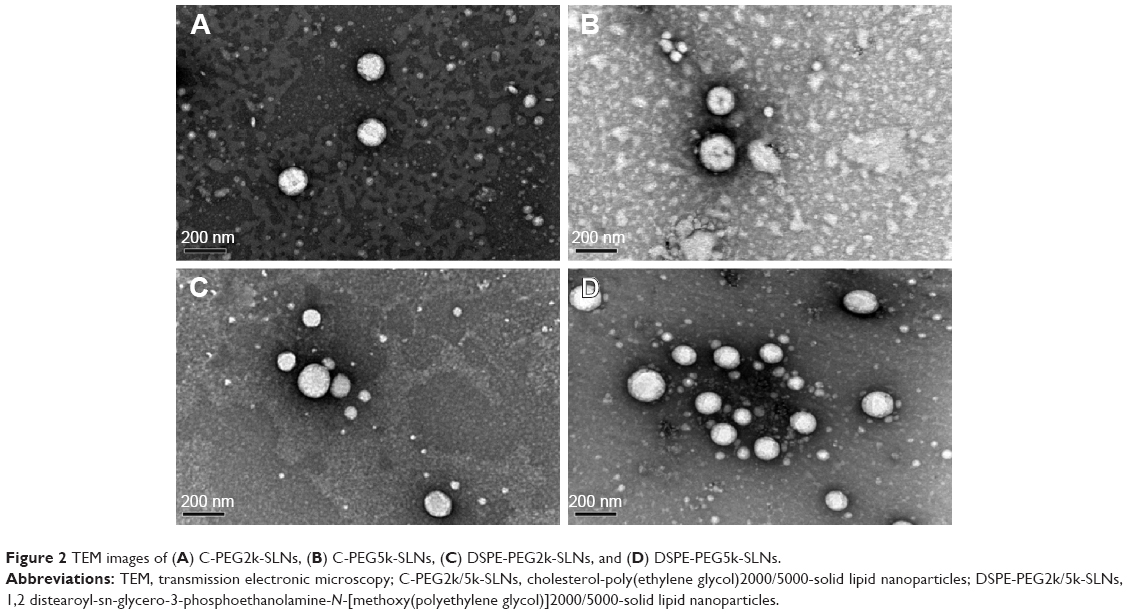 | Figure 2 TEM images of (A) C-PEG2k-SLNs, (B) C-PEG5k-SLNs, (C) DSPE-PEG2k-SLNs, and (D) DSPE-PEG5k-SLNs. |
Drug release behavior
The pH targeting nanotechnology has been widely applied in pH-sensitive drug carrier systems such as polymeric micelles and nanogels.38,39 To overcome MDR, drug carrier systems aiming at increasing intracellular drug concentration against the active transport by P-gp have been developed.40 In this work, the drug release behavior of the SLNs was studied at pH characteristic of the blood circulation (pH 7.4), the acidic tumor microenvironment (pH 6.0), and the subcellular acidic organelles (4.7) with succinate and PBS buffers.41 As shown in Figure 3, all SLNs tested exhibited pH-dependent drug release behaviors, with the highest drug release of ~55% at pH 4.7, followed by a moderate drug release of ~40% at pH 6.0, and the lowest drug release of ~25% at pH 7.4. The release of DOX under acidic conditions was facilitated by the protonation of the carboxyl group of laurate (pKa =5.34), leading to the reduction of the electrostatic attractions between the negatively charged laurate and the positively charged DOX. The high drug release at low medium pH can also be partly attributed to the increase in DOX solubility. The high drug release at pH 4.7 and low drug release at pH 7.4 suggest that enhanced drug release can be achieved within endosome/lysosome and the tumor microenvironment with minimal release in bloodstream.
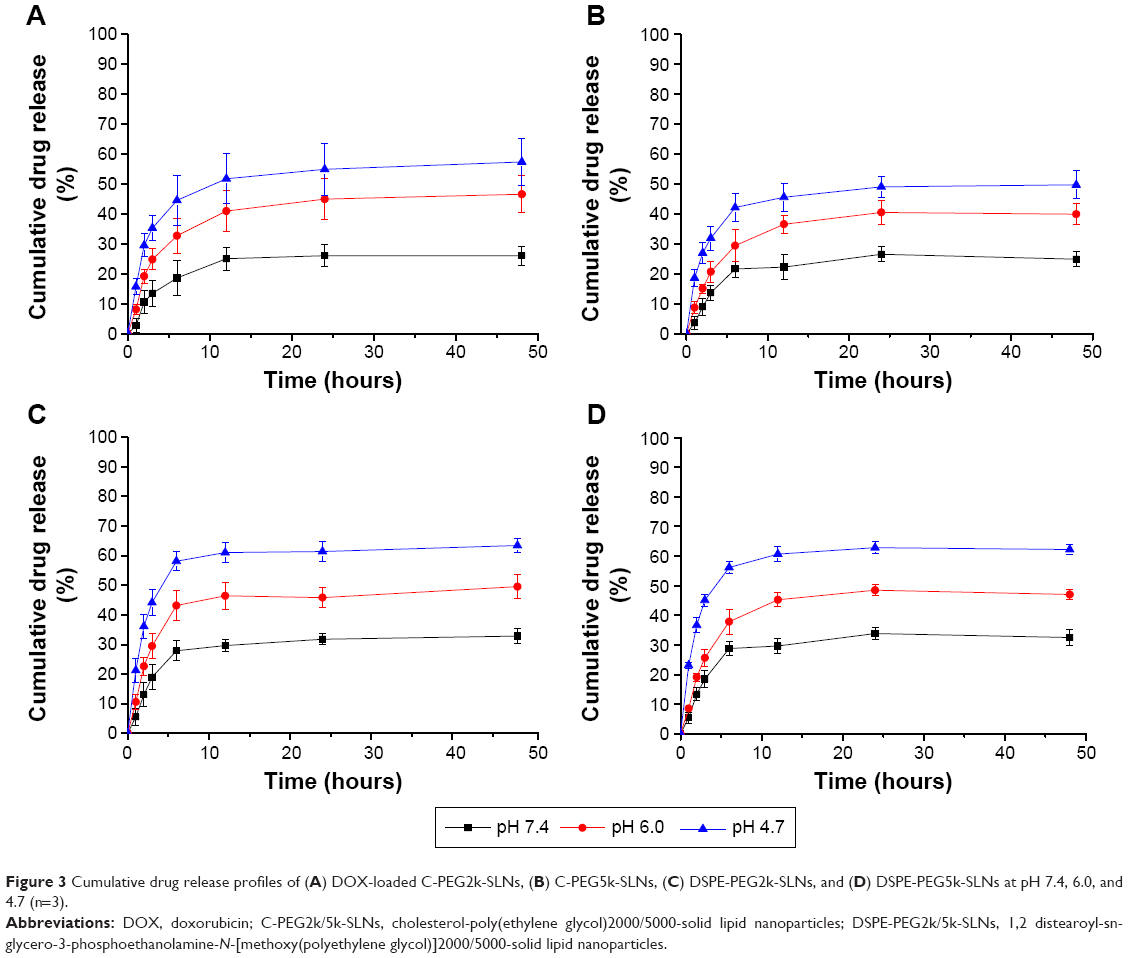 | Figure 3 Cumulative drug release profiles of (A) DOX-loaded C-PEG2k-SLNs, (B) C-PEG5k-SLNs, (C) DSPE-PEG2k-SLNs, and (D) DSPE-PEG5k-SLNs at pH 7.4, 6.0, and 4.7 (n=3). |
Although 45% of DOX remained bound in the hydrophobic core of the SLNs, composed mainly of trilaurin, it might be possible to achieve higher release efficiency in vivo upon endocytosis. It has been reported that the release of drug from nanoparticles prepared with ester-bond compounds, such as poly(epsilon-caprolactone) and triacylglycerol, could be promoted upon the lipase-catalyzed hydrolysis of nanoparticles.42,43 Since endosome and lysosome are known to contain lipases,44–46 the effect of lipase incubation on the structure of the SLNs was studied. It was found that the derived count rate of SLNs from the DLS measurement after lipase treatment decreased from ~15,000 to 3,500 (Table S1), indicating the disruption of SLNs by lipases. It is thus possible to achieve a higher release level of DOX from the SLNs in vivo due to the lipase-mediated disruption of the lipid nanoparticles.
Cytotoxicity study
The in vitro cytotoxicity of DOX-loaded SLNs against MCF-7 cells was determined by using Alamar Blue assay. As shown in Figure 4A, no significant reduction in cell viability was observed until the DOX concentration reached 1.25 μM, beyond which the viability dropped steadily to ~40% at a DOX concentration of 80 μM. Since pristine SLNs are nontoxic to cancer cells and normal cells (Figure S5), the cytotoxicity of DOX-loaded SLNs against MCF-7 cells can be solely attributed to the released DOX species instead of the lipid carriers. To assess the efficacy of DOX-loaded SLNs against MDR cancer cells, MCF-7/MDR cells overexpressing P-gp, as confirmed by Western blot assay illustrated in Figure S6, were treated with free DOX, DOX-loaded C-PEG-SLNs, and DSPE-PEG-SLNs. Figure 4B shows that free-DOX-treated MCF-7/MDR exhibited high cell viability (~75.9%) even at the highest DOX concentration (80 μM). Similar results were attained in groups treated with DOX-loaded DSPE-PEG2k/5k-SLNs with cell viabilities of 69.0% and 71.6%, respectively. The low cytotoxicity can be attributed to the excretion of DOX by the overexpressed P-gp. In contrast, high cytotoxicity toward MCF-7/MDR cells was observed for groups treated with DOX-loaded C-PEG2k/5k SLNs. At a DOX concentration of 80 μM, the cell viabilities for groups treated with DOX-loaded C-PEG2k/5k-SLNs, ~25%, were approximately threefold lower than that with free DOX and DOX-loaded DSPE-PEG-SLNs. Since all the SLNs were of identical lipid composition except for the surface PEG derivatives, the difference in drug efficacy against MCF-7/MDR cells can be ascribed to the inhibitory effect of C-PEG on the activity of P-gp. Nanocarriers with other PEG derivatives, such as TPGS and Brij, capable of overcoming MDR have also been reported in the literature.11,18 The 50% cellular growth inhibition (IC50) values of C-PEG2k/5k-SLNs against MCF-7/MDR were 9.2 and 8.1 μM of the DOX concentrations, respectively. These values are slightly higher than that reported in the literature, ~1–3 μM for TPGS and disulfiram,8,18 due to the short co-incubation time, 6 hours instead of 48 hours, used in this study.
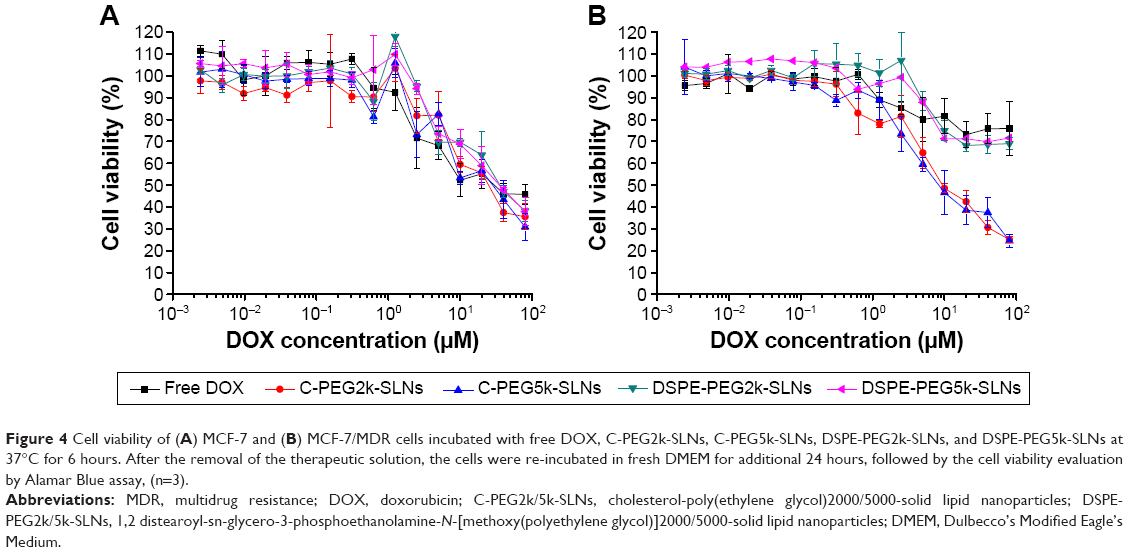 | Figure 4 Cell viability of (A) MCF-7 and (B) MCF-7/MDR cells incubated with free DOX, C-PEG2k-SLNs, C-PEG5k-SLNs, DSPE-PEG2k-SLNs, and DSPE-PEG5k-SLNs at 37°C for 6 hours. After the removal of the therapeutic solution, the cells were re-incubated in fresh DMEM for additional 24 hours, followed by the cell viability evaluation by Alamar Blue assay, (n=3). |
Cellular uptake
The cellular uptake of DOX-loaded C-PEG-SLNs and DSPE-PEG-SLNs in MCF-7 and MCF-7/MDR cells was characterized with the endocytic reactions for 3 and 6 hours. As shown in Figure 5A, significant accumulation of DOX in the nuclei was observed for MCF-7 cells treated with free DOX and DOX-loaded PEG-derivative SLNs, suggesting that the decline in cell viability was caused as reported by the inhibition of DNA replication and production of reactive oxygen species due to the accumulation of DOX in nuclei and the subsequent inactivation of topoisomerase II, eventually leading to the apoptosis of cells.47 On the contrary, for MCF-7/MDR cells treated with free DOX, the DOX species accumulated mainly around the cell membrane with only trace amount of DOX in the cytoplasm (Figure 5B), indicating the exclusion of DOX molecules by P-gp overexpressed in MCF-7/MDR cells, as reported elsewhere.18,48 Nevertheless, for groups treated with C-PEG-SLNs, essentially all the DOX molecules accumulated in the nuclei of MCF-7/MDR. Such a preferential distribution of DOX in the nuclei was not observed for groups treated with DSPE-PEG-SLNs. Similar results were also attained from the co-incubation of MCF-7 and MCF-7/MDR, separately, with SLNs for 3 hours (Figure S7).
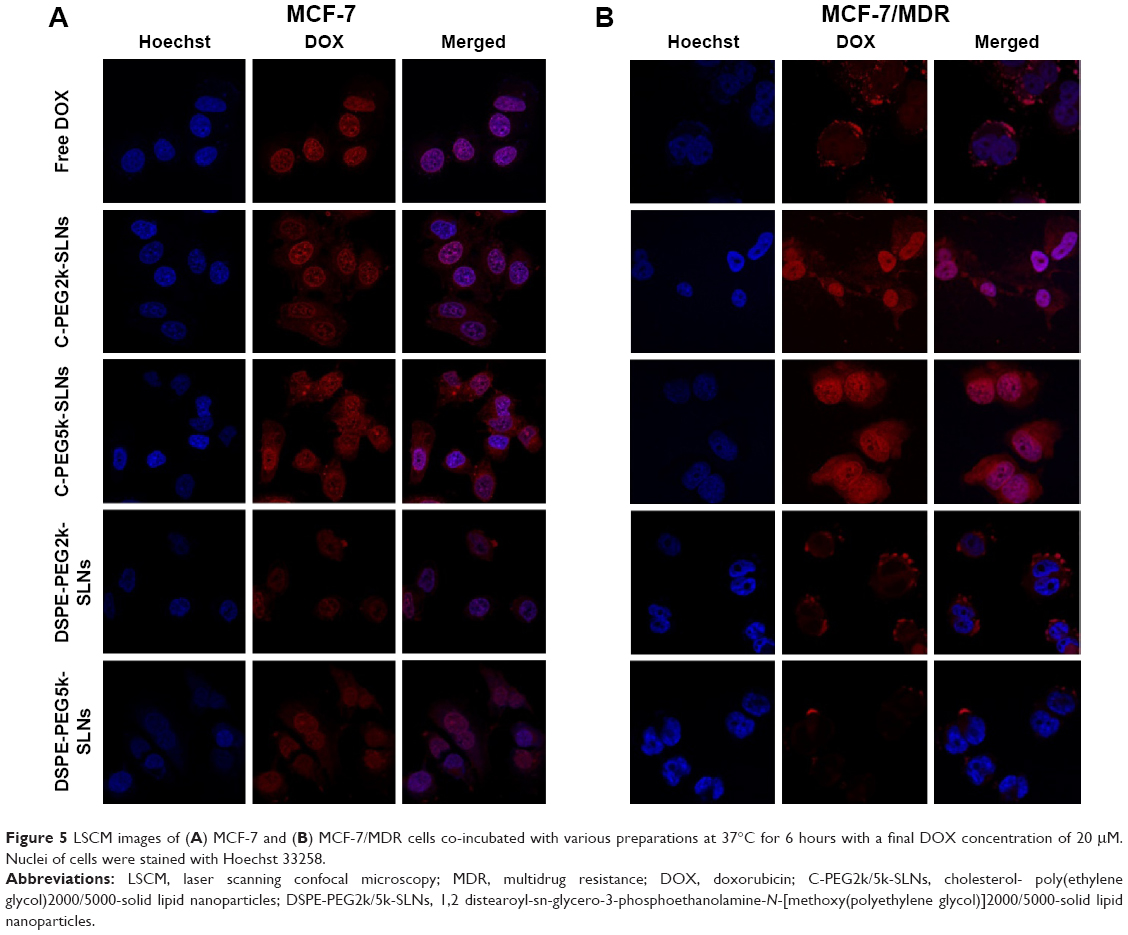 | Figure 5 LSCM images of (A) MCF-7 and (B) MCF-7/MDR cells co-incubated with various preparations at 37°C for 6 hours with a final DOX concentration of 20 μM. Nuclei of cells were stained with Hoechst 33258. |
The amounts of DOX residing within MCF-7/MDR after the cellular internalization of C-PEG-SLNs via endocytosis were quantified by fluorescence analysis upon cell disruption. The MCF-7/MDR cells treated with free DOX (20 μM) containing 50 μM verapamil, a P-gp inhibitor,49,50 and those treated with free DOX alone were used as the positive and negative controls, respectively. As shown in Figure 6A, the amount of DOX accommodated intracellularly by the positive control, 1.86±0.12 μg, was twofold of the negative control, 0.95±0.05 μg. As expected, low DOX accumulation, essentially identical to the negative control, was observed for MCF-7/MDR treated with DSPE-PEG2k/5k-SLNs, 1.09±0.08 μg, and 1.00±0.05 μg, respectively. Enhanced intracellular DOX accumulation, on the other hand, equivalent to that for the positive control was observed for MCF-7/MDR treated with C-PEG2k/5k-SLNs, 1.60±0.03 μg, and 1.85±0.04 μg, respectively. A concentration-dependent P-gp inhibition and intracellular DOX accommodation were observed for C-PEG but not for DSPE-PEG (Figure 6B). These results imply that C-PEG-SLNs enhance DOX accumulation within MCF-7/MDR by inhibiting the ATP-mediated translocation activity of P-gp. To further validate the inhibitory effect of C-PEG on P-gp activity of MCF-7/MDR cells, the accumulation profiles of rhodamine 123, a well-known substrate of P-gp frequently used to evaluate the efficiency of P-gp-mediated drug efflux,51,52 were studied. As expected, enhanced intracellular rhodamine accumulation was only observed for MDR cancer cells treated with C-PEG but not those with DSPE-PEG (Figure S8).
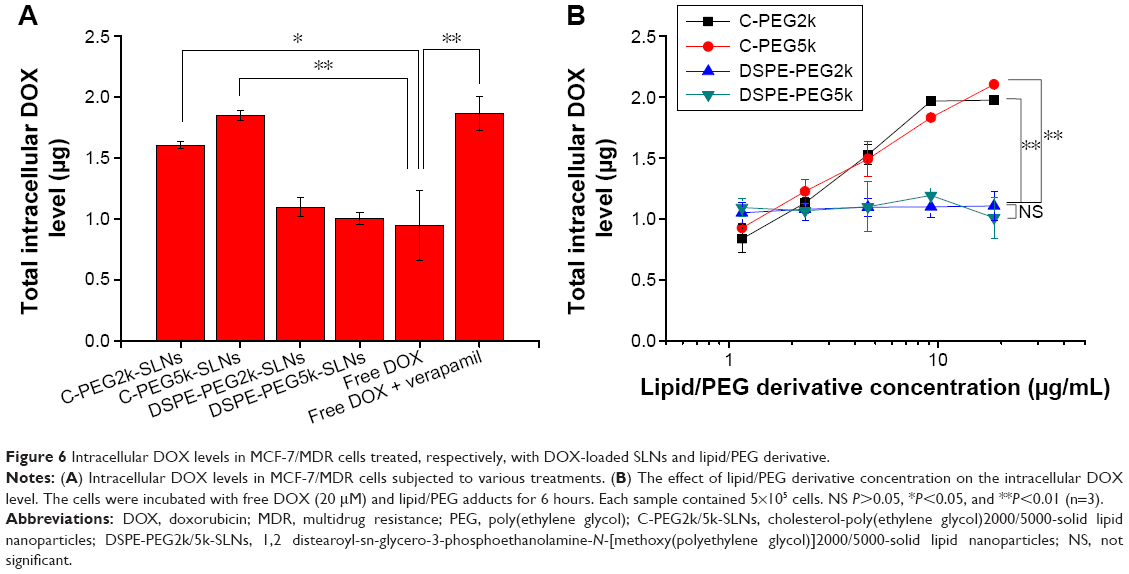 | Figure 6 Intracellular DOX levels in MCF-7/MDR cells treated, respectively, with DOX-loaded SLNs and lipid/PEG derivative. |
To further elucidate the mechanism of C-PEG-mediated P-gp inhibition, C-PEG(2k/5k)-R adducts were used to investigate the location of C-PEG within MCF-7/MDR cells. It has been postulated that the inhibitory effects of PEG-derived amphiphiles on the activity of P-gp can be classified by the action on either cell or mitochondria membranes. For the latter, the disturbance of mitochondria membranes by the attachment of PEG-derivatives, such as Pluronic, interferes with the ATP production, thereby leading to the depletion of energy required to drive the P-gp activity by ATPase. In contrast, the association of lipid/PEG adducts with cell membranes alters not only the membrane fluidity but also the lipid composition of lipid rafts intimately related to the function of P-gp, which could lead to the perturbation of membrane structure, and thus allosteric inhibition of ATPase and/or modulation of P-gp activity.26,53 As seen in Figure 7, the DiO-stained cell membrane exhibited a strong rhodamine fluorescence intensity, confirming that the C-PEG derivatives adopted herein accumulated on cell membrane. Since it has been shown that cholesterol in endosome is returned to cell membrane by the endocytic recycling compartment (ERC) transport,54 we believe that C-PEG released from SLNs in endosome was delivered via the endocytic recycling compartment pathway to cell membranes at which the allosteric inhibition of ATPase domain associated with P-gp thus developed. Although DSPE-PEG may also be returned to cell membrane by the endocytic recycling compartment transport as C-PEG, the structural similarity between DSPE and phospholipid of cell membrane allows DSPE-PEG to be better accommodated in cell membrane with limited perturbation on cell membrane fluidity and thus P-gp activity.
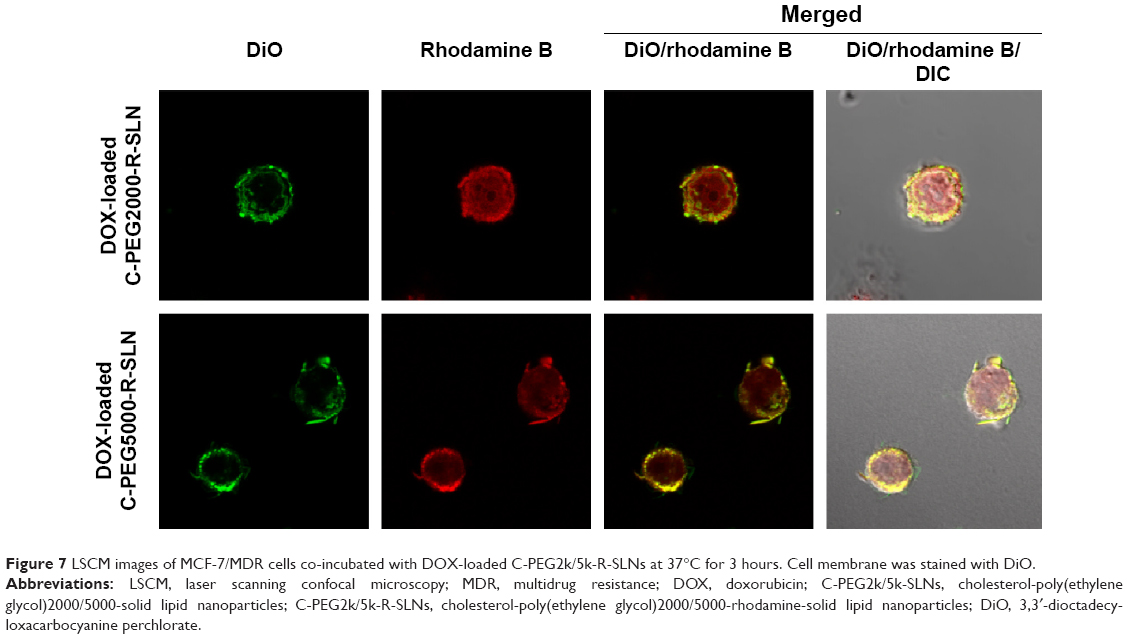 | Figure 7 LSCM images of MCF-7/MDR cells co-incubated with DOX-loaded C-PEG2k/5k-R-SLNs at 37°C for 3 hours. Cell membrane was stained with DiO. |
Biodistribution
To evaluate in vivo DOX delivery efficiency of the drug-loaded SLNs, administered via the tail vein, the MCF-7/MDR-bearing nude mice were used as the xenograft tumor model. Figure 8A shows the DOX fluorescence intensity of ex vivo organs and tumors by IVIS spectra. For groups treated with C-PEG-SLNs and DSPE-PEG-SLNs, high DOX fluorescence intensities were detected in the tumors with relatively low DOX intensities in the organs, indicating that large amounts of DOX were delivered preferentially to the tumor tissue possibly via the EPR effect. The DOX fluorescence intensities of the regions of interest of the selected organs/tumor were further quantified along with the background intensity correction from the PBS control and normalized to total corrected DOX fluorescence intensity of all isolated organs and tumor. As shown in Figure 8B, the percentages of DOX fluorescence intensities of livers, 14.1% and 14.5%, for C-PEG2k/5k-SLNs were significantly lower than that in the tumors, 45.6% and 50.6%. Although slightly lower, comparable accumulation of DOX in the tumors was also observed in the group treated with DSPE-PEG-SLNs. The results further confirm the accumulation of DOX in tumor by the passive tumor homing property of SLNs. It has been reported in the literature that surface-active PEGylated nanocarriers are capable of evading cellular uptake by the reticuloendothelial system present in liver and spleen.55 These results thus imply that the surface activities of C-PEG2k/5k and DSPE-PEG2k/5k are capable of protecting the SLNs from the reticuloendothelial system clearance.
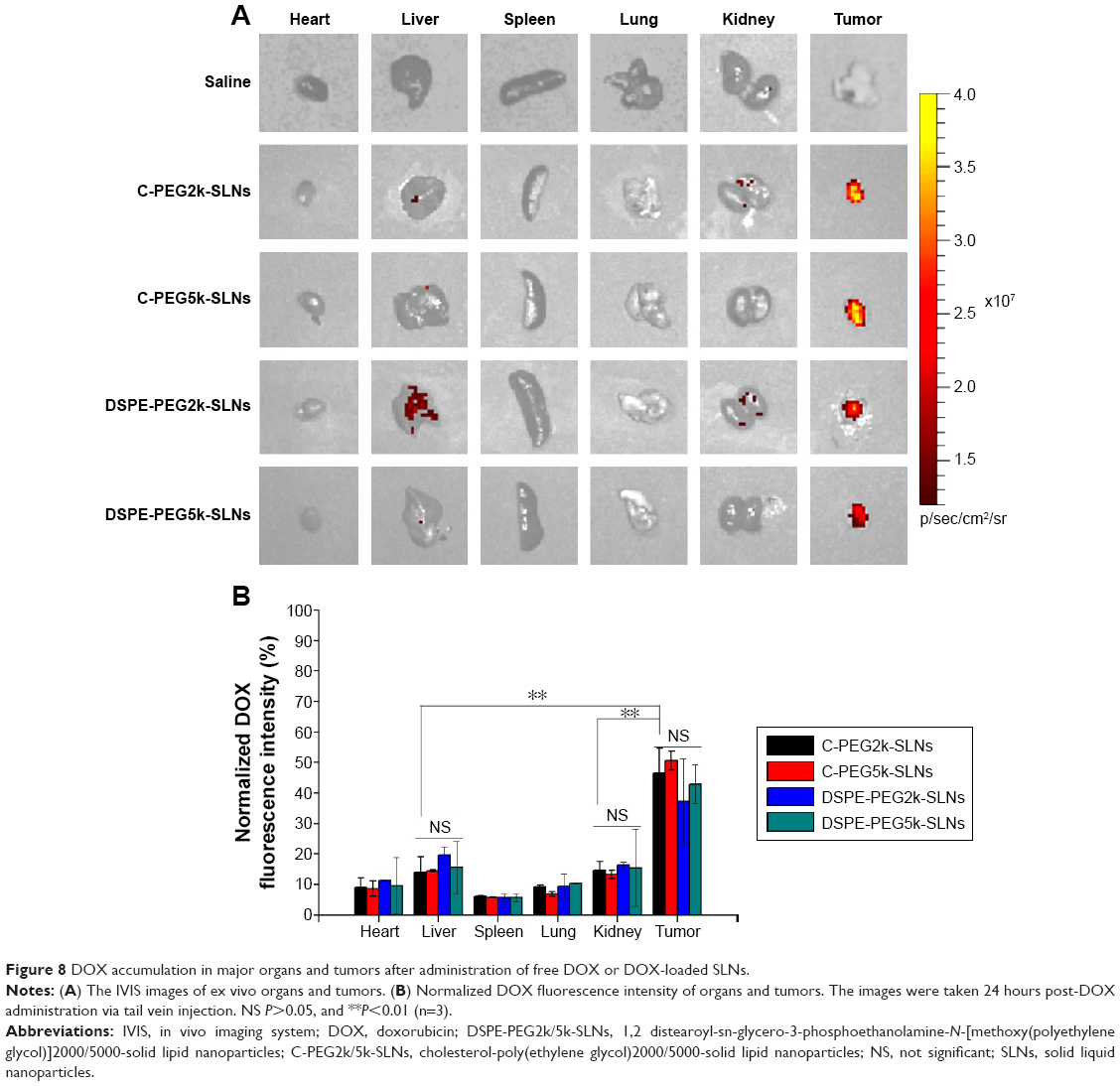 | Figure 8 DOX accumulation in major organs and tumors after administration of free DOX or DOX-loaded SLNs. |
Tumor growth inhibition
The MCF-7/MDR-bearing Balb/c nude mice were used as an animal tumor model to evaluate the tumor inhibition effect of SLNs. Based on the tumor growth inhibition curves shown in Figure 9, the tumor volumes of the group treated with free DOX, over 1,150 mm3, were close to those of the group treated with PBS, indicating that free DOX lacked tumor-targeting capability and was actively pumped out by the overexpressed P-gp in MDR xenograft tumor cells. The groups treated with DSPE-PEG2k/5k-SLNs exhibited medium inhibition efficiency, giving an average tumor volume of approximately 810 mm3, 30% lower than that of the group treated with free DOX, suggesting that the DSPE-PEG-SLNs accumulated to the tumor site via EPR effect. However, upon cellular endocytosis of SLNs, most of the DOX released in endosome/lysosome was actively excluded by the overexpressed P-gp, leaving a fraction of DOX in cancer cells, and thus giving a moderate inhibition efficiency. As expected, the growth of tumor was significantly depressed in the groups treated with C-PEG2k/5k-SLNs, giving an average tumor volume of ~400 mm3, one-third of the control. These results, in conjunction with the in vivo and in vitro data shown earlier, confirm the therapeutic efficacy of the C-PEG-SLNs in overcoming MDR of cancer cells.
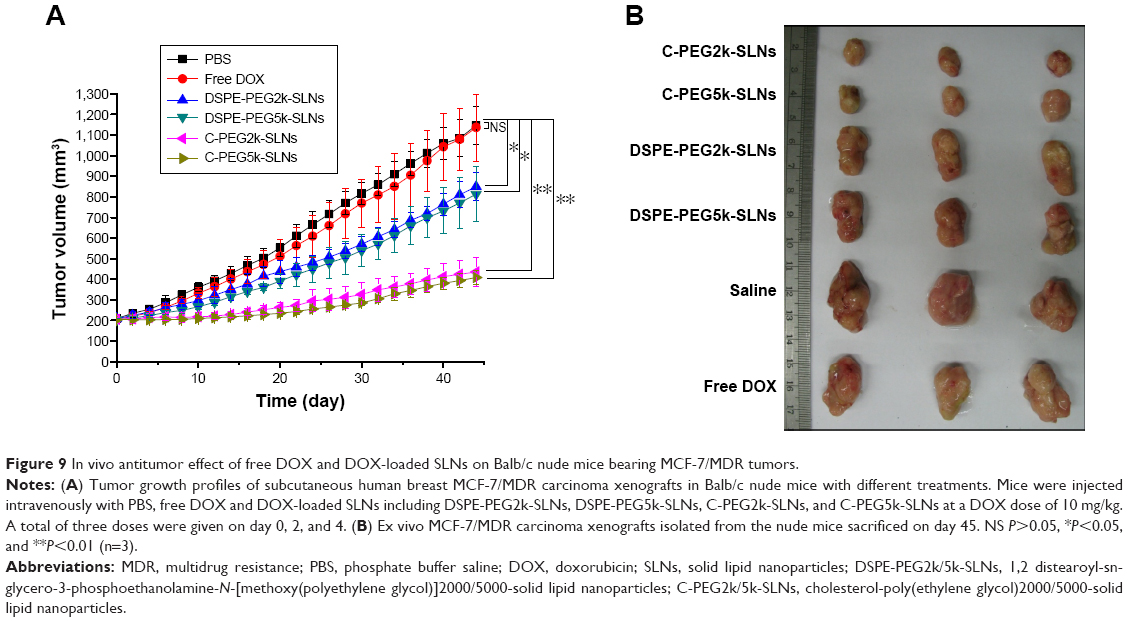 | Figure 9 In vivo antitumor effect of free DOX and DOX-loaded SLNs on Balb/c nude mice bearing MCF-7/MDR tumors. |
The variation of the body weights of mice after intravenous administration of DOX-loaded SLNs is shown in Figure S9. Eight days after the administration of free DOX, 15% reduction in body weight was observed. In contrast, no apparent change in body weight was observed in the groups treated with DOX-loaded SLNs, indicating that the SLNs effectively reduced the adverse effect of DOX to normal tissues. Serious damages to heart and liver were observed in the group treated with free DOX (highlighted by green arrows) in the histology images of organs and tumors, shown in Figure 10. In conjunction with the decrease in body weight described earlier, such damages indicate that the free DOX could induce detrimental side effects to nude mice. In contrast, while tumor necrosis of the groups treated with DOX-loaded C-PEG-SLNs was observed, no damage was observed in heart, lung, liver, or kidney, implying that C-PEG-SLNs can lower the inherent DOX side effects to normal tissues by transporting the DOX preferentially to tumor sites.
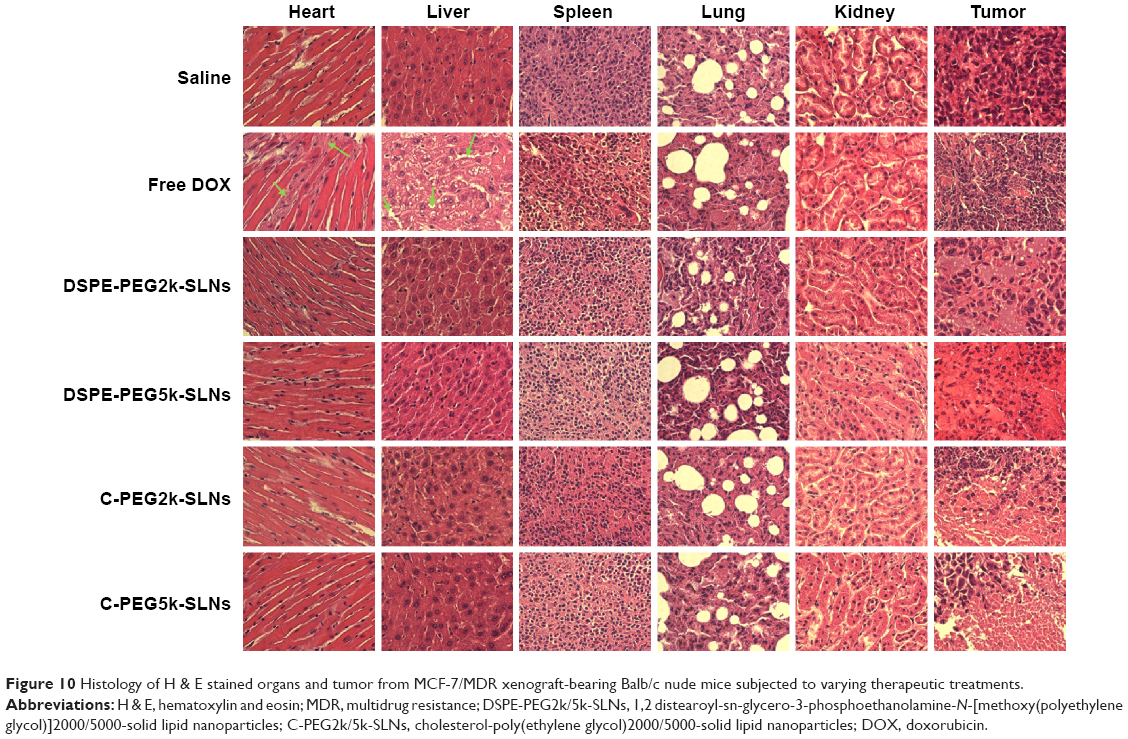 | Figure 10 Histology of H & E stained organs and tumor from MCF-7/MDR xenograft-bearing Balb/c nude mice subjected to varying therapeutic treatments. |
Conclusion
In this work, pH-responsive PEG-coated SLNs as nanocarriers for DOX were developed for improving the efficacy of MDR cancer chemotherapy. Through the pH-responsive electrostatic attractions between laurate and DOX, selective release of DOX in the microenvironment of tumors and in the acidic compartments of lysosomes and endosomes of cancer cells was achieved. The SLNs exhibited sound in vitro stability in FBS-rich medium and had a pertinent size for passive targeting via EPR effects in vivo, implying that the SLNs are stable in bloodstream and suitable for intravenous injection. The DOX-loaded C-PEG-SLNs displayed a superior capability of inhibiting the proliferation of MDR cancer cells due to the combined effects of intracellular pH-triggered drug release and the P-gp inhibition activity of C-PEG. Furthermore, the C-PEG-SLNs showed significant tumor inhibition in nude mice bearing human MDR breast cancer with no observable side effects. These results indicate the pH-responsive C-PEG-SLNs are promising nanocarriers for improving the efficacy of chemotherapy for MDR cancers. We believe that by incorporating additional targeting moieties and therapeutic agents, nanoparticles with enhanced efficacy and minimal side effects for MDR cancer chemotherapy can be developed based on the platform presented in this study.
Acknowledgments
This work is supported by the Ministry of Science and Technology (NSC 102-2221-E-007-133-MY3), National Tsing Hua University (101N7046E1) and National Taiwan University Hospital, Hsinchu Branch (HCH103-036), Taiwan.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161–171. | ||
Mitra S, Gaur U, Ghosh PC, Maitra AN. Tumour targeted delivery of encapsulated dextran-doxorubicin conjugate using chitosan nanoparticles as carrier. J Control Release. 2001;74:317–323. | ||
Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Del Rev. 2011;63:170–183. | ||
Potineni A, Lynn DM, Langer R, Amiji MM. Poly(ethylene oxide)-modified poly(beta-amino ester) nanoparticles as a pH-sensitive biodegradable system for paclitaxel delivery. J Control Release. 2003;86:223–234. | ||
Leslie EM, Deeley RG, Cole SPC. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. 2005;204:216–237. | ||
vanHelvoort A, Smith AJ, Sprong H, et al. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell. 1996;87:507–517. | ||
Iffert T, Soldan M, Moeller A, Maser E. Modulation of daunorubicin toxicity by liposomal encapsulation and use of specific inhibitors in vitro. Toxicology. 2000;144:189–195. | ||
Duan XP, Xiao JS, Yin Q, et al. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. ACS Nano. 2013;7:5858–5869. | ||
Lipman J, Jardine I, Roos C, Dreosti L. Intravenous calcium-chloride as an antidote to verapamil-induced hypotension. Intens Care Med. 1982;8:55–57. | ||
Berdeaux A, Coutte R, Giudicelli JF, Boissier JR. Effects of verapamil on regional myocardial blood-flow and ST segment – role of induced bradycardia. Eur J Pharmacol. 1976;39:287–294. | ||
Tang JL, Wang YJ, Wang D, et al. Key structure of brij for overcoming multidrug resistance in cancer. Biomacromolecules. 2013;14:424–430. | ||
Wu H, Hait WN, Yang JM. Small interfering RNA-induced suppression of MDR1 (P-glycoprotein) restores sensitivity to multidrug-resistant cancer cells. Cancer Res. 2003;63:1515–1519. | ||
Meng HA, Liong M, Xia TA, et al. Engineered design of mesoporous silica nanoparticles to deliver doxorubicin and P-glycoprotein siRNA to overcome drug resistance in a cancer cell line. ACS Nano. 2010;4:4539–4550. | ||
Navarro G, Sawant RR, Biswas S, Essex S, de Ilarduya CT, Torchilin VP. P-glycoprotein silencing with siRNA delivered by DOPE-modified PEI overcomes doxorubicin resistance in breast cancer cells. Nanomedicine. 2012;7:65–78. | ||
Saad M, Garbuzenko OB, Minko T. Co-delivery of siRNA and an anticancer drug for treatment of multidrug-resistant cancer. Nanomedicine. 2008;3:761–776. | ||
Luo Y, Chen DW, Ren LX, Zhao XL, Qin J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J Control Release. 2006;114:53–59. | ||
Minko T, Batrakova EV, Li S, et al. Pluronic block copolymers alter apoptotic signal transduction of doxorubicin in drug-resistant cancer cells. J Control Release. 2005;105:269–278. | ||
Shieh MJ, Hsu CY, Huang LY, Chen HY, Huang FH, Lai PS. Reversal of doxorubicin-resistance by multifunctional nanoparticles in MCF-7/ADR cells. J Control Release. 2011;152:418–425. | ||
Grant RL, Yao C, Gabaldon D, Acosta D. Evaluation of surfactant cytotoxicity potential by primary cultures of ocular-tissues: 1. characterization of rabbit corneal epithelial-cells and initial injury and delayed toxicity studies. Toxicology. 1992;76:153–176. | ||
Bansal T, Akhtar N, Jaggi M, Khar RK, Talegaonkar S. Novel formulation approaches for optimising delivery of anticancer drugs based on P-glycoprotein modulation. Drug Discov Today. 2009;14:1067–1074. | ||
Wempe MF, Wright C, Little JL, et al. Inhibiting efflux with novel non-ionic surfactants: rational design based on vitamin E TPGS. Int J Pharm. 2009;370:93–102. | ||
Ma Y, Liu D, Wang D, et al. Combinational delivery of hydrophobic and hydrophilic anticancer drugs in single nanoemulsions to treat MDR in cancer. Mol Pharm. 2014;11(8):2623–2630. | ||
Zhu H, Chen H, Zeng X, et al. Co-delivery of chemotherapeutic drugs with vitamin E TPGS by porous PLGA nanoparticles for enhanced chemotherapy against multi-drug resistance. Biomaterials. 2014;35:2391–2400. | ||
Hong W, Chen D, Zhang X, et al. Reversing multidrug resistance by intracellular delivery of Pluronic® P85 unimers. Biomaterials. 2013;34:9602–9614. | ||
Garrigues A, Escargueil AE, Orlowski S. The multidrug transporter, P-glycoprotein, actively mediates cholesterol redistribution in the cell membrane. P Natl Acad Sci U S A. 2002;99:10347–10352. | ||
dos Santos SM, Weber CC, Franke C, Muller WE, Eckert GP. Cholesterol: coupling between membrane microenvironment and ABC transporter activity. Biochem Biophys Res Commun. 2007;354:216–221. | ||
Collnot EM, Baldes C, Wempe MF, et al. Mechanism of inhibition of P-glycoprotein mediated efflux by vitamin E TPGS: influence on ATPase activity and membrane fluidity. Mol Pharm. 2007;4:465–474. | ||
Xu H, Deng YH, Chen DW, Hong WW, Lu Y, Dong XH. Esterase-catalyzed depegylation of pH-sensitive vesicles modified with cleavable peg-lipid derivatives. J Control Release. 2008;130:238–245. | ||
Harris JM, Struck EC, Case MG, Paley MS, Vanalstine JM, Brooks DE. Synthesis and characterization of poly(ethylene glycol) derivatives. J Polym Sci Pol Chem. 1984;22:341–352. | ||
Chiang WH, Huang WC, Chang CW, et al. Functionalized polymersomes with outlayered polyelectrolyte gels for potential tumor-targeted delivery of multimodal therapies and MR imaging. J Control Release. 2013;168:280–288. | ||
Lee ES, Na K, Bae YH. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J Control Release. 2005;103:405–418. | ||
Ruan JL, Tulloch NL, Muskheli V, et al. An improved cryosection method for polyethylene glycol hydrogels used in tissue engineering. Tissue Eng Part C Methods. 2013;19:794–801. | ||
Hollis CP, Weiss HL, Leggas M, Evers BM, Gemeinhart RA, Li TL. Biodistribution and bioimaging studies of hybrid paclitaxel nanocrystals: lessons learned of the EPR effect and image-guided drug delivery. J Control Release. 2013;172:12–21. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. | ||
Duhem N, Danhier F, Preat V. Vitamin E-based nanomedicines for anti-cancer drug delivery. J Control Release. 2014;182:33–44. | ||
Nair HB, Sung BY, Yadav VR, Kannappan R, Chaturvedi MM, Aggarwal BB. Delivery of antiinflammatory nutraceuticals by nanoparticles for the prevention and treatment of cancer. Biochem Pharmacol. 2010;80:1833–1843. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–515. | ||
Lee ES, Gao ZG, Bae YH. Recent progress in tumor pH targeting nanotechnology. J Control Release. 2008;132:164–170. | ||
Park SY, Baik HJ, Oh YT, Oh KT, Youn YS, Lee ES. A smart polysaccharide/drug conjugate for photodynamic therapy. Angew Chem Int Ed Engl. 2011;50:1644–1647. | ||
Oh KT, Baik HJ, Lee AH, Oh YT, Youn YS, Lee ES. The reversal of drug-resistance in tumors using a drug-carrying nanoparticular system. Int J Mol Sci. 2009;10:3776–3792. | ||
Kievit FM, Wang FY, Fang C, et al. Doxorubicin loaded iron oxide nanoparticles overcome multidrug resistance in cancer in vitro. J Control Release. 2011;152:76–83. | ||
Chawla JS, Amiji MM. Biodegradable poly(epsilon-caprolactone) nanoparticles for tumor-targeted delivery of tamoxifen. Int J Pharm. 2002;249:127–138. | ||
Muller RH, Ruhl D, Runge SA. Biodegradation of solid lipid nanoparticles as a function of lipase incubation time. Int J Pharm. 1996;144:115–121. | ||
Fu DD, Hornick CA. Modulation of lipid-metabolism at rat hepatic subcellular sites by female sex-hormones. Biochim Biophys Acta. 1995;1254:267–273. | ||
Schweitzer JK, Sedgwick AE, D’Souza-Schorey C. ARF6-mediated endocytic recycling impacts cell movement, cell division and lipid homeostasis. Semin Cell Dev Biol. 2011;22:39–47. | ||
Liscum L, Sturley SL. Intracellular trafficking of Niemann-Pick C proteins 1 and 2: obligate components of subcellular lipid transport. Biochim Biophys Acta. 2004;1685:22–27. | ||
Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65:157–170. | ||
You HY, Jin J, Shu HQ, et al. Small interfering RNA targeting the subunit ATP6L of proton pump V-ATPase overcomes chemoresistance of breast cancer cells. Cancer Lett. 2009;280:110–119. | ||
Bansal T, Jaggi M, Khar RK, Talegaonkar S. Emerging significance of flavonoids as P-glycoprotein inhibitors in cancer chemotherapy. J Pharm Pharm Sci. 2009;12:46–78. | ||
Schwab D, Fischer H, Tabatabaei A, Poli S, Huwyler J. Comparison of in vitro P-glycoprotein screening assays: recommendations for their use in drug discovery. J Med Chem. 2003;46:1716–1725. | ||
Chaudhary PM, Roninson IB. Expression and activity of p-glycoprotein, a multidrug efflux pump, in human hematopoietic stem-cells. Cell. 1991;66:85–94. | ||
O’Connor R, Ooi MG, Meiller J, et al. The interaction of bortezomib with multidrug transporters: implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemoth Pharm. 2013;71:1357–1368. | ||
Zastre J, Jackson JK, Wong W, Burt HM. Methoxypolyethylene glycol-block-polycaprolactone diblock copolymers reduce P-glycoprotein efflux in the absence of a membrane fluidization effect while stimulating P-glycoprotein ATPase activity. J Pharm Sci. 2007;96:864–875. | ||
Soccio RE, Breslow JL. Intracellular cholesterol transport. Arterioscl Throm Vas. 2004;24:1150–1160. | ||
Li SD, Huang L. Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. Biochim Biophys Acta. 2009;1788:2259–2266. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.