Back to Journals » Therapeutics and Clinical Risk Management » Volume 10
Personalized therapeutics for levofloxacin: a focus on pharmacokinetic concerns
Authors Gao C, Yu L, Zeng S, Huang Y, Zhou Q ![]()
Received 12 December 2013
Accepted for publication 11 February 2014
Published 27 March 2014 Volume 2014:10 Pages 217—227
DOI https://doi.org/10.2147/TCRM.S59079
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Chu-Han Gao,1 Lu-Shan Yu,2 Su Zeng,2 Yu-Wen Huang,1 Quan Zhou1
1Department of Pharmacy, the Second Affiliated Hospital, School of Medicine, 2Department of Pharmaceutical Analysis and Drug Metabolism, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, Zhejiang Province, People's Republic of China
Background: Personalized medicine should be encouraged because patients are complex, and this complexity results from biological, medical (eg, demographics, genetics, polypharmacy, and multimorbidities), socioeconomic, and cultural factors. Levofloxacin (LVX) is a broad-spectrum fluoroquinolone antibiotic. Awareness of personalized therapeutics for LVX seems to be poor in clinical practice, and is reflected in prescribing patterns. Pharmacokinetic–pharmacodynamic studies have raised concerns about suboptimal patient outcomes with the use of LVX for some Gram-negative infections. Meanwhile, new findings in LVX therapeutics have only been sporadically reported in recent years. Therefore, an updated review on personalized LVX treatment with a focus on pharmacokinetic concerns is necessary.
Methods: Relevant literature was identified by performing a PubMed search covering the period from January 1993 to December 2013. We included studies describing dosage adjustment and factors determining LVX pharmacokinetics, or pharmacokinetic–pharmacodynamic studies exploring how best to prevent the emergence of resistance to LVX. The full text of each included article was critically reviewed, and data interpretation was performed.
Results: In addition to limiting the use of fluoroquinolones, measures such as reducing the breakpoints for antimicrobial susceptibility testing, choice of high-dose short-course of once-daily LVX regimen, and tailoring LVX dose in special patient populations help to achieve the validated pharmacokinetic–pharmacodynamic target and combat the increasing LVX resistance. Obese individuals with normal renal function cleared LVX more efficiently than normal-weight individuals. Compared with the scenario in healthy subjects, standard 2-hour spacing of calcium formulations and oral LVX was insufficient to prevent a chelation interaction in cystic fibrosis patients. Inconsistent conclusions were derived from studies of the influence of sex on the pharmacokinetics of LVX, which might be associated with sample size and administration route. Children younger than 5 years cleared LVX nearly twice as fast as adults. Patients in intensive care receiving LVX therapy showed significant pharmacokinetic differences compared with healthy subjects. Creatinine clearance explained most of the population variance in the plasma clearance of LVX. Switching from intravenous to oral delivery of LVX had economic benefits. Addition of tamsulosin to the LVX regimen was beneficial for patients with bacterial prostatitis because tamsulosin could increase the maximal concentration of LVX in prostatic tissue. Coadministration of multivalent cation-containing drugs and LVX should be avoided. For patients receiving warfarin and LVX concomitantly, caution is needed regarding potential changes in the international normalized ratio; however, it is unnecessary to seek alternatives to LVX for the sake of avoiding drug interaction with warfarin. It is unnecessary to proactively reduce the dose of cyclosporin or tacrolimus when comedicated with LVX. Transporters such as organic anion-transporting polypeptide 1A2, P-glycoprotein, human organic cation transporter 1, and multidrug and toxin extrusion protein 1 are involved in the pharmacokinetics of LVX.
Conclusion: Personalized LVX therapeutics are necessary for the sake of better safety, clinical success, and avoidance of resistance. New findings regarding individual dosing of LVX in special patient populations and active transport mechanisms in vivo are opening up new horizons in clinical practice.
Keywords: drug interactions, fluoroquinolone resistance, individual dosing, patient complexity, personalized medicine, pharmacokinetics, pharmacodynamics, therapeutics
Introduction
Patients are complex, and further complexity results from factors such as biological, medical (demographics, genetics, polypharmacy, multimorbidities, medication adherence, dietary habits), socioeconomic, and cultural factors.1,2 Therefore, personalized medicine should be encouraged in clinical practice.
According to the fourth edition of the Joint Commission International accreditation standards, appropriateness of physician orders or prescriptions should be evaluated by trained pharmacists prior to dispensing. Pharmacists should not only audit the medication information (eg, dose, administration route, dose frequency, and current medications), but also check key patient information (eg, age, sex, body weight, body surface area, current diagnosis, allergy history, nutrition status, and clinical laboratory examination results, such as hepatic and renal function, international normalized ratio [INR], routine blood tests, and serum drug levels).3–5 The process of review of appropriateness fully reflects the attention to personalized therapeutics.
Levofloxacin (LVX) is a broad-spectrum antibiotic of the fluoroquinolone drug class. It is rapidly and completely absorbed after oral administration.6 Peak plasma concentration (Cmax) is usually attained 1–2 hours after oral dosing. The plasma concentration profile of LVX after intravenous administration is comparable in area under the concentration-time curve (AUC) to that observed for oral tablets when equal doses are administered. LVX is excreted largely (87%) as unchanged drug in the urine. The mean terminal plasma elimination half-life (t1/2) of LVX ranges from approximately 6 to 8 hours.6,7
LVX should be administered only as described within the dosage guidelines table found within the package insert. However, a recent survey on appropriateness of physician orders relating to LVX in our hospital indicated no individual patient tailoring for administration of LVX. All patients received oral or intravenous LVX at a set dose of 500 mg regardless of patient complexity, indicating poor awareness of individual dosing of LVX in clinical practice. Prajapati and Ganguly assessed the accuracy of drug dosing and frequency in patients with renal dysfunction in a tertiary care hospital.8 Of the total drugs (1,338) prescribed to the patients, 180 (13.45%) drugs needed dose adjustment according to creatinine clearance (CrCl). Of these 180 drugs, 146 (81.11%) were not adjusted according to CrCl and were rated as “inappropriate”. LVX was found to be the most inappropriately prescribed individual drug. It seems that there is an underestimation or lack of knowledge of the importance regarding CrCl in determining the appropriate drug dose.
Meanwhile, in recent years, new findings concerning the pharmacokinetics and therapeutic effects of LVX have been sporadically reported. Pharmacokinetic–pharmacodynamic (PK–PD) studies have raised concerns about suboptimal patient outcomes with the use of LVX for certain Gram-negative infections. To our knowledge, no up-to-date review is available on personalized LVX therapeutics with a focus on pharmacokinetic concerns. Therefore, we now present a review on this issue so as to enhance the awareness of patient-tailored LVX dosing and guide rational use of this drug.
Methods
Relevant literature was identified by performing a PubMed search covering the period from January 1993 (the year LVX was launched) to December 2013, using the search terms “levofloxacin” and “pharmacokinetics” and additional filters (language, English; species, human). Three hundred and ninety-seven articles were identified. We considered studies that described dosage adjustment of LVX and factors determining the pharmacokinetics of LVX, and PK–PD studies that explored how best to prevent emergence of resistance to LVX. Thirty-nine papers met our inclusion/exclusion criteria and were finally included. The full text of each paper was critically reviewed, and valuable information was summarized by interpretation of the data.
Results
Prevention of emergence of resistance to LVX from the PK–PD modeling perspective
The fluoroquinolones show concentration-dependent killing and a postantibiotic effect.9 Two PK–PD indices commonly used to evaluate fluoroquinolone dosing and response are the ratio of the area under the concentration-time curve for 24 hours (AUC0–24) to the minimum inhibitory concentration (MIC) (AUC0–24/MIC) and the ratio of peak concentration to MIC (Cmax/MIC).10 The AUC0–24/MIC predicts microbiological outcomes for patients treated with LVX and Cmax/MIC is an indicator for selection of LVX resistance.
Multiple studies have documented the declining susceptibilities of Gram-negative isolates to the fluoroquinolones.10 In addition to limiting the use of fluoroquinolones, increasing fluoroquinolone doses is an option to improve the outcomes for patients with Gram-negative infections. However, this may put some patients at undue risk for increased adverse events (eg, those with low MICs who can attain PK–PD targets with normal dosages). The maximum steady-state AUC value in healthy volunteers after intravenous administration of LVX 750 mg every 24 hours is 108 mg · hr/L. In order to attain an AUC/MIC of ≥125 in the blood at this AUC, the MIC value for LVX must remain at ≤0.86 mg/mL, which is considerably lower than the level deemed to indicate susceptibility according to Clinical and Laboratory Standards Institute standards (2 mg/mL for LVX).11 Defife et al stratified patients into three groups, ie, low MIC (≤0.25 mg/L), intermediate MIC (0.5 mg/L), and high MIC (1–2 mg/L),12 and found that a high MIC was associated with a significant (5.7 days) increase in length of stay post-culture relative to the mean length of stay post-culture in the low MIC group (P=0.02). The study suggests that higher MICs may be associated with poorer clinical outcomes even when these MICs are in the “susceptible” range. Therefore, another option for combating the rising resistance to LVX is to reduce the breakpoints for antimicrobial susceptibility testing, which could increase the likelihood of attaining current PK–PD targets for LVX and reduce the need to increase the dose.10
Compared with the well-established regimen of LVX 500 mg once daily for 10 days in patients with community-acquired pneumonia, acute bacterial sinusitis, complicated urinary tract infection, and acute pyelonephritis, the high-dose (750 mg) short course (5 days) of once-daily LVX regimen maximizes its concentration-dependent bactericidal activity and may reduce the potential for resistance to emerge. In addition, this regimen lends itself to better compliance due to the shorter duration of treatment and the convenient once-daily administration schedule.13,14
Pharmacokinetic alterations and tailoring the LVX dose in special patient populations
Pharmacokinetic alterations and individual dosing of LVX in special patient populations is summarized in Table 1.15–21
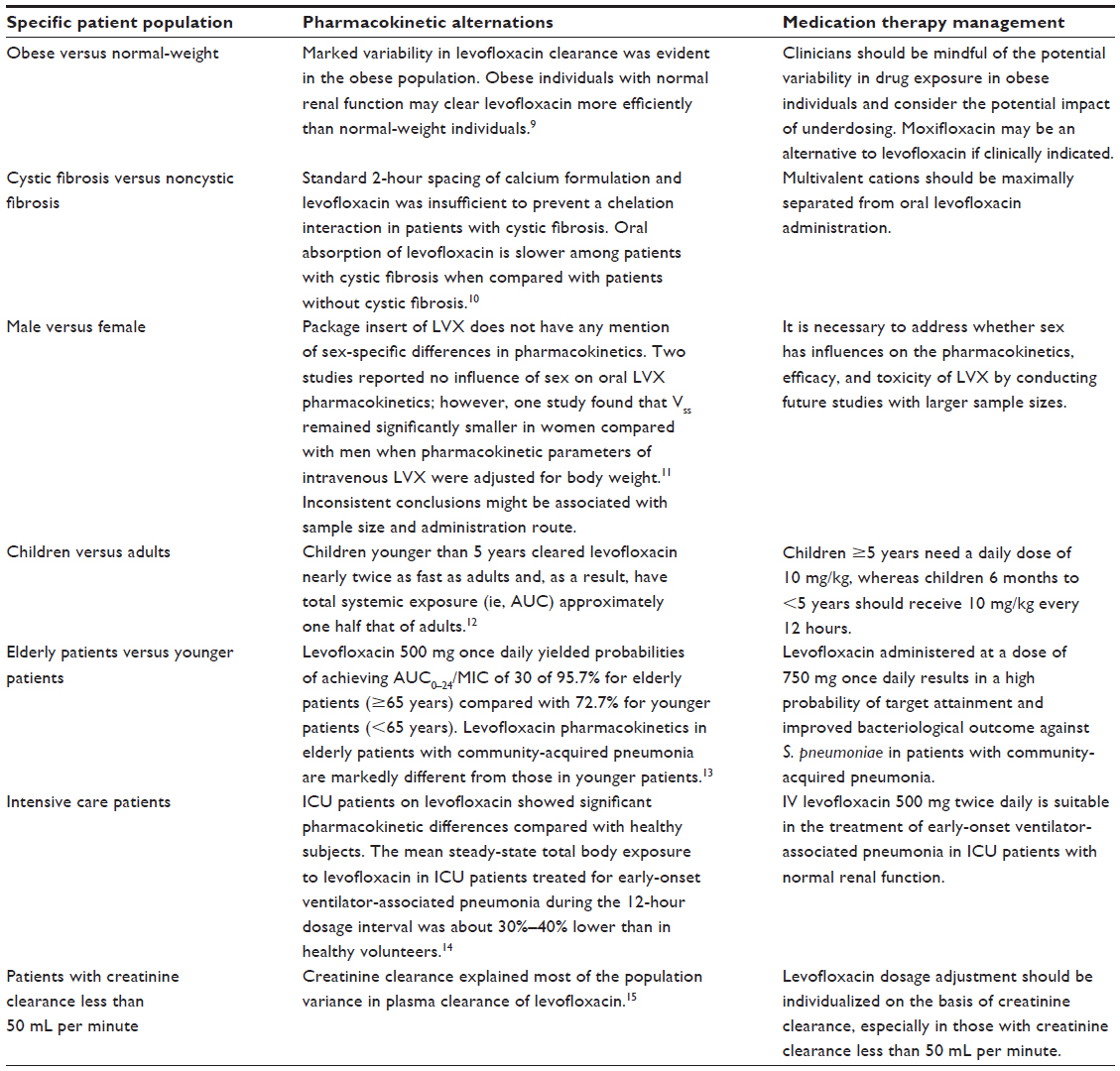 | Table 1 Pharmacokinetic concerns in personalized therapeutics for levofloxacin in specific patient populations |
Obese individuals versus normal-weight individuals
People are considered obese when the body mass index exceeds 30 kg/m2. Obesity causes a number of changes, including an increase in volume of distribution and changes in hepatic metabolism and renal excretion.22 Cook et al described the effects of obesity on LVX pharmacokinetics in a study where a single 750 mg intravenous dose of LVX was given to both hospitalized and ambulatory obese individuals.15 All participants had a mean body mass index approaching 50 kg/m2. The Cmax of LVX in these obese individuals appeared to be similar to that of normal-weight individuals. However, marked variability in LVX clearance was seen in this obese population. Ambulatory obese individuals showed markedly increased LVX clearance, resulting in a much lower AUC than would be expected in normal-weight individuals. Obese individuals with normal renal function clear LVX more efficiently than normal-weight individuals.
Although the target AUC0–24/MIC ratio likely depends on the causative pathogen, the most accurate predictor of fluoroquinolone success is the AUC0–24/MIC. Clinicians should be mindful of the potential variability in drug exposure in obese individuals and consider the potential impact of underdosing when evaluating the response to infection. Luque et al have published a case report of LVX weight-adjusted dosing and pharmacokinetic disposition in a morbidly obese patient (weight, 179 kg; body mass index, 56.2 kg/m2).23 An intravenous LVX dose of 750 mg (4 mg/kg) was given to this patient at 12-hourly intervals and achieved double the adult exposure following a standard dose of 750 mg per 24 hours to nonobese healthy subjects. Additionally, a longer t1/2 was observed due to an increase in steady-state volume of distribution (Vss), indicating that it would be appropriate to administer an initial loading dose to achieve steady state rapidly, followed by administration of doses less frequently than 12 hourly in order to avoid potential drug accumulation.
Kees et al assessed the pharmacokinetics of oral versus intravenous moxifloxacin in morbidly obese patients. Their study results indicated that the pharmacokinetics of moxifloxacin were not significantly affected by morbid obesity.24 No dose adjustment seems to be necessary for moxifloxacin in this particular population, which supports the use of moxifloxacin as an alternative fluoroquinolone if clinically indicated.
Cystic fibrosis versus noncystic fibrosis
Cystic fibrosis is associated with numerous pathological changes that can alter the disposition of drugs. The mean gastric emptying and small intestinal transit times may be twice as long in patients with cystic fibrosis compared with healthy subjects, and the rate of drug absorption may be affected.25
Fluoroquinolones are commonly used to treat lung infections in patients with cystic fibrosis. These patients are susceptible to lung infection with common bacteria such as Staphylococcus aureus and Haemophilus influenzae, but are also prone to infection by opportunistic bacteria, including Pseudomonas aeruginosa.26 Lee et al investigated the pharmacokinetics of LVX in adults with cystic fibrosis.27 The steady-state pharmacokinetics of LVX in patients with cystic fibrosis receiving 500 mg once daily orally were similar to those in subjects without cystic fibrosis when corrected for body mass, except for the time to Cmax (Tmax). Patients with cystic fibrosis have a longer Tmax probably due to prolonged gastric emptying (cystic fibrosis versus non-cystic fibrosis: 2.20±0.99 hours versus 1.1±0.4 hours, respectively, P<0.01). Pai et al characterized the steady-state pharmacokinetics of LVX 750 mg given orally with and without 2-hour spaced calcium carbonate in patients with cystic fibrosis and compared them with matched healthy volunteers.16 There was no significant interaction between LVX and calcium carbonate in healthy volunteers; however, when patients with cystic fibrosis received LVX with 2-hour spaced calcium, the Cmax decreased by 19% and Tmax increased by 37% (P<0.05), indicating that standard 2-hour spacing between the calcium formulation and LVX was insufficient to prevent a chelation interaction. Patients with cystic fibrosis are usually prescribed calcium carbonate to prevent osteoporosis and may be at risk for this interaction. Other cations, such as aluminum, iron, and magnesium, may have greater impact on the absorption of LVX, given that they have a higher chelation potential than calcium. Therefore, multivalent cations should be maximally separated from oral LVX administration to further optimize the antibiotic efficacy and decrease the potential for development of resistance in patients with cystic fibrosis.
MP-376 is a novel solution formulation for aerosol administration of LVX, and was developed for patients with cystic fibrosis who have chronic infections due to P. aeruginosa. In one study, this product was used at three doses (120 mg every day, 240 mg every day, 240 mg twice a day) for 28 days, and was well tolerated and demonstrated significant clinical efficacy in heavily treated patients with cystic fibrosis and pulmonary P. aeruginosa infection.28 All doses of MP-376 resulted in reduced density of P. aeruginosa in sputum at day 28, with MP-376 240 mg twice a day showing a 0.96 log difference compared with placebo (P=0.001). There was a dose-dependent increase in the forced expiratory volume in one second (FEV1) for MP-376, with a difference of 8.7% in FEV1 between the 240 mg twice a day group and placebo (P=0.003). Significant reductions (61%–79%) in the need for other antimicrobials to treat P. aeruginosa were observed with all MP-376 treatment groups when compared with placebo. From this perspective, use of MP-376 may circumvent the special administration requirements when comedication of oral LVX and multivalent cation drugs is unavoidable.
Males versus females
Almeida et al compared the bioavailability of two tablet formulations of LVX and evaluated the effect of sex on analysis of bioequivalence. The use of sex-related model effects showed that sex was a significant factor for the AUC; however, when the parameters were normalized by dose adjusted to body weight, none of the tested model effects were significant. Possible differences in pharmacokinetic parameters between males and females may be related to differences in body weight.29 Chien et al investigated the influence of sex on the pharmacokinetics of LVX in healthy subjects receiving a single oral 500 mg dose of LVX, and concluded that dose adjustment based on sex alone was not necessary given that the differences in LVX kinetics between males and females are minimal and mainly related to renal function.30
Overholser et al compared the pharmacokinetics of intravenous LVX after a single 500 mg dose in men and women who showed no significant difference in body mass index, and observed that women had a 24% greater exposure (AUC) to LVX, with a significantly smaller Vss (P<0.01) and a slower clearance (P<0.01).17 When pharmacokinetic parameters were adjusted for both lean and total body weight, the difference in clearance was attenuated, but Vss remained significantly smaller in women than in men, indicating a sex difference in Vss after intravenous administration of LVX. The investigators concluded that intravenous LVX dosage adjustments based on sex should be considered on an individual basis and that women may have an increased risk of fluoroquinolone toxicity than men whereas men may need higher doses to achieve similar drug efficacy than women. However, dosing recommendations in the LVX product labeling approved by the US Food and Drug Administration do not mention any sex-specific differences in pharmacokinetics.31 Inconsistent conclusions derived from studies of the influence of sex on the pharmacokinetics of LVX may be associated with sample size and administration route. It is worth noting that Bailey et al have demonstrated male sex to be a significant risk factor for resistance to LVX (P<0.05).32 Therefore, it is necessary to address whether sex has influences on the pharmacokinetics, efficacy, and toxicity of LVX by conducting further studies with larger sample sizes.
Children versus adults
Fluoroquinolones, including LVX, are not recommended for use in children, mainly because studies in juvenile laboratory animals suggest that there may be an increased risk of fluoroquinolone-associated cartilage lesions. However, in May 2008, LVX was approved by the US Food and Drug Administration as a treatment for children following inhalational exposure to anthrax. The risk–benefit assessment indicated that administration of LVX to pediatric patients with inhalational anthrax (post-exposure) is appropriate.31 According to the prescribing information for LVX,31 the dosage in pediatric patients ≥6 months of age is as follows: 500 mg per 24 hours for 60 days (>50 kg bodyweight) and 8 mg/kg (not to exceed 250 mg per dose) per 12 hours for 60 days (<50 kg bodyweight).
Chien et al conducted a single-dose, multicenter pharmacokinetic study in 85 children in five age groups (6 months to <2 years, 2 to <5 years, 5 to <10 years, 10 to <12 years, and 12–16 years) and described the basis of the pediatric dose recommendation for LVX in the prescribing information.18 Each child received a single 7 mg/kg dose of LVX intravenously or orally. LVX absorption (reflected by Cmax and Tmax) and distribution in children were similar to that in adults, whereas elimination of LVX (reflected by t1/2 and clearance) was age-dependent. Children younger than 5 years cleared LVX nearly twice as fast as adults and had a systemic exposure level about one half that of adults. To provide compatible LVX exposures associated with clinical effectiveness and safety in adults, the investigators recommended that children aged 5 years and older need a daily dose of 10 mg/kg, whereas children 6 months to <5 years should receive 10 mg/kg every 12 hours.
Elderly patients versus younger patients
A population pharmacokinetics study of LVX in 522 subjects, including normal individuals and patients with infectious diseases, reported that elderly subjects (≥65 years) had a 32% reduction in clearance and a 6% greater apparent volume of distribution.33
Chien et al investigated the influence of age on the pharmacokinetics of LVX in healthy subjects receiving a single oral 500 mg dose of LVX and found that the difference in LVX kinetics between the young and the elderly was minimal.30 Noreddin et al determined the target attainment potential of LVX administered at various dosing regimens according to age in inpatients with community-acquired pneumonia (ie, elderly [≥65 years] versus younger [<65 years] patients).19 Dosing regimens for LVX were evaluated as 500 mg, 750 mg, and 1,000 mg once daily administered to both elderly patients and younger patients. Monte Carlo simulation using LVX 500 mg once daily yielded probabilities of achieving a free-drug AUC0–24/ MIC of 30 of 95.7% for elderly patients compared with 72.7% for younger patients. LVX 750 mg and 1,000 mg once daily had probabilities of achieving free-drug AUC0–24/MIC of 30 of 98.1% and 99.2% and of 90.1% and 95.2% for elderly patients and younger patients, respectively. The pharmacokinetics of LVX in elderly patients with community-acquired pneumonia were markedly different from those in younger patients. Higher AUC and longer t1/2 values in elderly patients with community-acquired pneumonia compared with younger patients indicate a higher probability of PK–PD target attainment and improved bacteriological outcome in patients with S. pneumoniae.
Intensive care patients
In general, the pharmacokinetics of antibiotics in intensive care patients are known to differ from those in healthy subjects. Tayab et al assessed the relationship between patient covariates and the pharmacokinetic parameters of LVX in critically ill patients and found that CrCl was a statistically significant predictor of variability in total LVX clearance.34
Intensive care patients on LVX therapy showed significant differences in pharmacokinetics compared with healthy subjects. Patients with a high renal clearance who receive a standard dose of 500 mg of LVX may not achieve a high enough AUC/MIC ratio, and thus may show a decreased antibiotic effect. Conversely, patients with reduced renal clearance may have a greater exposure of LVX because of a larger AUC/MIC.35 This may have significant implications in clinical practice (eg, avoiding the potential of significant drug accumulation and any undesired effects) for identifying optimal dosage regimens for intensive care patients, especially for those with impaired kidney function. Sánchez Navarro et al evaluated the influence of clinical circumstances in intensive care units on the pharmacokinetic behavior of LVX 500 mg/day by multiple regression analysis.36 CrCl was found to be the only one independent variable, explaining more than 85% of the observed variability in AUC. The apparent volume of distribution value showed a statistically significant correlation with severity of illness on the Simplified Acute Physiology Score II.36 CrCl, as well as total bodyweight and severity of illness, might be considered when designing the dosage regimen of LVX to achieve AUC/MIC and Cmax/MIC values with higher probabilities of clinical success and avoidance of resistance.36
Pea et al investigated the pharmacokinetics of LVX and the PK–PD appropriateness of its total body exposure in intensive care patients treated with intravenous LVX 500 mg twice daily for early-onset ventilator-associated pneumonia.20 The mean steady-state total body exposure to LVX in these patients during the 12-hour dosage interval was about 30%–40% lower than in healthy volunteers. This reduced exposure might be the consequence of a much greater mean LVX clearance in these patients when compared with that in healthy volunteers (3.40 versus 2.42 mL/min/kg, respectively), resulting in a shorter t1/2 (5.2 hours versus 7.6 hours). More unmetabolized LVX was excreted by these intensive care patients than by healthy subjects (76% versus 68%, respectively). Coadministered drugs used to treat underlying diseases (eg, dopamine, furosemide, mannitol) might partially account for the enhanced elimination in critically ill patients. These findings support the suitability of intravenous LVX 500 mg twice daily in the treatment of early-onset ventilator-associated pneumonia in intensive care patients with normal renal function since the regimen ensures appropriate Cmax/MIC and AUC/MIC values against methicillin-sensitive S. aureus and H. influenzae.
Patients with creatinine clearance less than 50 mL per minute
Preston et al developed a population model of the disposition of LVX and found that CrCl explained most of the population variance in plasma clearance of LVX.21 LVX dosage adjustment should be individualized on the basis of the CrCl, especially in those with CrCl less than 50 mL per minute. For patients with a CrCl of 20–49 mL per minute, a 500 mg dose should be followed by 250 mg every 24 hours. For those with a CrCl less than 20 mL per minute, the same initial dose should be followed by 250 mg every 48 hours. This extended interval also applies to patients on hemodialysis or chronic ambulatory peritoneal dialysis. For patients with a CrCl of 20–49 mL per minute, an initial dose of 750 mg should be followed by 750 mg every 48 hours. For those with a CrCl less than 20 mL per minute, including patients on hemodialysis or chronic ambulatory peritoneal dialysis, the initial 750 mg dose should be followed by 500 mg every 48 hours.7,31
Intravenous versus oral administration
The interchangeability of the oral and intravenous routes of LVX administration is supported by randomized, double-blind, parallel-design, placebo-controlled studies. No significant differences in the pharmacokinetics of once-daily oral LVX or intravenous LVX were found in a study of 40 healthy male volunteers.37 LVX was rapidly and completely absorbed from the oral tablets, with a mean Tmax of approximately 1.5 hours and a mean absolute bioavailability of ≥99%. Yen et al investigated the clinical and economic impact of a pharmacist-managed intravenous to oral conversion service for LVX in Taiwan. This service not only reduced the duration of hospital stay (27.2 days versus 16.1 days, P=0.001), but also yielded significant cost savings with regard to total inpatient expenditure ($6,096±$5,164 versus $3,649.6±$3,740.4, preintervention period versus intervention period, respectively, P=0.017).38
Dose frequency
Deguchi et al evaluated the relationship between dosing frequency of LVX and AUC0–24/MIC and Cmax/MIC in complicated urinary tract infections with Escherichia coli or P. aeruginosa.39 Patients received 500 mg once daily, 100 mg three times daily, 200 mg twice daily, or 200 mg three times daily dose regimens of LVX. Monte Carlo simulation was performed for 10,000 cases using the pharmacokinetic data of patients with complicated urinary tract infections and the LVX MIC distributions for clinical strains of E. coli or P. aeruginosa. For the probabilities of achieving the Cmax/MIC targets that prevent emergence of fluoroquinolone resistance, the once-daily dose regimen (66.8%) did not differ from the other multiple-dose regimens (62.3%–66.2%) in E. coli, whereas the former regimen (44.2%) was superior to the latter regimens (10.8%–31.7%) in P. aeruginosa. The 500 mg once-daily dose regimen of LVX could produce a larger AUC0–24and higher Cmax, thus ensuring effective eradication of uropathogens and reducing the risk of fluoroquinolone resistance in complicated urinary tract infections.
Food-drug interactions
The overall bioavailability of LVX following a high-fat meal was not altered despite the fact that absorption of LVX was slightly delayed by food.40 However, a study by Amsden et al concluded a lack of bioequivalence of LVX when administered in a fasting state as compared with administration with a typical breakfast of calcium-fortified orange juice and ready-to-eat cereal.41 Wallace et al demonstrated that orange juice with or without calcium fortification decreased Cmax values for LVX by 14%–18% and prolonged the Tmax by approximately 50%.42 The underlying mechanism for such an interaction may be that LVX and components of orange juice compete for intestinal transport mechanisms, such as P-glycoprotein and organic anion-transporting polypeptides, rather than a chelation interaction.
Drug–drug interactions
Tamsulosin–LVX
Altintas et al retrospectively investigated the clinical outcomes in patients with type III inflammatory chronic prostatitis who were treated with fluoroquinolones with and without tamsulosin. The patients were divided into six groups as follows: group 1 (ciprofloxacin), group 2 (ofloxacin), group 3 (LVX), group 4 (ciprofloxacin + tamsulosin), group 5 (ofloxacin + tamsulosin), and group 6 (LVX + tamsulosin). The combination therapies were better than antibiotic therapies alone, and the best result was obtained using the LVX + tamsulosin combination.43 The enhanced efficacy may be explained by the finding that tamsulosin could alter the pharmacokinetics of LVX in prostate tissue derived from rats with acute bacterial prostatitis.44 Ninety-six rats with experimental bacterial prostatitis were randomly assigned to two groups, ie, an experimental group (treated with both tamsulosin and LVX, n=48) and a control group (treated with LVX and solvents, n=48). Tamsulosin increased the Cmax, prolonged the t1/2, and decreased the clearance of LVX (P<0.05) in the prostatic tissue, despite there being no obvious differences (P>0.05) between the two model rat groups in terms of the major pharmacokinetic parameters of LVX in plasma or in the hepatic and kidney tissues. These results indicate that tamsulosin may enhance the effect of LVX in the treatment of bacterial prostatitis without changing the LVX concentration in the liver or kidney. Thus, physicians may consider adding tamsulosin to the levofloxacin regimen for patients with bacterial prostatitis in view of the synergistic pharmacodynamic and pharmacokinetic effects of these drugs.
Multivalent cation-containing drugs–LVX
Coadministration of oral divalent or trivalent cation-containing medications and oral fluoroquinolones may impair fluoroquinolone absorption. Among 3,134 patients who received a course of oral LVX, coadministration was significantly associated with subsequent identification of an LVX-resistant isolate.45 Strategies to curb the emergence of LVX resistance should include avoiding coadministration of divalent or trivalent cation-containing compounds and LVX.
Warfarin–LVX
Case reports and retrospective evaluations have indicated that LVX may significantly potentiate the anticoagulation effect of warfarin and lead to an increase in INR.46–48 Moffett et al evaluated readmissions for warfarin-related bleeding in pediatric patients after hospital discharge and found that LVX at discharge (odds ratio 8.3, P<0.01) was a significant risk factor for readmission for bleeding.47 Concomitant use of other antibiotics, such as amoxicillin, azithromycin, ciprofloxacin, and moxifloxacin, could also result in a significantly increased INR in patients ≥65 years on stable warfarin therapy.49 Clark et al investigated the interactions between warfarin and antibiotics in the ambulatory care setting and concluded that acute upper respiratory tract infection could increase the risk of excessive anticoagulation independent of antibiotic use and antibiotics could also increase the risk. However, most patients on previously stable warfarin therapy will not experience clinically relevant increases in INR following exposure to antibiotic or acute upper respiratory tract infection.50 For patients receiving warfarin and LVX concomitantly, caution is advised regarding potential changes in INR values; however, it is not necessary to seek alternatives to LVX for the sake of avoiding a drug interaction with warfarin.
Cyclosporin/tacrolimus–LVX
Comedication with fluoroquinolones and immunosuppressive agents such as cyclosporin and tacrolimus are possible in clinical practice. Cyclosporin and tacrolimus are two immunosuppressive agents with similar mechanisms of action and are widely used in kidney transplantation. They are extensively metabolized by the liver via the cytochrome P450 (CYP) enzyme, the most important isoenzyme of which is CYP3A4. Ciprofloxacin and norfloxacin can increase blood concentrations of cyclosporin because they are metabolized by the liver through the same enzymatic pathway as cyclosporin. However, this drug–drug interaction does not indicate a “class effect”. A placebo-controlled, randomized, double-blind, two-phase crossover study in healthy subjects showed that the pharmacokinetics of cyclosporin were not significantly different in the absence and presence of LVX.51 Federico et al investigated the pharmacokinetic interaction between LVX and cyclosporin or tacrolimus in kidney transplant recipients. LVX administered at a dose of 500 mg twice daily for 6 days significantly increased the mean AUC, Cmax, average blood concentration (Cav), and blood concentration measured at the end of a dosing interval at steady state (Ctrough) of cyclosporin by about 25%, and increased the mean AUC and Cav of tacrolimus by 27%.52 Nevertheless, no adverse effects were observed, and no supratherapeutic concentrations of cyclosporin or tacrolimus were found. Reduction of the dosage of cyclosporin or tacrolimus should only be considered when clinical and/or laboratory signs of toxicity appear.
Relationship between transporters and LVX pharmacokinetics
Fluoroquinolones exist as charged molecules in blood and urine, making their absorption, distribution, and elimination likely to be influenced by active transport mechanisms. Greater understanding of in vivo fluoroquinolone clearance mechanisms should help improve the predictability of drug–drug interactions and enhance the clinical safety and efficacy of such agents.53 Sporadic reports in the literature show that drug transporters are involved in the pharmacokinetics of LVX. Maeda et al identified the influx transporters of LVX in a human colon cancer (Caco-2) cell line.54 Organic anion transporting polypeptide 1A2 was concluded to transport LVX, with a Michaelis constant (Km) value of 136 μM, and the results of this study suggest that active influx transport could at least partially explain the high membrane permeability of the fluoroquinolone in various tissues.
Naruhashi et al investigated the transport of fluoroquinolones and the contribution of the secretory transporter P-glycoprotein by using a P-glycoprotein-overexpressing cell line, LLC-GA5-COL150, and P-glycoprotein gene-deficient mice (mdr1a/1b double knockout mice). The secretory-directed transport (basal to apical) of LVX was detected in LLC-GA5-COL150 cells, indicating that P-glycoprotein contributed to disposition of LVX.55 Ito et al also confirmed that P-glycoprotein is involved in the renal tubular secretion of LVX. The apparent Km value for the saturable transcellular transport of LVX from the basolateral to apical side in LLC-GA5-COL150 monolayers was 3.0 mM. The increased basolateral-to-apical transport in LLC-GA5-COL150 monolayers was completely inhibited by P-glycoprotein inhibitors such as cyclosporin and quinidine.56
Mulgaonkar et al reported that human organic cation transporter 1 in the sinusoidal membrane of hepatocytes and the basolateral membrane of proximal tubule cells is likely to play a role in the disposition of LVX since LVX achieved statistically significant (P<0.01) but weak inhibition of human organic cation transporter 1 transport.57 Using rat multidrug and toxin extrusion protein (MATE) 1 expressed in MDCKII cells, Ohta et al demonstrated that MATE1 is also involved in the renal tubular secretion of fluoroquinolones, including LVX.58
Single nucleotide polymorphisms have been identified in the SLCO1A2 gene encoding organic anion transporting polypeptide 1A2, the ABCB1 gene encoding P-glycoprotein, and the SLC47A1 gene encoding MATE1.59–62 It would be worthwhile performing studies to determine whether these polymorphisms contribute to intersubject variation in LVX pharmacokinetics.
Conclusion
Personalized therapeutics of LVX are necessary for the sake of better safety, clinical efficacy, and avoidance of resistance. We specially addressed this topic with a focus on pharmacokinetic concerns. New findings regarding individual dosing of LVX in special patient populations and in vivo active transport mechanisms open up new horizons in clinical practice. Personalized therapeutics would go deeper into routine practice and improve patient-specific outcomes if clinical practitioners performed comprehensive interventions, such as seeking online information to assist dose guidelines, pharmacokinetic dose consultation services provided by pharmacy, daily attendance of a clinical pharmacist during ward rounds, and prospective review of the appropriateness of physician orders by pharmacists.
Acknowledgments
This work was supported by the Zhejiang Provincial Bureau of Health (grant 2012KYA090), the National Natural Science Foundation of China (grant 81373488), and the National Major Projects of China (grants 2012ZX09506001–004).
Disclosure
The authors report no conflicts of interest in this work.
References
International Pharmaceutical Federation. Towards a future vision for complex patients. Available from: http://www.fip.org/dublin2013/Dublin/2041/ProGramme/. Accessed December 4, 2013. | |
Morello CM, Hirsch JD, Lee KC. Navigating complex patients using an innovative tool: the MTM Spider Web. J Am Pharm Assoc (2003). 2013;53:530–538. | |
Joint Commission International. JCI accreditation standards for hospitals. Oak Brook, IL, USA: Joint Commission International; 2010. Available from: http://www.jointcommissioninternational.org/common/pdfs/jcia/IAS400_Standards_Lists_Only.pdf. Accessed March 20, 2013. | |
Li W, Zhou Q. Patient diagnosis on electronic prescription orders: a key element for appropriateness review by pharmacists. Res Social Adm Pharm. 2013;9:493–494. | |
Zhu LL, Zhou Q. Intervention for improving the appropriateness of physician orders for oral medications in geriatric VIP patients during the journey to JCI accreditation. Ther Clin Risk Manag. 2013;9:273–275. | |
McGregor JC, Allen GP, Bearden DT. Levofloxacin in the treatment of complicated urinary tract infections and acute pyelonephritis. Ther Clin Risk Manag. 2008;4:843–853. | |
Giordano P, Weber K, Gesin G, Kubert J. Skin and skin structure infections: treatment with newer generation fluoroquinolones. Ther Clin Risk Manag. 2007;3:309–317. | |
Prajapati A, Ganguly B. Appropriateness of drug dose and frequency in patients with renal dysfunction in a tertiary care hospital: a cross-sectional study. 2013;5:136–140. | |
Nicolau DP. Predicting antibacterial response from pharmacodynamic and pharmacokinetic profiles. Infection. 2001;29 Suppl 2:11–15. | |
Labreche MJ, Frei CR. Declining susceptibilities of Gram-negative bacteria to the fluoroquinolones: effects on pharmacokinetics, pharmacodynamics, and clinical outcomes. Am J Health Syst Pharm. 2012;69:1863–1870. | |
Drusano GL, Preston SL, Fowler C, Corrado M, Weisinger B, Kahn J. Relationship between fluoroquinolone area under the curve: minimum inhibitory concentration ratio and the probability of eradication of the infecting pathogen, in patients with nosocomial pneumonia. J Infect Dis. 2004;189:1590–1597. | |
Defife R, Scheetz MH, Feinglass JM, Postelnick MJ, Scarsi KK. Effect of differences in MIC values on clinical outcomes in patients with bloodstream infections caused by Gram-negative organisms treated with levofloxacin. Antimicrob Agents Chemother. 2009; 53:1074–1079. | |
Anderson VR, Perry CM. Levofloxacin: a review of its use as a high-dose, short-course treatment for bacterial infection. Drugs. 2008;68:535–565. | |
Noreddin AM, Elkhatib WF, Cunnion KM, Zhanel GG. Cumulative clinical experience from over a decade of use of levofloxacin in community-acquired pneumonia: critical appraisal and role in therapy. Drug Healthc Patient Saf. 2011;3:59–68. | |
Cook AM, Martin C, Adams VR, Morehead RS. Pharmacokinetics of intravenous levofloxacin administered at 750 milligrams in obese adults. Antimicrob Agents Chemother. 2011;55:3240–3243. | |
Pai MP, Allen SE, Amsden GW. Altered steady state pharmacokinetics of levofloxacin in adult cystic fibrosis patients receiving calcium carbonate. J Cyst Fibros. 2006;5:153–157. | |
Overholser BR, Kays MB, Lagvankar S, Goldman M, Mueller BA, Sowinski KM. Pharmacokinetics of intravenously administered levofloxacin in men and women. Pharmacotherapy. 2005;25:1310–1318. | |
Chien S, Wells TG, Blumer JL, et al. Levofloxacin pharmacokinetics in children. J Clin Pharmacol. 2005;45:153–160. | |
Noreddin AM, Hoban DJ, Zhanel GG. Comparison of gatifloxacin and levofloxacin administered at various dosing regimens to hospitalised patients with community-acquired pneumonia: pharmacodynamic target attainment study using North American surveillance data for Streptococcus pneumoniae. Int J Antimicrob Agents. 2005;26:120–125. | |
Pea F, Di Qual E, Cusenza A, Brollo L, Baldassarre M, Furlanut M. Pharmacokinetics and pharmacodynamics of intravenous levofloxacin in patients with early-onset ventilator-associated pneumonia. Clin Pharmacokinet. 2003;42:589–598. | |
Preston SL, Drusano GL, Berman AL, et al. Levofloxacin population pharmacokinetics and creation of a demographic model for prediction of individual drug clearance in patients with serious community-acquired infection. Antimicrob Agents Chemother. 1998;42:1098–1104. | |
Janson B, Thursky K. Dosing of antibiotics in obesity. Curr Opin Infect Dis. 2012;25:634–649. | |
Luque S, Grau S, Valle M, Colino CI, Ferrer A. Levofloxacin weight-adjusted dosing and pharmacokinetic disposition in a morbidly obese patient. J Antimicrob Chemother. 2011;66:1653–1654. | |
Kees MG, Weber S, Kees F, Horbach T. Pharmacokinetics of moxifloxacin in plasma and tissue of morbidly obese patients. J Antimicrob Chemother. 2011;66:2330–2335. | |
Rey E, Tréluyer JM, Pons G. Drug disposition in cystic fibrosis. Clin Pharmacokinet. 1998;35:313–329. | |
Hurley M, Smyth A. Fluoroquinolones in the treatment of bronchopulmonary disease in cystic fibrosis. Ther Adv Respir Dis. 2012;6:363–373. | |
Lee CK, Boyle MP, Diener-West M, Brass-Ernst L, Noschese M, Zeitlin PL. Levofloxacin pharmacokinetics in adult cystic fibrosis. Chest. 2007;131:796–802. | |
Geller DE, Flume PA, Staab D, Fischer R, Loutit JS, Conrad DJ; Mpex 204 Study Group. Levofloxacin inhalation solution (MP-376) in patients with cystic fibrosis with Pseudomonas aeruginosa. Am J Respir Crit Care Med. 2011;183:1510–1516. | |
Almeida S, Filipe A, Almeida A, Wong H, Caparrós N, Tanguay M. Comparative bioavailability of two formulations of levofloxacin and effect of sex on bioequivalence analysis. Data from a randomised, 2 × 2 crossover trial in healthy volunteers. Arzneimittelforschung. 2005;55:414–419. | |
Chien SC, Chow AT, Natarajan J, et al. Absence of age and gender effects on the pharmacokinetics of a single 500-milligram oral dose of levofloxacin in healthy subjects. Antimicrob Agents Chemother. 1997;41:1562–1565. | |
Food and Drug Administration. Levaquin medication guide. Available from: http://www.fda.gov/downloads/Drugs/.../UCM133684.pdf. Accessed December 4, 2013. | |
Bailey AM, Weant KA, Baker SN. Prevalence and risk factor analysis of resistant Escherichia coli urinary tract infections in the emergency department. Pharm Pract (Granada). 2013;11:96–101. | |
Tanigawara Y, Nomura H, Kagimoto N, Okumura K, Hori R. Premarketing population pharmacokinetic study of levofloxacin in normal subjects and patients with infectious diseases. Biol Pharm Bull. 1995;18:315–320. | |
Tayab ZR, Hochhaus G, Kaufmann S, Jäger D, Barth J. Do intensive care patients need an individualized dosing regimen for levofloxacin? Int J Clin Pharmacol Ther. 2006;44:262–269. | |
Rebuck JA, Fish DN, Abraham E. Pharmacokinetics of intravenous and oral levofloxacin in critically ill adults in a medical intensive care unit. Pharmacotherapy. 2002;22:1216–1225. | |
Sánchez Navarro A, Colino Gandarillas CI, Alvarez Lerma F, Menacho YA, Domínguez-Gil A. Pharmacokinetics and pharmacodynamics of levofloxacin in intensive care patients. Clin Pharmacokinet. 2005;44:627–635. | |
Chien SC, Rogge MC, Gisclon LG, et al. Pharmacokinetic profile of levofloxacin following once-daily 500-milligram oral or intravenous doses. Antimicrob Agents Chemother. 1997;41:2256–2260. | |
Yen YH, Chen HY, Wuan-Jin L, Lin YM, Shen WC, Cheng KJ. Clinical and economic impact of a pharmacist-managed iv-to-po conversion service for levofloxacin in Taiwan. Int J Clin Pharmacol Ther. 2012;50:136–141. | |
Deguchi T, Seike K, Yasuda M, Matsumoto T. Evaluation by Monte Carlo simulation of levofloxacin dosing for complicated urinary tract infections caused by Escherichia coli or Pseudomonas aeruginosa. J Infect Chemother. 2011;17:726–730. | |
Lee LJ, Hafkin B, Lee ID, Hoh J, Dix R. Effects of food and sucralfate on a single oral dose of 500 milligrams of levofloxacin in healthy subjects. Antimicrob Agents Chemother. 1997;41:2196–2200. | |
Amsden GW, Whitaker AM, Johnson PW. Lack of bioequivalence of levofloxacin when coadministered with a mineral-fortified breakfast of juice and cereal. J Clin Pharmacol. 2003;43:990–995. | |
Wallace AW, Victory JM, Amsden GW. Lack of bioequivalence when levofloxacin and calcium-fortified orange juice are coadministered to healthy volunteers. J Clin Pharmacol. 2003;43:539–544. | |
Altintas R, Oguz F, Beytur A, Ediz C, Gunes A, Ozer A. Comparison of results after flouroquinolones and combination therapies in type IIIA chronic prostatitis. Actas Urol Esp. 2013;37:619–624. | |
Qin GD, Xiao MZ, Zhou YD, et al. Tamsulosin alters levofloxacin pharmacokinetics in prostates derived from rats with acute bacterial prostatitis. Asian J Androl. 2013;15:254–260. | |
Cohen KA, Lautenbach E, Weiner MG, Synnestvedt M, Gasink LB. Coadministration of oral levofloxacin with agents that impair absorption: impact on antibiotic resistance. Infect Control Hosp Epidemiol. 2008;29:975–977. | |
Vadlamudi RS, Smalligan RD, Ismail HM. Interaction between warfarin and levofloxacin: case series. South Med J. 2007;100:720–724. | |
Moffett BS, Kim S, Bomgaars LR. Readmissions for warfarin-related bleeding in pediatric patients after hospital discharge. Pediatr Blood Cancer. 2013;60:1503–1506. | |
Mercadal Orfila G, Gracia García B, Leiva Badosa E, Perayre Badía M, Reynaldo Martínez C, Jodar Masanés R. Retrospective assessment of potential interaction between levofloxacin and warfarin. Pharm World Sci. 2009;31:224–229. | |
Ghaswalla PK, Harpe SE, Tassone D, Slattum PW. Warfarin-antibiotic interactions in older adults of an outpatient anticoagulation clinic. Am J Geriatr Pharmacother. 2012;10:352–360. | |
Clark NP, Delate T, Riggs CS, et al. Warfarin interactions with antibiotics in the ambulatory care setting. JAMA Intern Med. 2014;174:409–416. | |
Doose DR, Walker SA, Chien SC, Williams RR, Nayak RK. Levofloxacin does not alter cyclosporine disposition. J Clin Pharmacol. 1998;38:90–93. | |
Federico S, Carrano R, Capone D, Gentile A, Palmiero G, Basile V. Pharmacokinetic interaction between levofloxacin and ciclosporin or tacrolimus in kidney transplant recipients: ciclosporin, tacrolimus and levofloxacin in renal transplantation. Clin Pharmacokinet. 2006;45:169–175. | |
Mulgaonkar A, Venitz J, Sweet DH. Fluoroquinolone disposition: identification of the contribution of renal secretory and reabsorptive drug transporters. Expert Opin Drug Metab Toxicol. 2012;8:553–569. | |
Maeda T, Takahashi K, Ohtsu N, et al. Identification of influx transporter for the quinolone antibacterial agent levofloxacin. Mol Pharm. 2007;4:85–94. | |
Naruhashi K, Tamai I, Inoue N, et al. Active intestinal secretion of new quinolone antimicrobials and the partial contribution of P-glycoprotein. J Pharm Pharmacol. 2001;53:699–709. | |
Ito T, Yano I, Tanaka K, Inui KI. Transport of quinolone antibacterial drugs by human P-glycoprotein expressed in a kidney epithelial cell line, LLC-PK1. J Pharmacol Exp Ther. 1997;282:955–960. | |
Mulgaonkar A, Venitz J, Gründemann D, Sweet DH. Human organic cation transporters 1 (SLC22A1), 2 (SLC22A2), and 3 (SLC22A3) as disposition pathways for fluoroquinolone antimicrobials. Antimicrob Agents Chemother. 2013;57:2705–2711. | |
Ohta KY, Imamura Y, Okudaira N, Atsumi R, Inoue K, Yuasa H. Functional characterization of multidrug and toxin extrusion protein 1 as a facilitative transporter for fluoroquinolones. J Pharmacol Exp Ther. 2009;328:628–634. | |
Franke RM, Scherkenbach LA, Sparreboom A. Pharmacogenetics of the organic anion transporting polypeptide 1A2. Pharmacogenomics. 2009;10:339–344. | |
Gong IY, Kim RB. Impact of genetic variation in OATP transporters to drug disposition and response. Drug Metab Pharmacokinet. 2013;28:4–18. | |
Zhou Q, Ruan ZR, Yuan H, Xu DH, Zeng S. ABCB1 gene polymorphisms, ABCB1 haplotypes and ABCG2 c.421c > A are determinants of inter-subject variability in rosuvastatin pharmacokinetics. Pharmazie. 2013;68:129–134. | |
Christensen MM, Pedersen RS, Stage TB, et al. A gene-gene interaction between polymorphisms in the OCT2 and MATE1 genes influences the renal clearance of metformin. Pharmacogenet Genomics. 2013;23:526–534. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.