Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 8
Personalized antiplatelet and anticoagulation therapy: applications and significance of pharmacogenomics
Authors Beitelshees A, Voora D, Lewis J
Received 13 May 2014
Accepted for publication 17 June 2014
Published 9 February 2015 Volume 2015:8 Pages 43—61
DOI https://doi.org/10.2147/PGPM.S52900
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Amber L Beitelshees,1,* Deepak Voora,2,* Joshua P Lewis,1,*
1Program for Personalized and Genomic Medicine and Division of Endocrinology, Diabetes, and Nutrition, University of Maryland School of Medicine, Baltimore, MD, USA; 2Center for Applied Genomics & Precision Medicine, Department of Medicine, Duke School of Medicine, Durham, NC, USA
*All authors contributed equally to this work
Abstract: In recent years, substantial effort has been made to better understand the influence of genetic factors on the efficacy and safety of numerous medications. These investigations suggest that the use of pharmacogenetic data to inform physician decision-making has great potential to enhance patient care by reducing on-treatment clinical events, adverse drug reactions, and health care-related costs. In fact, integration of such information into the clinical setting may be particularly applicable for antiplatelet and anticoagulation therapeutics, given the increasing body of evidence implicating genetic variation in variable drug response. In this review, we summarize currently available pharmacogenetic information for the most commonly used antiplatelet (ie, clopidogrel and aspirin) and anticoagulation (ie, warfarin) medications. Furthermore, we highlight the currently known role of genetic variability in response to next-generation antiplatelet (prasugrel and ticagrelor) and anticoagulant (dabigatran) agents. While compelling evidence suggests that genetic variants are important determinants of antiplatelet and anticoagulation therapy response, significant barriers to clinical implementation of pharmacogenetic testing exist and are described herein. In addition, we briefly discuss development of new diagnostic targets and therapeutic strategies as well as implications for enhanced patient care. In conclusion, pharmacogenetic testing can provide important information to assist clinicians with prescribing the most personalized and effective antiplatelet and anticoagulation therapy. However, several factors may limit its usefulness and should be considered.
Keywords: pharmacogenetics, clopidogrel, warfarin, anticoagulant, aspirin, precision medicine
Introduction
In recent years, the use of pharmacogenetic information to help guide physician decision-making and to enhance patient care has been an area of emphasis in scientific and health communities worldwide. The rapid generation of pharmacogenetic data has greatly facilitated our ability to better understand the impact of genetic variation on therapeutic response and clinical endpoints. In addition, national and international regulatory agencies such as the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have focused on the potential impact of genetic variability on the efficacy and safety of numerous therapeutic agents. In fact, to date, the FDA has added pharmacogenetic information to the approved labels of over 125 drugs, including the most widely used antiplatelet and anticoagulant medications, in order to highlight that genotype may be useful in determining optimal dose and/or choice of drug.
Antiplatelet and anticoagulant therapy is critical in the primary and secondary prevention of a wide range of cardiovascular disorders characterized by pathological prothrombotic states. Despite the general effectiveness of current antiplatelet and anticoagulant agents, recurrent thrombotic events and associated adverse events such as bleeding remain an important clinical problem in cardiovascular medicine. Significant effort in the past 10 years has been dedicated to understanding the clinical factors that affect variability in drug response, and while the determinants of both antiplatelet and anticoagulant response are multifactorial, there is a large body of evidence to suggest that genetic predisposition substantially influences drug pharmacokinetics and pharmacodynamics, thereby resulting in altered drug response and increases in the rate of adverse cardiovascular events.
The purpose of this review is to summarize the transport, metabolism, and action of some of the most commonly used antiplatelet (aspirin and clopidogrel) and anticoagulant (warfarin) medications in the context of currently available pharmacogenetic data. In addition, we briefly highlight the currently known role of genetic variability in response to next-generation antiplatelet (prasugrel and ticagrelor) and anticoagulant (eg, dabigatran) agents. Given these data, we also discuss the development of new diagnostic targets and therapeutic strategies, current barriers to implementation of antiplatelet and anticoagulant pharmacogenetics, and implications for enhanced patient care. Although this review focuses primarily on DNA-based variations that underlie drug response, we acknowledge the burgeoning field of other “omic” technologies (eg, transcriptomics, proteomics, and metabolomics) that are increasingly being used to characterize drug responses.
Antiplatelet pharmacogenomics
Dual antiplatelet therapy with aspirin and clopidogrel is currently the standard of care for treating patients with coronary artery disease (CAD) and/or acute coronary syndrome (ACS) undergoing percutaneous coronary intervention (PCI). The next-generation thienopyridine prasugrel, as well as the cyclopentyltriazolopyrimidine ticagrelor, have recently been approved and increasingly serve as alternative antiplatelet agents to clopidogrel. While all of these medications are generally effective, wide interindividual variation in response to these agents, defined by either laboratory response (ie, ex vivo measures of platelet aggregation) or clinical response (cardiovascular endpoints), have been documented. For each of these medications, variable response is, at least in part, heritable. For example, reported heritability estimates suggest that approximately 70% of the variability observed in clopidogrel response, as measured by adenosine diphosphate (ADP)-stimulated platelet aggregation, is attributed to genetic factors.1 Similarly, it has been shown in both Caucasian and African Americans that heritable factors significantly contribute to on-aspirin platelet responsiveness in molecular pathways directly and indirectly related to cyclooxygenase-1 (COX-1).2 While, to our knowledge, no large-scale investigation to date has evaluated the heritability of prasugrel or ticagrelor response, several candidate gene studies have revealed polymorphisms that influence response to these agents (see the “Next-Generation Thienopyridine” section), suggesting a nontrivial genetic component. Therefore, identifying the genetic variants that influence response to these medications provides important information regarding pharmacokinetics and pharmacodynamics of these agents and also offers critical insights concerning the potential use of genotype information in prescribing the most effective and individualized antiplatelet therapy.
Clopidogrel
Given its effectiveness and relatively low cost compared to its alternatives, clopidogrel remains one of the most widely prescribed antiplatelet medications to prevent recurrent ischemic events in patients with ACS, myocardial infarction, and/or who are undergoing PCI. Despite its wide use, however, it has been consistently shown that approximately 4%–30% of patients do not respond adequately to this drug, resulting in high on-treatment platelet reactivity (HTPR) and increased rates of cardiovascular events.3,4 In fact, pharmacodynamic investigations have shown that laboratory measures of ex vivo ADP-stimulated platelet reactivity, a widely used surrogate marker of clopidogrel response, vary substantially among clopidogrel-treated patients.3,5 Moreover, PCI patients on clopidogrel who exhibit HTPR are more likely to experience a recurrent adverse clinical event, seemingly regardless of clinical presentation.6 While several clinical and demographic factors that influence clopidogrel efficacy have been determined (eg, age, body mass index, diabetes, diet, smoking, drug–drug interactions [eg, proton pump inhibitors], etc),7 the proportion of variation in drug response they collectively account for is relatively modest. In order to better understand variability in clopidogrel response, genetic evaluation of multiple candidate genes has been conducted, revealing several single nucleotide polymorphisms (SNPs) that may significantly influence clopidogrel response.
The genes responsible for clopidogrel transport, metabolism, and action were obvious choices for pharmacogenetic candidate gene studies (Figure 1). Clopidogrel is an oral, second-generation thienopyridine prodrug that, following rapid absorption by the duodenum, is metabolized through a two-step conversion by hepatic cytochrome P450 enzymes, primarily CYP2C19, resulting in a biologically active thiol metabolite. Approximately 15% of clopidogrel prodrug is converted into the active metabolite, while ~85% is degraded into inactive carboxylic acid derivatives by hepatic esterases, most notably carboxylesterase 1 (CES1). In circulation, the active thiol metabolite irreversibly binds to and inactivates the P2Y12 receptor on the surface of platelets, leading to inhibition of ADP-induced platelet activation and aggregation. In the following sections, we briefly describe the role of genetic variants in genes responsible for clopidogrel transport (ATP-binding cassette subfamily B member 1 [ABCB1]), metabolism (CYP enzymes, paraoxonase 1 [PON1], CES1), and action (P2Y12) on clopidogrel pharmacodynamics, pharmacokinetics, and cardiovascular event rates.
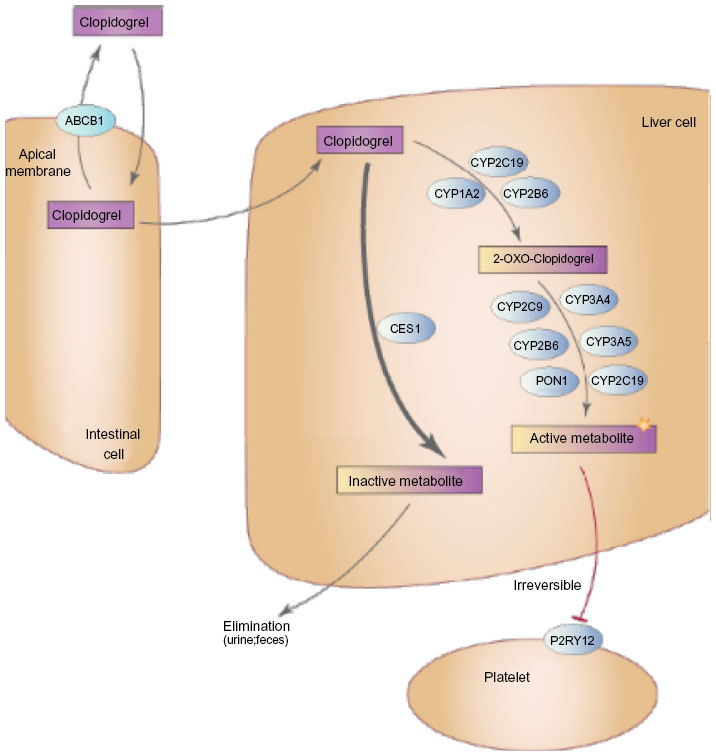 | Figure 1 Major proteins involved in clopidogrel transport and metabolism. |
ABCB1
The ABCB1 gene encodes the well-described multidrug resistance protein 1 (MDR1), an ATP-dependent efflux transporter important in the bioavailability of multiple endogenous and xenobiotic compounds including clopidogrel. As clopidogrel is absorbed from the intestinal lumen via duodenal enterocytes, MDR1 immediately transports a portion of the drug back into the lumen, resulting in decreased clopidogrel bioavailability. While ABCB1 is highly polymorphic, significant attention has focused on the effect of a three-SNP haplotype, tagged by the C3435T SNP (rs1045642), on clopidogrel metabolite level, platelet reactivity, and cardiovascular events. Prior investigations have shown that the T-allele of the C3435T variant is relatively common (allele frequency ranges from 10%–60% depending on race/ethnicity) and results in increased MDR1 expression, thereby potentially leading to increased clopidogrel extrusion.8
Early investigations of the ABCB1 C3435T variant revealed that PCI patients who were homozygous for the T-allele had significantly less clopidogrel prodrug and active metabolite levels compared to C-allele carriers when given either a 300 or 600 mg loading dose.9 Simon et al subsequently showed in 2,208 acute myocardial infarction (MI) patients that ABCB1 3435 T-allele homozygotes were significantly more likely to experience a cardiovascular event at 1 year compared to patients that carried the C-allele (hazard ratio [HR] =1.72, 95% confidence interval [CI]: 1.20–2.47).10 Similarly, in clopidogrel-treated ACS PCI patients of the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel -Thrombolysis in Myocardial Infarction (TRITON-TIMI) 38 trial, ABCB1 T-allele homozygotes had a 72% increased risk of a composite endpoint consisting of cardiovascular death, MI, or stroke.11 However, a recent meta-analysis consisting of over 10,000 clopidogrel-treated patients, primarily with ACS (89%) and/or undergoing PCI (74%), was conducted in order to evaluate the effect of the C3435T variant on several cardiovascular outcomes.12 When comparing 3435 T-allele homozygotes to C-allele carriers, while there was moderate evidence of a relationship between this variant and short-term (<30 days) recurrent ischemic events (P=0.02, odds ratio [OR] =1.41, 95% CI: 1.06–1.87), no evidence of association was observed between the C3435T variant and overall recurrent events (P=0.07, OR =1.15, 95% CI: 0.99–1.33), stent thrombosis (P=0.37, OR =0.79, 95% CI: 0.47–1.32), or bleeding (P=0.82, OR =0.98, 95% CI: 0.79–1.21). In contrast, a second meta-analysis published by Su et al showed a significant relationship between the ABCB1 C3435T variant and both short- as well as long-term cardiovascular events (P=0.02, OR =1.48, 95% CI: 1.06–2.06 and P=0.01, OR =1.39, 95% CI: 1.08–1.79, respectively).13 It should be noted, however, that, in contrast to the investigations highlighted above, the meta-analysis by Su et al reported the results of a dominant (TC/TT versus CC) instead of recessive (TT versus TC/CC) genetic model due to the high heterogeneity observed in this study. Taken together, while there is a growing evidence base supporting a potential role for the ABCB1 C3435T variant in clopidogrel efficacy, the inconsistencies of these findings make the use of this variant in genotype-directed therapy or other clinical applications, at this time, premature.
CYP2C19
After its absorption, several enzymes contribute to hepatic metabolism of clopidogrel, resulting in both biologically active and inactive derivatives (Figure 1). While we discuss some of these enzymes in greater detail in the current section, the “PON1” section, the “CES1” section, and the “Other variants” section, previous investigations have shown that CYP2C19 is the major contributor regarding generation of the bioactive metabolite.14 Consistent with this observation, both loss-of-function (LOF) and gain-of-function (GOF) genetic variants in CYP2C19 have been most consistently associated with clopidogrel efficacy. There are several LOF variants in CYP2C19 that contribute to altered clopidogrel response. CYP2C19*2 (rs4244285), which results in a cryptic splice site in exon 5 leading to a premature stop codon, is the most common of these variants, with approximately 20%–30% of Caucasians and Africans and 60% of Asians carrying at least one copy of this allele.15 Other LOF variants (ie, CYP2C19*3–*8) are generally rare in most populations, thus limiting their potential clinical utility, with perhaps the exception of CYP2C19*3 (rs4986893), which is substantially more common in Asian populations, with an allele frequency ranging from 5%–9%. The most comprehensively evaluated GOF variant is CYP2C19*17 (rs12248560). CYP2C19*17 resides in the promoter region of this gene and has been implicated in altered clopidogrel response and increased bleeding risk (see the last two paragraphs of the current section) through its ability to increase transcription of CYP2C19.16
There is now a convincingly large body of evidence to suggest that the CYP2C19*2 variant significantly impacts clopidogrel pharmacokinetics and pharmacodynamics. Hulot et al published the first of these investigations in 2006, revealing that, in a small sample of healthy subjects (N=28), individuals who carried the CYP2C19*2 variant had significantly poorer clopidogrel response as assessed by ADP-stimulated platelet reactivity.17 It was later shown, again in healthy volunteers, that this CYP2C19*2-specific higher platelet aggregation post-clopidogrel exposure was due to altered clopidogrel metabolism resulting in a reduction of circulating clopidogrel active metabolite.18,19 These findings have been subsequently replicated in multiple investigations of patients with cardiovascular disease, leaving little doubt regarding the relationship between CYP2C19*2 and clopidogrel pharmacokinetics as well as pharmacodynamics.20–22 A more comprehensive list of studies that have evaluated the role of CYP2C19*2 on clopidogrel active metabolite levels and platelet reactivity has been published previously.23,24
The impact of the CYP2C19*2 variant on cardiovascular endpoints has also been well documented in recent years. Some of the earliest of these investigations reported that clopidogrel-treated patients who carried at least one copy of the CYP2C19*2 allele were significantly more likely to experience a major adverse cardiovascular event (MACE) compared to individuals who were homozygous for the CYP2C19*1 allele.25,26 In these studies, the authors also observed an approximately two- to threefold increase in the incidence of stent thrombosis in CYP2C19*2 allele carriers compared to noncarriers, an observation that has subsequently been replicated by other groups.10,27 Given the potential clinical utility of these findings, multiple investigations were subsequently performed and revealed a generally consistent relationship between CYP2C19*2 genotype and on-clopidogrel MACE.24 However, in 2010, data from two independent clinical trials (Clopidogrel in Unstable Angina to Prevent Recurrent Event [CURE] and Atrial Fibrillation Clopidogrel Trial with Irbesartan for Prevention of Vascular Events - Aspirin [ACTIVE-A]) challenged the significance of the pharmacogenetic relationship between CYP2C19*2 genotype and on-clopidogrel cardiovascular events.28 In a genetic substudy of the CURE trial, which primarily consisted of non-ST segment elevation MI patients who did not undergo PCI, no evidence of association was observed between CYP2C19 LOF alleles and cardiovascular events.28 Similarly, in clopidogrel-treated atrial fibrillation patients of the ACTIVE-A trial, no difference in the rate of cardiovascular events was observed between carriers and noncarriers of CYP2C19 LOF alleles.28 Together, the results observed in the CURE and ACTIVE-A trials were seemingly in direct contrast with the findings of several previous investigations and led to some initial confusion regarding the impact of CYP2C19 genotype on cardiovascular outcomes in clopidogrel-treated patients.10,25–27
In the last few years, several meta-analyses have revealed important insights regarding the effect of the CYP2C19*2 variant in clopidogrel-treated patients.29–33 Furthermore, these studies may explain, at least in part, the inconsistencies observed in the investigations described in the previous paragraph. Together, these results suggest that the effect of the CYP2C19*2 variant on clinical outcomes may be indication-specific. It is well established that high-risk patients who undergo PCI derive the most benefit from clopidogrel therapy compared to other indications. Therefore, it is reasonable to speculate that the impact of the CYP2C19*2 variant may be more pronounced in PCI patients, particularly in those who may experience stent thrombosis. Indeed, as reviewed in depth previously,24,34 nearly all meta-analyses conducted to date have shown a strong association between CYP2C19*2 genotype and risk of stent thrombosis. On the other hand, large-scale evaluations of lower-risk and/or non-PCI patients, such as the populations used in the CURE and ACTIVE-A trials, have not reproducibly shown an effect of CYP2C19*2 on cardiovascular outcomes. In a controversial meta-analysis by Holmes et al,35 the authors concluded that CYP2C19*2 genotype was not a significant predictor of cardiovascular outcomes in clopidogrel-treated patients and that small study bias of treatment-only investigations influenced initial effect size estimates. However, others contest that the patients used in the analysis by Holmes et al, particularly those in the larger studies, in which no effect of CYP2C19*2 genotype was observed, were those who receive little-to-no benefit from clopidogrel therapy.36,37 Taken together, the results of these meta-analyses have 1) provided robust evidence of a relationship between CYP2C19*2 genotype and risk of stent thrombosis, and 2) highlighted the importance of clinical indication in studies of CYP2C19 genetic variability and clopidogrel efficacy.
While the CYP2C19*2 variant has been the most extensively studied polymorphism with regard to clopidogrel response variability, substantial effort has been made to understand the impact of the relatively common CYP2C19*17 GOF allele. One of the first positive associations between the CYP2C19*17 variant and on-clopidogrel platelet reactivity was observed by Frére et al in 2009.38 In a retrospective study consisting of 598 non-ST elevation clopidogrel-treated ACS patients, the authors found that individuals who carried the CYP2C19*17 variant had significantly better clopidogrel response as assessed by vasodilator stimulated-phosphoprotein (VASP) platelet reactivity index. However, subsequent follow-up investigations regarding the impact of this variant on the formation of clopidogrel active metabolite, platelet aggregation, and cardiovascular outcomes have had mixed results.39 Similarly, currently available meta-analysis data are also inconsistent. For example, two meta-analyses performed in 2012 provided evidence that the CYP2C19*17 was significantly associated with decreased rates of adverse clinical outcomes but increased rates of adverse bleeding.33,40 In contrast, an independent systematic review and meta-analysis by Bauer et al revealed that carriers of the CYP2C19*17 variant did not significantly influence risk of experiencing a composite cardiovascular endpoint or stent thrombosis.32
While it is possible that differences in study design, patient population, and statistical methodologies may explain the discrepancy in results between investigations of CYP2C19*17 and clopidogrel efficacy, recent work by our group may suggest an alternative explanation.39 The CYP2C19*2 and *17 variants are in relatively close proximity on chromosome 10q24. Thus, it is possible that the effects of these variants are not independent of each other but instead are linked through linkage disequilibrium. Indeed, we observed that the CYP2C19*17 and *2 variants are in high linkage disequilibrium with each other (D’=1, r2=0.07), such that CYP2C19*2 and CYP2C19*17 alleles are rarely located on the same chromosome together. Therefore, while single-SNP association analyses of CYP2C19*17 suggest significant associations with both generation of clopidogrel active metabolite and ADP-stimulated platelet aggregation, when these analyses were adjusted for the effect of CYP2C19*2, no association was observed. Furthermore, evaluation of the various two-SNP haplotypes revealed that the CYP2C19*17 variant had little to no effect on clopidogrel response.39 Therefore, in order to better assess the true effect of the CYP2C19*17 variant in clopidogrel-treated patients, we feel that adjustment for the CYP2C19*2 variant is critical.
PON1
In 2011, Bouman et al published a high-profile investigation suggesting that a genetic variant in paraoxonase 1 (PON1), the common Q192R missense variant (rs662), is a major determinant of clopidogrel efficacy.41 In fact, they observed that PON1, a well-described hepatic esterase, was critical in converting 2-oxo-clopidogrel into the bioactive thiol metabolite and that the 192Q allele was significantly associated with reduced clopidogrel pharmacokinetics and increased occurrence of stent thrombosis in CAD PCI patients (OR =3.6, 95% CI: 1.6–7.9, P=0.003). They extended these findings in an independent prospective cohort consisting of 1,982 ACS patients and found that PON1 192Q-allele homozygotes were significantly more likely to experience fatal or nonfatal definite stent thrombosis (HR =10.2, 95% CI: 4.3–71.4, P<0.001) as well as a composite cardiovascular endpoint consisting of vascular death, nonfatal MI, and nonfatal stroke (OR =3.9, 95% CI: 2.1–7.2, P<0.001). Consistent with decreased clopidogrel response, it was also observed that PON1 192Q-allele homozygotes had lower risk of major bleeding (HR =0.4, 95% CI: 0.2–0.8, P=0.006).41
As a result of this investigation, several groups, including our own,42 evaluated the role of the PON1 Q192R variant on clopidogrel efficacy. Interestingly, however, nearly all replication efforts failed to observe an association between the Q192R variant and clopidogrel response, as assessed by either platelet reactivity or occurrence of on-treatment cardiovascular events. Given that several factors, including statistical power may explain, at least in part, the discrepant results observed between the investigation by Bouman et al and subsequent replication studies, members of the PON1 Study Group recently conducted a systematic review and meta-analysis of summarized data in order to evaluate the impact of PON1 Q192R on platelet reactivity and recurrent ischemic events.43 Consistent with the findings of the individual replication efforts, no evidence of association was observed between PON1 Q192R and platelet reactivity, regardless of the laboratory method used (global mean standardized difference =0.10, 95% CI: −0.06 to 0.25, P=0.22). Moreover, analysis of eleven independent investigations that evaluated the impact of this polymorphism on MACE risk revealed no difference in event rate by Q912R genotype (OR =1.28, 95% CI: 0.97–1.68, P=0.08).43 While more recent investigations suggest that PON1 Q192R influences relative platelet inhibition instead of on-clopidogrel platelet reactivity44 and that genetic variability in this gene is associated with clinical outcomes in PCI patients through mechanisms independent of clopidogrel treatment,45 currently available information do not convincingly support a role of PON1 in clopidogrel pharmacogenetics.
CES1
CES1 is the primary enzyme responsible for converting clopidogrel, 2-oxo-clopidogrel, and the bioactive thiol metabolite into biologically inactive carboxylic acid derivatives.41 In fact, up to 85% of therapeutically administered clopidogrel may be degraded by CES1 activity in the liver. Given the important role of this enzyme in clopidogrel metabolism, it is not difficult to speculate that variability in CES1 function and/or expression may have important clinical implications. To date, however, few studies have evaluated the effect of genetic variants in CES1 on clopidogrel response. In 566 participants of the Pharmacogenomics of Anti-Platelet Intervention (PAPI) Study, we observed that an LOF missense polymorphism resulting in glycine-to-glutamic acid substitution at position 143 (G143E, rs71647871) significantly impacts clopidogrel pharmacokinetics and pharmacodynamics.46 In fact, compared to CES1 143G allele homozygotes, individuals who carried the 143E allele had significantly higher circulating levels of clopidogrel active metabolite (19.0 versus 30.3 ng/mL, respectively, P=0.001) as well as greater inhibition of ADP-stimulated platelet aggregation (43% versus 29% of baseline, respectively, P=0.003). We extended these findings in 350 CAD patients and observed that CES1 143E allele carriers had significantly better clopidogrel response as assessed by ADP-stimulated platelet aggregation compared to 143G homozygotes (on-clopidogrel maximal platelet aggregation =25% and 45%, respectively, P=0.03). Furthermore, 0% of CES1 143E allele carriers experienced a cardiovascular event at 1 year compared to 13.7% in patients who carried two copies of the 143G allele; however, this comparison was not statistically significantly (P=0.44), possibly due to the relatively low power of the analysis.46 At approximately the same time we published our findings, Zhu et al showed, in human liver s9 fractions, that the G143E variant completely inhibited the hydrolysis of both clopidogrel and 2-oxo-clopidogrel, consistent with our data.47 In addition, the latter investigation also evaluated the role of other genetic variants on CES1 enzymatic function and it was observed that the D260fs mutation significantly impacted CES1 activity, while the G18V, S82L, and A269S variants did not.47 It is important to note that, while these initial investigations evaluating the role of genetic variation in CES1 on clopidogrel response have yielded some interesting results, CES1 is a highly polymorphic gene. Given its critical role in clopidogrel metabolism, further studies that more comprehensively evaluate the effect of genetic variation in this gene on clopidogrel response seem warranted.
Other variants
While CYP2C19 is the most important enzyme in the bioactivation of clopidogrel, several other CYP enzymes contribute to its metabolism, including CYP2B6, CYP2C9, CYP3A4/5, and CYP1A2, albeit to a lesser extent.14 In some early studies of clopidogrel pharmacogenetics, nominal evidence of association was observed between polymorphisms in these genes (eg, CYP1A2*1F and CYP2C9*2/3) and clopidogrel response.48,49 However, subsequent replication efforts have had mixed results. Given the lesser role of genetic variation in these enzymes in clopidogrel activation, it has been speculated that redundant mechanisms of metabolism make the overall effect of these variants relatively small.50 These gene variants have been reviewed in detail previously.24,50 Taken together, while genetic variation in these genes may contribute to variable clopidogrel response, the current evidence is not strong enough to support the use of genotype information in other CYP genes for the purpose of tailoring antiplatelet therapy.
Genetic variants in genes responsible for clopidogrel pharmacodynamics have also been implicated in altered clopidogrel response. Previous investigations suggest that a two-SNP haplotype in the P2RY12 gene consisting of the G52T (rs2046934) and T744C (rs2046934) leads to higher P2Y12 receptor expression and higher platelet reactivity in clopidogrel-treated subjects.51 However, inconsistent findings in replication efforts have called the validity of this association into question.10 Similarly, polymorphisms in the ITGB3 gene, which encodes the beta subunit of the well-described glycoprotein IIb/IIIa receptor, have been associated with clopidogrel response and cardiovascular events including stent thrombosis in some investigations52 but not others.10,53 Taken together, these investigations suggest that, if variants in P2RY12 or ITGB3 are truly associated with clopidogrel response, their effects are small and not likely to be of clinical utility.
Aspirin
Aspirin is the mainstay antiplatelet agent used for the primary and secondary prevention of MI, stroke, and death. While generally effective, non-responsiveness to aspirin has been well-documented and occurs in approximately 6%–60% of individuals, depending on how it is defined.54 Aspirin irreversibly inhibits prostaglandin G/H synthase 1 (PTGS1, or COX-1) and the conversion of arachidonic acid to thromboxane. Platelet function assays specific for aspirin’s effects on platelet COX-1 include serum thromboxane B2 and arachidonic acid-induced platelet aggregation. With adequate dosing and compliance, aspirin is capable of completely inhibiting COX-1 using such assays in >99% of individuals; thus true “aspirin resistance” is rare.55,56 However, alternate agonists, such as ADP, collagen, and epinephrine, can produce robust aggregation in the face of complete COX-1 inhibition.57 These platelet aggregation pathways are sensitive to the effects of aspirin because thromboxane serves in a positive-feedback loop, thus amplifying the downstream signals of these agonists. However, these pathways are not completely dependent on the generation of thromboxane. As a consequence, these “non-COX-1-dependent” platelet function assays demonstrate wide interindividual variability before and after aspirin exposure.58 Direct measures of platelet COX-1 on aspirin demonstrate little variability and heritability;2 however, indirect or non-COX-dependent pathways demonstrate significant heritability within families.2,59 These findings suggest that there is a significant genomic contribution to the observed variability in platelet aggregation responses to aspirin. Furthermore, the observation that individuals with high levels of residual platelet aggregation on aspirin are also at heightened risk for cardiovascular events60,61 suggests that these non-COX-dependent measures of aspirin response may also be clinically significant.
Aspirin is rapidly absorbed after oral administration and has a half-life of 15–20 minutes.62 With typical daily aspirin dosing (ie, <100 mg/day), there is nearly uniform inhibition of platelet COX-1,56 suggesting that, for the vast majority of individuals, these dosages are sufficient to inhibit platelet COX-1. However, there are certain populations in which higher aspirin doses may be required.57 Aspirin undergoes hydrolysis in the plasma63 as well as in erythrocytes64 with significant interindividual variability. To identify genetic determinants of plasma hydrolytic activity, a genome-wide association study (GWAS) was performed that identified a genetic variant (rs6445035) in proximity to the butyrylcholinesterase (BCHE) gene at genome-wide level of significance, such that each additional copy of the minor allele was associated with a 1.2 nmol/mL/min reduction in aspirin hydrolytic activity but explained 3% of the overall variability in response.63 Therefore, genetic variation at BCHE is unlikely to explain much of the observed variability in aspirin response.
Several observations have been made that suggest that the response to surgical procedures, specifically coronary artery bypass grafting, results in a transient decrease in the in vitro response to aspirin.65 This transient reduction in the effects of aspirin may be explained by increased transcription of ABCC4, which is expressed in platelets and can extrude acetylsalicylate (an organic anion derived from aspirin) out of platelets, thus limiting the amount within platelets that is available to inhibit platelet COX-1.66 Although genetic variation in ABCC4 has not been linked to variation in the response to aspirin, expression profiling (ie, RNA or protein levels) may be more suitable biomarkers by which to identify individuals with high levels of this transporter.
There is a long history of largely small and underpowered studies aimed at identifying genetic variation that underlies the platelet aggregation response to aspirin. For the most part, these are candidate gene studies of platelet-related genes (PEAR1, ITGB3, VAV3, GP6, F2R, and GP1BA) with low-level evidence of association with the response to aspirin.67–70 One variant with moderate evidence is rs5918 in ITGB3, where carriers of the risk allele have heightened platelet function on aspirin.71–73 Several groups have recently taken a genome-wide approach to identifying determinants of platelet function in response to aspirin. The first of of these studies focused on native platelet function, which is a large determinant of platelet function in response to aspirin.74 Using imputation and genome-wide meta-analysis, several loci have emerged as being associated with native platelet function: rs12041331 in PEAR1; rs6943029 in SHH; rs1874445 in MRVI1; rs869244 in ADRA2A; rs2893923 in JMJD1C; and rs1671152 in GP6.74 Subsequent resequencing of the PEAR1 locus confirmed the association of rs12041331 such that, for each copy of the minor allele, there are lower levels of platelet function before and after aspirin exposure.75 The findings surrounding PEAR1 are notable, because an independent group also identified PEAR1 through GWAS of platelet function in response to aspirin.76 The PEAR1 gene is expressed in platelets and megakaryocytes and participates in megakaryopoiesis77 as well as platelet activation.78 Carriers of the intronic variant rs12041331 have higher PEAR1 gene expression as well as platelet PEAR1 protein content, suggesting a functional role for this variant.75 Additional genome-wide approaches using gene expression analysis have identified a “signature” associated with platelet function in response to aspirin. Using peripheral blood gene expression, our group identified the “aspirin response signature” (ARS), which is a set of 62 coexpressed genes, primarily of platelet origin, whose aggregate expression was reproducibly associated with platelet function in response to aspirin.79 Several genes represented by the ARS ascribe new roles to many well-known platelet genes – ITGA2B, ITGB3, MYL9, CLU, PPKAR2B, TREML1, and CTTN – with respect to platelet function on aspirin. Taking the above findings together, genetic variation or gene expression of several platelet genes is emerging as a novel marker of aspirin’s ability to inhibit platelet function.
In an effort to translate findings related to platelet function measured in the laboratory to clinical outcomes of patients taking aspirin, several groups have attempted to link genetic data with long-term clinical outcomes. In two large-scale investigations, most genetic associations with laboratory outcomes did not translate to differences in risk in clinical outcomes,80,81 though these variants were selected prior to recent GWAS findings. In a more contemporary study, the rs12041331 PEAR1 variant was studied in two independent populations and, whereas carriers of the minor allele had lower levels of platelet function on aspirin, carriers of the minor allele were consistently at higher risk for cardiovascular events.76 Further, the risk conferred by the rs12041331 PEAR1 variant seemed to depend on aspirin use, such that the risk in carriers of the minor allele was greatest in those who reported aspirin use.76 These divergent associations between platelet aggregation results and clinical outcomes with respect to PEAR1 demonstrate the complexity surrounding use of a laboratory-based assay for studying drug responses and our lack of understanding of the biological processes that occur in vivo. Further, the apparent statistical interaction between aspirin use and PEAR1 genetic variation demonstrates the complexity of translating genetic findings into clinical outcomes.
A complementary approach to identifying genetic variants that may predict the risk of cardiovascular outcomes in patients treated with aspirin comes from studies of LPA. LPA codes for apolipoprotein (a), which forms Lp(a) when linked with low-density lipoprotein particles. Lp(a) is known to be associated with the development of CAD, though it is not known to affect platelet function. A rare variant in LPA (rs3798220) was associated with higher concentrations of Lp(a),82 and carriers had a more than twofold reduction in the risk for cardiovascular disease with aspirin, whereas noncarriers (>95% of Caucasians) had no reduction in a large, placebo-controlled clinical trial.83 This suggests that the benefits of aspirin in primary prevention may be concentrated in carriers of this rare allele. Thus, this marker could be used to identify individuals who would benefit from low-dose aspirin in the primary prevention of cardiovascular disease.
Despite the recent advances in the genetics underlying the response to aspirin, there are many hurdles to overcome before these can be implemented in clinical practice. For example, although the associations with PEAR1 and platelet function are sound, the divergent results with clinical outcomes demands further explanation. Gene expression profiling of peripheral blood RNA is now a clinically available diagnostic test,84 thus facilitating measurement of genes represented by the ARS; however, it is not yet clear how treatment would be modified on the basis of these data to improve clinical outcomes. Direct platelet function testing using point-of-care devices is available, and although these testing platforms can detect inadequate levels of platelet inhibition on aspirin, no study to date has consistently shown an association of test results with cardiovascular events on aspirin. Therefore, although aspirin has been a mainstay of treatment for patients at risk for cardiovascular disease, there will continue to be uncertainty regarding the tailoring of aspirin therapy in the clinical setting.
Next-generation P2Y12 inhibitors (prasugrel and ticagrelor)
The known variability in the response to clopidogrel spurred the development of novel P2Y12 inhibitors to overcome this limitation of clopidogrel. Prasugrel, which, like clopidogrel, is administered as an inactive prodrug that must be converted to an active metabolite (R-138727), also irreversibly inhibits the platelet P2Y12 receptor.85 However, unlike clopidogrel, due to its chemical structure, the vast majority of the parent compound is converted into an active metabolite, thus achieving higher concentrations of active metabolite compared to clopidogrel and greater platelet P2Y12 receptor inhibition.86,87 Ticagrelor is a novel, non-thienopyridine, P2Y12 receptor antagonist that is administered as an orally active drug, thereby overcoming the bioactivation process.88 In addition, ticagrelor, a cyclopentyltriazolopyrimidine, is the first in its class of non-thienopyridines that reversibly inhibits the platelet P2Y12 receptor. Ticagrelor can also be metabolized into an active metabolite (AR-C124910XX) and has concentrations that are roughly one-third that of the parent compound.89
Though both prasugrel and ticagrelor achieve consistently high levels of inhibition of the platelet P2Y12 receptor, there remains significant variability when examining agonists other than ADP, such as collagen.90 Furthermore, in patients treated with prasugrel, variability in ex vivo platelet function is associated with the risk of cardiovascular events.91 This suggests that, as with clopidogrel, ex vivo platelet function testing may be a useful biomarker for identifying those who may (or may not) receive the full benefits of these novel agents. Although there have not been genome-wide approaches applied to the response to prasugrel, several candidate gene approaches have been used. Because prasugrel is a prodrug that is dependent on hepatic CYPs for bioactivation, several genetic association studies involving CYP2C19, CYP2C9, CYP2B6, and CYP1A2 have been performed. While most suggest that genetic variation at these loci are not important for prasugrel response,18,92,93 another study has shown that CYP2C19*2 and CYP2C19*17 significantly impact platelet reactivity index VASP in prasugrel-treated patients undergoing coronary stenting.94 Based on the findings surrounding PEAR1, this gene was resequenced in healthy individuals exposed to prasugrel with measurement of response using the VerifyNow® (Accumetrics, San Diego, CA, USA) platform. Two regions of the PEAR1 locus appear to be associated with the extent of platelet inhibition after prasugrel exposure.95 These findings have not yet been replicated or extended to clinical outcomes in patients treated with prasugrel. Because ticagrelor is administered as an orally active drug, we should not expect genetic variation in CYP2C19 to influence the response to ticagrelor. To confirm this, investigators have shown that CYP2C19*2 does not influence platelet aggregation96 or clinical outcomes97 in patients treated with ticagrelor, unlike patients treated with clopidogrel. In an effort to identify genetic variants beyond CYP2C19 that are associated with the response to ticagrelor, a recent GWAS was conducted on levels of ticagrelor and its active metabolite from the PLATO clinical trial.98 Genetic variants in linkage disequilibrium with the SLC01B1*5 loss of function variant were associated with higher ticagrelor and active metabolite levels. Further, a genetic variant (rs61361928) in UGT2B7 was associated with ticagrelor active metabolite levels. None of the variants in SLCO1B1 nor UGT2B7, however, were associated with bleeding or ischemic events in the ticagrelor-treated arm.99
Anticoagulant pharmacogenetics
Warfarin
Warfarin has been the mainstay of oral anticoagulant therapy for many years. However, it has significant variability in pharmacological response among individuals, with doses varying by up to a factor of 10;100 has a narrow therapeutic index; and requires frequent monitoring in order to maintain a therapeutic international normalized ratio (INR). In addition, variation in clinical response to warfarin and other coumarin derivatives is consistently implicated among the leading causes of hospitalization from adverse drug events such as bleeding.101,102 Warfarin is given as a racemic mixture, with S-warfarin being 3–5 times more potent than R-warfarin. Numerous retrospective studies have found that the enzyme responsible for metabolizing S-warfarin, CYP2C9, and the gene that encodes warfarin’s target, VKORC1, are associated with warfarin dose requirements; these have been reviewed previously in detail.34,103–106 Multivariable models have found that CYP2C9 and VKORC1 variants account for about 50% of the variability in dose requirements.100 Variants in other genes including CYP4F2 have also been reproducibly associated with warfarin dose requirements, although to a smaller extent, explaining an additional 2%–3% of the variability in addition to clinical factors and CYP2C9 and VKORC1 genotype.107,108
The field of anticoagulant pharmacogenetics has recently been complicated by the simultaneous publication of three randomized clinical trials comparing pharmacogenetic algorithms incorporating CYP2C9 and VKORC1 genotypes with either standard of care or a clinical algorithm for dose selection of vitamin K antagonist.109–111 The United States-based study, Clarification of Optimal Anticoagulation through Genetics (COAG),110 randomized 1,015 patients to receive warfarin dose according to an algorithm that contained genotype and clinical variables versus a dosing algorithm that contained only clinical variables, including early INR data. The primary endpoint was percent of time that INR was in the therapeutic range day 4 or 5 through day 28. In the COAG study, no significant difference was found between the two dosing algorithms, with time in the therapeutic range of 45.2% in the genotype-guided arm and 45.4% in the clinically guided group. The investigators also noted a significant interaction between race and algorithm, whereby black patients did significantly worse in the genotype-guided strategy. Time in the therapeutic range in black patients assigned to the genotype-guided dosing was 35.2%, compared to 43.5% in the clinically dosed algorithm (P=0.01). Among nonblack patients, there was a trend toward improvement, with time in the therapeutic range of 48.8% in the genotype-guided group compared to 46.1% in the clinically guided group (P=0.15). The study was not powered to look at bleeding events, but, interestingly, all clinically relevant bleeding events were greater in the clinically guided group, particularly when evaluated from randomization to the end of follow-up.
In the European study from the European Pharmacogenetics of AntiCoagulant Therapy (EU-PACT) group, which evaluated warfarin, 455 patients were randomized to a genotype-guided group or control.109 The control arm in this study consisted of a standard (ie, fixed-dose) 3-day loading dose regimen. The primary outcome was percent of time in therapeutic range during the 12 weeks after warfarin initiation. The trial found that genotype-guided dosing was superior to standard dosing, with a mean percent of time in the therapeutic range of 67.4% in the genotype-guided group compared to 60.3% in the control group (P<0.001). Patients in the genotype group were also significantly less likely to have an INR ≥4 (27% versus 36.6%, P=0.03) and had a significantly shorter time to reach therapeutic INR (21 days versus 29 days, P<0.001). There were no major bleeding events in the study.
The second EU-PACT study evaluated acenocoumarol and phenprocoumon, agents not available in the United States.111 A total of 548 patients were randomized to genotype-guided dosing or dosing with a clinical algorithm that included early INR data. There was no significant difference in the primary outcome (percent of time in therapeutic range during the 12 weeks of coumarin initiation) between the two dosing algorithms (61.6% in the genotype-guided dosing arm and 60.2% in the clinically guided dosing arm, P=0.52). During the first 4 weeks of initiation of treatment (similar to the primary outcome in the COAG study), the percent of time in the therapeutic range was greater for the genotype-guided group compared to the clinically guided group (52.8% versus 47.5% [P=0.02], respectively). Some studies have reported that CYP2C9 variants had less of an influence on the pharmacokinetics of phenprocoumon than warfarin or acenocoumarol.112 Nonetheless, several observational studies have found associations between CYP2C9 and phenprocoumon dose requirements.113,114 Thus, it is likely that the high performance of the clinical algorithm used in the comparator group is the reason for the small nonsignificant difference between the two dosing strategies. We will focus the remaining discussion of these studies on warfarin, given that it is the only one of these drugs available in the United States.
The impact of race on genotype-guided warfarin dosing merits further discussion. The EU-PACT studies included almost exclusively white patients, but nearly one-third of the COAG study population consisted of black patients, and the genotype dosing algorithm did not improve the amount of time spent in the therapeutic range in these patients.109–111 The genotype dosing algorithms from all three studies included the CYP2C9*2 and *3 alleles and VKORC1 rs9923231 (3673G→A), all of which are much less frequent in individuals of African descent compared to those of European descent.109–111 Consequently, these alleles explain less of the variation in warfarin dose requirements in African Americans than in populations in which they are more common.115 The CYP2C9*8 allele, which is far more common in African Americans, was not part of the algorithm. In addition, a novel locus in the CYP2C cluster which contributes to warfarin dose requirements independently of CYP2C9 and VKORC1 in African Americans and improved the amount of variability explained by about 20% was not discovered in time to be included in the COAG study.116 Therefore, it remains to be determined whether the inclusion of genotype dosing algorithms incorporating appropriate race-specific alleles would have improved outcomes in the COAG trial.
In trying to sort out these contradictory findings, several points should be considered. First is the difference in duration of these studies. Genotype would be expected to have the biggest impact in the first 1–2 months of therapy because INR itself will provide more information as time passes. Therefore, it is possible that the COAG study missed some value of genotype by only having data in the first 4 weeks compared to 12 weeks in EU-PACT.109,111 A second difference between COAG and EU-PACT that could account for their different findings is the use of a loading dose in the EU-PACT study. It is possible that the difference between groups in the EU-PACT study was due to systematically larger loading doses in the genotype-guided group compared to the standard-dose group. Next, it is not currently standard practice to dose warfarin via a clinical algorithm. Additionally, the study protocols required intensive INR measurement and frequent dose adjustments which are not reflective of how the majority of patients are managed outside of research studies. This frequent monitoring may have obscured detectible differences between the groups. Given the differences in the comparator groups (ie, clinical algorithm in COAG versus standard dose in EU-PACT warfarin), the COAG study could be considered an efficacy study, whereas the EU-PACT warfarin study could be considered an effectiveness study. Therefore, we can conclude that genotype-guided warfarin dosing may improve the amount of time spent in the therapeutic range compared to more real-world settings utilizing standard empiric dosing (eg, 10 mg, 5 mg, 5 mg loading regimen) but not compared to idealized settings which already incorporate personalized dosing strategies using clinical variables. In terms of other hard outcomes, such as bleeding or thromboembolic events, the randomized prospective studies were not powered for these. An ongoing study (Genetics Informatics Trial [GIFT] of Warfarin to Prevent DVT, NCT01006733), however, will be powered to detect bleeding and thrombotic outcomes.
Novel oral anticoagulants
Several novel oral anticoagulants have been approved or are nearing approval for the treatment of thromboembolic disorders, including dabigatran, which is a direct thrombin inhibitor, and rivaroxaban and apixaban, which are direct factor Xa inhibitors. These agents are given as fixed doses and do not require monitoring of INR, so offer some advantages over warfarin in terms of convenience.
Much less is known about the pharmacogenomic determinants of response to these agents than with warfarin, but one study has been published with regard to dabigatran.117 Dabigatran is a prodrug which requires conversion by esterases in the liver to be activated. Considerable variability in blood concentrations of the active metabolite has been observed. The drug is primarily renally eliminated and is not metabolized by CYP450 enzymes. Genetic samples were collected from a subset of patients enrolled in the Randomized Evaluation of Long-term Anticoagulation Therapy (RE-LY), which compared two doses of dabigatran with warfarin in patients with atrial fibrillation.118 The investigators performed a GWAS to look for variants associated with dabigatran peak and trough concentrations in 1,490 patients of European ancestry and then evaluated the top SNPs for association with bleeding events and thromboembolic events in 1,694 dabigatran-treated patients. One SNP in CES1 (rs4148738) and one SNP in ABCB1 (rs8192935) met genome-wide significance for association with peak concentrations, and two SNPs in linkage disequilibrium at the CES1 locus (rs2244613 and rs4580160) were genome-wide significant for trough concentrations. It was estimated that individuals with combinations of the variant alleles could have from a 29.6% decrease up to a 20.2% increase in drug exposure compared to individuals without any copies of the variant alleles. The two peak SNPs and the lead trough SNPs were then tested for association with outcomes. Neither of the two SNPs associated with peak concentrations were associated with bleeding or ischemic events. The CES1 SNP, rs2244613, that was associated with trough concentrations was associated with any bleeding (OR =0.67, 95% CI: 0.55–0.82, P=7×10−5), minor bleeding (OR =0.70, 95% CI: 0.57–0.85, P=4×10−4), and trended toward significance with major bleeding, (OR =0.66, 95% CI: 0.43–1.01, P=0.06) on dabigatran. There was no such association between rs2244613 and bleeding events in warfarin-treated patients leading to a significant SNP by treatment interaction (P=0.002). These findings have not yet been replicated outside of the RE-LY study.
Clinical application
For drugs such as aspirin, the newer antiplatelet agents, and novel anticoagulants, the data are not yet mature enough to suggest clinical implementation of pharmacogenetic information. For warfarin and clopidogrel, however, there are sufficient data to justify using genotype information to guide therapeutic strategies in a limited manner. The roadmap for implementation of pharmacogenetics into the clinical environment for warfarin or clopidogrel is not a straightforward approach. Unlike the large effect sizes observed with genetic variants in the HLA locus and adverse effects of carbamazepine and abacavir, for example, the potential benefits for clopidogrel or warfarin are likely to be more modest. However, given the large number of patients prescribed these agents, modest gains may lead to large effects at the population level. The subtleties of clopidogrel genetic associations suggest that the context and indication for which clopidogrel is being used are critical to an implementation strategy. Similarly, the same may be true of warfarin. In fact, it may be that clopidogrel and warfarin pharmacogenetics are not appropriate for routine management of these medications but instead in selected high-risk patients/conditions (Figure 2).
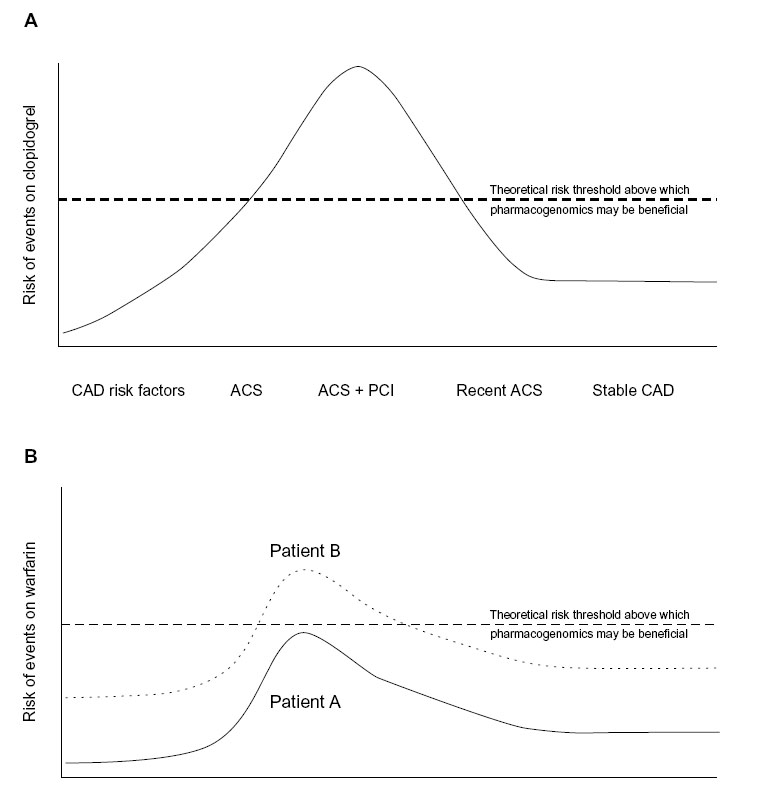 | Figure 2 Theoretical implementation of clopidogrel and warfarin pharmacogenomics. |
The available evidence for clopidogrel demonstrates consistent associations for variants in CYP2C19 in ACS patients receiving PCI who are treated with clopidogrel (see “CYP2C19” section). Furthermore, for this patient population, ticagrelor and prasugrel are two FDA-approved alternatives to clopidogrel for this indication. Currently, physicians caring for ACS patients after PCI have few clinical tools to choose from among the available P2Y12 inhibitors. Often, clinical characteristics, such as age, body size, diabetes, or ST segment elevation MI presentation, are used to guide clinical decisions. In this context, CYP2C19*2 could be used as an adjunct datum to assist with clinical decision-making. Physicians caring for patients who carry the CYP2C19*2 allele could be advised of the heightened risk of stent thrombosis on clopidogrel and to consider prasugrel or ticagrelor instead. The increasing use of electronic medical records and clinical decision support could facilitate communication, education, and ordering. Given that the average length of hospital stay for ACS patients is ~2–3 days, genotyping could be initiated at the time of presentation (ie, in the emergency room or catheterization laboratory), such that results are available by the time of discharge. Alternatively, rapid point-of-care genotyping platforms could be utilized. Outside of the high-risk patient populations, there are minimal data to suggest that genotype-guided clopidogrel therapy is justified and/or is expected to improve patient outcomes.
The use of genetic data to guide warfarin therapy is less clear given the results of recent clinical trials and the availability of novel anticoagulants that are noninferior and/or superior relative to warfarin.119,120 One potential approach could target patients when there are no alternatives to warfarin (eg, mechanical valve replacement). This approach is facilitated by the fact that many valve repair surgeries are scheduled procedures, thus allowing several days for the return of genotype results. Clinical decision support embedded within an electronic medical record could facilitate interpretation of genetic data into warfarin dose recommendations. Another potential application would be to target patients/practices when there is difficulty in obtaining frequent INRs, where warfarin is not managed by an anticoagulation service, or in patients at high risk for bleeding-related outcomes (eg, concomitant dual antiplatelet therapy) (Figure 2). In these settings, genetically tailored warfarin dosing may be of benefit to patients over a fixed-dose algorithm.
Implications for enhanced patient care
While traditional medical approaches that typically rely on the application of a “one-size-fits-all” model of patient treatment are generally effective, the clinical utility of pharmacogenomic testing in cardiovascular medicine has the potential to substantially improve patient care. The most obvious benefits to such personalized approaches are the reductions in both recurrent thrombotic events and onset of secondary complications. Indeed, in a relatively small (N=200) prospective proof-of-concept trial of CYP2C19*2 genotype-directed antiplatelet therapy, Roberts et al observed that none of the patients who carried the CYP2C19*2 allele in the genotype-directed arm, and thus received prasugrel, had HTPR, whereas approximately 30% of CYP2C19*2 carriers in the standard treatment arm (ie, received clopidogrel) did experience HTPR.121 These results are consistent with the work of Xie et al, who observed that Chinese CAD patients undergoing PCI who received CYP2C19 testing had lower rates of stent thrombosis (0.66% versus 3.01%, P=0.03), MI (0.33% versus 3.01%, P=0.01), and death (0.33% versus 2.34%, P=0.01) than those who did not receive genetic testing.122 While these data alone cannot provide an estimate of the impact of genotype-guided therapy on hard cardiovascular outcomes, these initial findings are encouraging. At this time, the scientific community awaits the results of large-scale prospective randomized trials of genotype-directed antiplatelet therapy to determine the impact of genetic testing on cardiovascular event reduction.
Adverse drug reactions remain an important problem in both antiplatelet and anticoagulant therapy. As described in the “Warfarin” section, given the narrow therapeutic index of warfarin, patients who demonstrate genetic sensitivity have increased risk of pathological bleeding. Similarly, patients on clopidogrel who carry CYP2C19 GOF variants (eg, CYP2C19*17) are more likely to experience adverse bleeding events than those who do not. While better characterization of the genetic variants that predispose patients to such adverse drug reactions is needed, guidelines developed by the Clinical Pharmacogenetics Implementation Consortium (CPIC) have attempted to include such information in clinical algorithms used to aid in physician decision-making for both clopidogrel (Figure 3) and warfarin therapy.23,123 Through the use of such pharmacogenetic approaches, it may be possible to reduce adverse drug reactions, thereby improving drug adherence and ultimately further reducing the rate of recurrent events.
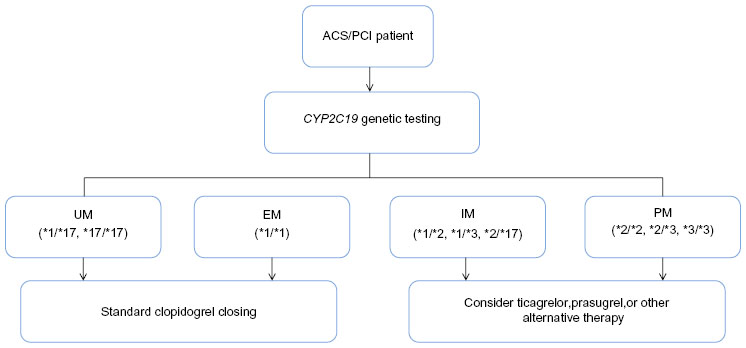 | Figure 3 Pharmacogenetic algorithm for initiation of antiplatelet therapy based on CYP2C19 genotype recommended by the Clinical Pharmacogenetics Implementation Consortium. |
In addition to the health-related benefits described in the previous paragraph, there are several economic mechanisms by which pharmacogenetics can result in enhanced patient care. Several studies have evaluated the cost-effectiveness of genotype-guided antiplatelet therapy versus various empiric treatment strategies.124–127 Using conservative estimates on the effect size of CYP2C19 genotype (HR =1.18, 95% CI: 1.09–1.28) in their base-case analysis, Kazi et al124 found that genotyping with ticagrelor as the alternative therapy yielded the lowest incremental cost-effectiveness ratio per quality-adjusted life year, at US$30,200. The use of ticagrelor in all individuals was the next most favorable strategy, at US$52,500 per quality-adjusted life year, relative to genotyping with ticagrelor in the base-case analysis. Sensitivity analyses incorporating larger CYP2C19 effect sizes (HR =1.57, 95% CI: 1.13–2.16) greatly increased the cost-effectiveness of genotyping. Several studies have been performed at different times (eg, before ticagrelor was approved or before clopidogrel was officially off-patent), but they have been fairly consistent in finding genotype-guided therapy to be cost-effective.125–127
Many cost-effectiveness studies evaluating genotype-guided anticoagulation have been published, and most have found genotyping to be modestly cost-effective, although these studies were conducted prior to the publication of COAG and the EU-PACT studies.128–135 Therefore, how relevant they are in light of the most recent data is unclear. That point aside, the studies have generally found that genotype-guided anticoagulation that reduces bleeding events or that obtains time in therapeutic range (TTR) of ~70% to be cost-effective. However, it is important to point out that TTR varies greatly depending on how long patients are followed and how strictly the therapeutic range is defined (eg, INR 2–3 or 1.8–3.2). For example, in COAG, TTR was 45% at 4 weeks with INR 2–3; 62% at 4 weeks with INR 1.8–3.2; 51% at 12 weeks with INR 2–3; and 68% at 12 weeks with INR 1.8–3.2.110
Conclusion
The use of pharmacogenetic data to tailor antiplatelet and anticoagulation regimens, particularly clopidogrel and warfarin, may significantly reduce on-treatment cardiovascular events and adverse drug reactions and result in enhanced patient care. In addition, while currently available data regarding the genetic determinants of response to next-generation agents such as prasugrel, ticagrelor, and dabigatran are limited at this time, initial investigations suggest that genetic variants may significantly impact efficacy of these medications. Despite these data, however, nontrivial barriers to implementation, including cost considerations, genotype turnaround time, and lack of randomized clinical trial data, currently limit the use of genetic testing in the clinical setting. Even in the absence of such barriers, recent investigations have highlighted that genetic testing may not be the best strategy for routine management for all patients, but should perhaps be considered in selected high-risk patients/conditions. Not surprisingly, careful evaluation of all clinical information (eg, cardiovascular indication, patient characteristics, prior medical history, etc) will be critical in selecting patients who may benefit from genetic testing. Future investigations aimed at overcoming the barriers to implementation, as well as defining the subset of patients who will receive benefit from genotype-directed therapy, will be critical in determining the impact of pharmacogenetics on antiplatelet and anticoagulation therapy.
Acknowledgments
This work was supported by National Institutes of Health grants U01 HL105198, K23 GM102678, K23HL091120, and R01HL118049.
Disclosure
The authors report no conflicts of interest in this work.
References
Shuldiner AR, O’Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA. 2009;302(8):849–857. | |
Faraday N, Yanek LR, Mathias R, et al. Heritability of platelet responsiveness to aspirin in activation pathways directly and indirectly related to cyclooxygenase-1. Circulation. 2007;115(19):2490–2496. | |
Gurbel PA, Bliden KP, Hiatt BL, O’Connor CM. Clopidogrel for coronary stenting: response variability, drug resistance, and the effect of pretreatment platelet reactivity. Circulation. 2003;107(23):2908–2913. | |
Gurbel PA, Cummings CC, Bell CR, Alford AB, Meister AF, Serebruany VL; Plavix Reduction Of New Thrombus Occurrence (PRONTO) trial. Onset and extent of platelet inhibition by clopidogrel loading in patients undergoing elective coronary stenting: the Plavix Reduction Of New Thrombus Occurrence (PRONTO) trial. Am Heart J. 2003;145(2):239–247. | |
Serebruany VL, Steinhubl SR, Berger PB, Malinin AI, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol. 2005;45(2):246–251. | |
Bonello L, Tantry US, Marcucci R, et al; Working Group on High On-Treatment Platelet Reactivity. Consensus and future directions on the definition of high on-treatment platelet reactivity to adenosine diphosphate. J Am Coll Cardiol. 2010;56(12):919–933. | |
Frelinger AL 3rd, Bhatt DL, Lee RD, et al. Clopidogrel pharmacokinetics and pharmacodynamics vary widely despite exclusion or control of polymorphisms (CYP2C19, ABCB1, PON1), noncompliance, diet, smoking, co-medications (including proton pump inhibitors), and pre-existent variability in platelet function. J Am Coll Cardiol. 2013;61(8):872–879. | |
Ameyaw MM, Regateiro F, Li T, et al. MDR1 pharmacogenetics: frequency of the C3435T mutation in exon 26 is significantly influenced by ethnicity. Pharmacogenetics. 2001;11(3):217–221. | |
Taubert D, von Beckerath N, Grimberg G, et al. Impact of P-glycoprotein on clopidogrel absorption. Clin Pharmacol Ther. 2006; 80(5):486–501. | |
Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med. 2009;360(4):363–375. | |
Mega JL, Close SL, Wiviott SD, et al. Genetic variants in ABCB1 and CYP2C19 and cardiovascular outcomes after treatment with clopidogrel and prasugrel in the TRITON-TIMI 38 trial: a pharmacogenetic analysis. Lancet. 2010;376(9749):1312–1319. | |
Luo M, Li J, Xu X, Sun X, Sheng W. ABCB1 C3435T polymorphism and risk of adverse clinical events in clopidogrel treated patients: a meta-analysis. Thromb Res. 2012;129(6):754–759. | |
Su J, Xu J, Li X, et al. ABCB1 C3435T polymorphism and response to clopidogrel treatment in coronary artery disease (CAD) patients: a meta-analysis. PLoS One. 2012;7(10):e46366. | |
Kazui M, Nishiya Y, Ishizuka T, et al. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos. 2010;38(1):92–99. | |
de Morais SM, Wilkinson GR, Blaisdell J, Nakamura K, Meyer UA, Goldstein JA. The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J Biol Chem. 1994;269(22):15419–15422. | |
Sim SC, Risinger C, Dahl ML, et al. A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther. 2006;79(1):103–113. | |
Hulot JS, Bura A, Villard E, et al. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood. 2006;108(7):2244–2247. | |
Brandt JT, Close SL, Iturria SJ, et al. Common polymorphisms of CYP2C19 and CYP2C9 affect the pharmacokinetic and pharmacodynamic response to clopidogrel but not prasugrel. J Thromb Haemost. 2007;5(12):2429–2436. | |
Kim KA, Park PW, Hong SJ, Park JY. The effect of CYP2C19 polymorphism on the pharmacokinetics and pharmacodynamics of clopidogrel: a possible mechanism for clopidogrel resistance. Clin Pharmacol Ther. 2008;84(2):236–242. | |
Varenhorst C, James S, Erlinge D, et al. Genetic variation of CYP2C19 affects both pharmacokinetic and pharmacodynamic responses to clopidogrel but not prasugrel in aspirin-treated patients with coronary artery disease. Eur Heart J. 2009;30(14):1744–1752. | |
Collet JP, Hulot JS, Anzaha G, et al; CLOVIS-2 Investigators. High doses of clopidogrel to overcome genetic resistance: the randomized crossover CLOVIS-2 (Clopidogrel and Response Variability Investigation Study 2). JACC Cardiovasc Interv. 2011;4(4):392–402. | |
Hulot JS, Collet JP, Cayla G, et al. CYP2C19 but not PON1 genetic variants influence clopidogrel pharmacokinetics, pharmacodynamics, and clinical efficacy in post-myocardial infarction patients. Circ Cardiovasc Interv. 2011;4(5):422–428. | |
Scott SA, Sangkuhl K, Stein CM, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther. 2013;94(3):317–323. | |
O’Connor SA, Hulot JS, Silvain J, Cayla G, Montalescot G, Collet JP. Pharmacogenetics of clopidogrel. Curr Pharm Des. 2012;18(33):5309–5327. | |
Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360(4):354–362. | |
Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet. 2009;373(9660):309–317. | |
Sibbing D, Stegherr J, Latz W, et al. Cytochrome P450 2C19 loss-of-function polymorphism and stent thrombosis following percutaneous coronary intervention. Eur Heart J. 2009;30(8):916–922. | |
Paré G, Mehta SR, Yusuf S, et al. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med. 2010;363(18):1704–1714. | |
Hulot JS, Collet JP, Silvain J, et al. Cardiovascular risk in clopidogrel-treated patients according to cytochrome P450 2C19*2 loss-of-function allele or proton pump inhibitor coadministration: a systematic meta-analysis. J Am Coll Cardiol. 2010;56(2):134–143. | |
Mega JL, Simon T, Collet JP, et al. Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: a meta-analysis. JAMA. 2010;304(16):1821–1830. | |
Sofi F, Giusti B, Marcucci R, Gori AM, Abbate R, Gensini GF. Cytochrome P450 2C19*2 polymorphism and cardiovascular recurrences in patients taking clopidogrel: a meta-analysis. Pharmacogenomics J. 2011;11(3):199–206. | |
Bauer T, Bouman HJ, van Werkum JW, Ford NF, ten Berg JM, Taubert D. Impact of CYP2C19 variant genotypes on clinical efficacy of antiplatelet treatment with clopidogrel: systematic review and meta-analysis. BMJ. 2011;343:d4588. | |
Zabalza M, Subirana I, Sala J, et al. Meta-analyses of the association between cytochrome CYP2C19 loss- and gain-of-function polymorphisms and cardiovascular outcomes in patients with coronary artery disease treated with clopidogrel. Heart. 2012;98(2):100–108. | |
Johnson JA, Cavallari LH. Pharmacogenetics and cardiovascular disease – implications for personalized medicine. Pharmacol Rev. 2013;65(3):987–1009. | |
Holmes MV, Perel P, Shah T, Hingorani AD, Casas JP. CYP2C19 genotype, clopidogrel metabolism, platelet function, and cardiovascular events: a systematic review and meta-analysis. JAMA. 2011;306(24):2704–2714. | |
Shuldiner AR, Vesely MR, Fisch A. CYP2C19 genotype and cardiovascular events. JAMA. 2012;307(14):1482; author reply 1484–1485. | |
Mega JL, Topol EJ, Sabatine MS. CYP2C19 genotype and cardiovascular events. JAMA. 2012;307(14):1482–1483; author reply 1484–1485. | |
Frére C, Cuisset T, Gaborit B, Alessi MC, Hulot JS. The CYP2C19*17 allele is associated with better platelet response to clopidogrel in patients admitted for non-ST acute coronary syndrome. J Thromb Haemost. 2009;7(8):1409–1411. | |
Lewis JP, Stephens SH, Horenstein RB, et al. The CYP2C19*17 variant is not independently associated with clopidogrel response. J Thromb Haemost. 2013;11(9):1640–1646. | |
Li Y, Tang HL, Hu YF, Xie HG. The gain-of-function variant allele CYP2C19*17: a double-edged sword between thrombosis and bleeding in clopidogrel-treated patients. J Thromb Haemost. 2012;10(2):199–206. | |
Bouman HJ, Schomig E, van Werkum JW, et al. Paraoxonase-1 is a major determinant of clopidogrel efficacy. Nat Med. 2011;17(1):110–116. | |
Lewis JP, Fisch AS, Ryan K, et al. Paraoxonase 1 (PON1) gene variants are not associated with clopidogrel response. Clin Pharmacol Ther. 2011;90(4):568–574. | |
Reny JL, Combescure C, Daali Y, Fontana P; PON1 Meta-Analysis Group. Influence of the paraoxonase-1 Q192R genetic variant on clopidogrel responsiveness and recurrent cardiovascular events: a systematic review and meta-analysis. J Thromb Haemost. 2012;10(7):1242–1251. | |
Li X, Zhang L, Chen X, et al. PON1 Q192R genotype influences clopidogrel responsiveness by relative platelet inhibition instead of on-treatment platelet reactivity. Thromb Res. 2013;132(4):444–449. | |
Park KW, Park JJ, Kang J, et al. Paraoxonase 1 gene polymorphism does not affect clopidogrel response variability but is associated with clinical outcome after PCI. PLoS One. 2013;8(2):e52779. | |
Lewis JP, Horenstein RB, Ryan K, et al. The functional G143E variant of carboxylesterase 1 is associated with increased clopidogrel active metabolite levels and greater clopidogrel response. Pharmacogenet Genomics. 2013;23(1):1–8. | |
Zhu HJ, Wang X, Gawronski BE, Brinda BJ, Angiolillo DJ, Markowitz JS. Carboxylesterase 1 as a determinant of clopidogrel metabolism and activation. J Pharmacol Exp Ther. 2013;344(3):665–672. | |
Harmsze A, van Werkum JW, Bouman HJ, et al. Besides CYP2C19*2, the variant allele CYP2C9*3 is associated with higher on-clopidogrel platelet reactivity in patients on dual antiplatelet therapy undergoing elective coronary stent implantation. Pharmacogenet Genomics. 2010;20(1):18–25. | |
Park KW, Park JJ, Jeon KH, et al. Enhanced clopidogrel responsiveness in smokers: smokers’ paradox is dependent on cytochrome P450 CYP1A2 status. Arterioscler Thromb Vasc Biol. 2011;31(3):665–671. | |
Fisch AS, Perry CG, Stephens SH, Horenstein RB, Shuldiner AR. Pharmacogenomics of anti-platelet and anti-coagulation therapy. Curr Cardiol Rep. 2013;15(7):381. | |
Bura A, Bachelot-Loza C, Ali FD, Aiach M, Gaussem P. Role of the P2Y12 gene polymorphism in platelet responsiveness to clopidogrel in healthy subjects. J Thromb Haemost. 2006;4(9):2096–2097. | |
Cayla G, Hulot JS, O’Connor SA, et al. Clinical, angiographic, and genetic factors associated with early coronary stent thrombosis. JAMA. 2011;306(16):1765–1774. | |
Lev EI, Patel RT, Guthikonda S, Lopez D, Bray PF, Kleiman NS. Genetic polymorphisms of the platelet receptors P2Y(12), P2Y(1) and GP IIIa and response to aspirin and clopidogrel. Thromb Res. 2007;119(3):355–360. | |
Mansour K, Taher AT, Musallam KM, Alam S. Aspirin resistance. Adv Hematol. 2009;2009:937352. | |
Becker DM, Segal J, Vaidya D, et al. Sex differences in platelet reactivity and response to low-dose aspirin therapy. JAMA. 2006;295(12):1420–1427. | |
Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, FitzGerald GA. Drug resistance and pseudoresistance: an unintended consequence of enteric coating aspirin. Circulation. 2013;127(3):377–385. | |
Gurbel PA, Bliden KP, DiChiara J, et al. Evaluation of dose-related effects of aspirin on platelet function: results from the Aspirin-Induced Platelet Effect (ASPECT) study. Circulation. 2007;115(25):3156–3164. | |
Voora D, Ortel TL, Lucas JE, Chi JT, Becker RC, Ginsburg GS. Time-dependent changes in non-COX-1-dependent platelet function with daily aspirin therapy. J Thromb Thrombolysis. 2012;33(3):246–257. | |
Shen H, Herzog W, Drolet M, et al. Aspirin resistance in healthy drug-naive men versus women (from the Heredity and Phenotype Intervention Heart Study). Am J Cardiol. 2009;104(4):606–612. | |
Frelinger AL 3rd, Li Y, Linden MD, et al. Association of cyclooxygenase-1-dependent and -independent platelet function assays with adverse clinical outcomes in aspirin-treated patients presenting for cardiac catheterization. Circulation. 2009;120(25):2586–2596. | |
Snoep JD, Hovens MM, Eikenboom JC, van der Bom JG, Huisman MV. Association of laboratory-defined aspirin resistance with a higher risk of recurrent cardiovascular events: a systematic review and meta-analysis. Arch Intern Med. 2007;167(15):1593–1599. | |
Benedek IH, Joshi AS, Pieniaszek HJ, King SY, Kornhauser DM. Variability in the pharmacokinetics and pharmacodynamics of low dose aspirin in healthy male volunteers. J Clin Pharmacol. 1995;35(12):1181–1186. | |
Zhou G, Marathe GK, Hartiala J, et al. Aspirin hydrolysis in plasma is a variable function of butyrylcholinesterase and platelet-activating factor acetylhydrolase 1b2 (PAFAH1b2). J Biol Chem. 2013;288(17):11940–11948. | |
Zhou G, Marathe GK, Willard B, McIntyre TM. Intracellular erythrocyte platelet-activating factor acetylhydrolase I inactivates aspirin in blood. J Biol Chem. 2011;286(40):34820–34829. | |
Zimmermann N, Wenk A, Kim U, et al. Functional and biochemical evaluation of platelet aspirin resistance after coronary artery bypass surgery. Circulation. 2003;108(5):542–547. | |
Mattiello T, Guerriero R, Lotti LV, et al. Aspirin extrusion from human platelets through multidrug resistance protein-4-mediated transport: evidence of a reduced drug action in patients after coronary artery bypass grafting. J Am Coll Cardiol. 2011;58(7):752–761. | |
Lepäntalo A, Mikkelsson J, Reséndiz JC, et al. Polymorphisms of COX-1 and GPVI associate with the antiplatelet effect of aspirin in coronary artery disease patients. Thromb Haemost. 2006;95(2):253–259. | |
Smith SM, Judge HM, Peters G, et al. PAR-1 genotype influences platelet aggregation and procoagulant responses in patients with coronary artery disease prior to and during clopidogrel therapy. Platelets. 2005;16(6):340–345. | |
Herrera JE, Qayyum R, Faraday N, et al. Abstract 1440: platelet response to aspirin is under polygenic control of variants in the VAV3 and phospholipase C gamma 2 (PLCG2) genes. Circulation. 2008;118:S_326. | |
Herrera-Galeano JE, Becker DM, Wilson AF, et al. A novel variant in the platelet endothelial aggregation receptor-1 gene is associated with increased platelet aggregability. Arterioscler Thromb Vasc Biol. 2008;28(8):1484–1490. | |
Undas A, Brummel K, Musial J, Mann KG, Szczeklik A. Pl(A2) polymorphism of beta(3) integrins is associated with enhanced thrombin generation and impaired antithrombotic action of aspirin at the site of microvascular injury. Circulation. 2001;104(22):2666–2672. | |
Cooke GE, Bray PF, Hamlington JD, Pham DM, Goldschmidt-Clermont PJ. PlA2 polymorphism and efficacy of aspirin. Lancet. 1998;351(9111):1253–1253. | |
Cooke GE, Liu-Stratton Y, Ferketich AK, et al. Effect of platelet antigen polymorphism on platelet inhibition by aspirin, clopidogrel, or their combination. J Am Coll Cardiol. 2006;47(3):541–546. | |
Johnson AD, Yanek LR, Chen MH, et al. Genome-wide meta-analyses identifies seven loci associated with platelet aggregation in response to agonists. Nat Genet. 2010;42(7):608–613. | |
Faraday N, Yanek LR, Yang XP, et al. Identification of a specific intronic PEAR1 gene variant associated with greater platelet aggregability and protein expression. Blood. 2011;118(12):3367–3375. | |
Lewis JP, Ryan K, O’Connell JR, et al. Genetic variation in PEAR1 is associated with platelet aggregation and cardiovascular outcomes. Circ Cardiovasc Genet. 2013;6(2):184–192. | |
Kauskot A, Vandenbriele C, Louwette S, et al. PEAR1 attenuates megakaryopoiesis via control of the PI3K/PTEN pathway. Blood. 2013;121(26):5208–5217. | |
Kauskot A, Di Michele M, Loyen S, Freson K, Verhamme P, Hoylaerts MF. A novel mechanism of sustained platelet alphaIIbbeta3 activation via PEAR1. Blood. 2012;119(17):4056–4065. | |
Voora D, Cyr D, Lucas J, et al. Aspirin exposure reveals novel genes associated with platelet function and cardiovascular events. J Am Coll Cardiol. 2013;62(14):1267–1276. | |
Voora D, Horton J, Shah SH, Shaw LK, Newby LK. Polymorphisms associated with in vitro aspirin resistance are not associated with clinical outcomes in patients with coronary artery disease who report regular aspirin use. Am Heart J. 2011;162(1):166–172. e161. | |
Le Hello C, Morello R, Lequerrec A, Duarte C, Riddell J, Hamon M. Effect of PlA1/A2 glycoprotein IIIa gene polymorphism on the long-term outcome after successful coronary stenting. Thromb J. 2007;5:19. | |
Clark R, Peden JF, Hopewell JC, et al; PROCARDIS Consortium. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518–2528 | |
Chasman DI, Shiffman D, Zee RY, et al. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis. 2009;203(2):371–376. | |
Thomas GS, Voros S, McPherson JA, et al. A blood-based gene expression test for obstructive coronary artery disease tested in symptomatic nondiabetic patients referred for myocardial perfusion imaging the COMPASS Study. Circ Cardiovasc Genet. 2013;6(2):154–162. | |
Angiolillo DJ, Bhatt DL, Gurbel PA, Jennings LK. Advances in antiplatelet therapy: agents in clinical development. Am J Cardiol. 2009;103(Suppl 3):40A–51A. | |
Wallentin L, Varenhorst C, James S, et al. Prasugrel achieves greater and faster P2Y12 receptor-mediated platelet inhibition than clopidogrel due to more efficient generation of its active metabolite in aspirin-treated patients with coronary artery disease. Eur Heart J. 2008;29(1):21–30. | |
Payne CD, Li YG, Small DS, et al. Increased active metabolite formation explains the greater platelet inhibition with prasugrel compared to high-dose clopidogrel. J Cardiovasc Pharmacol. 2007;50(5):555–562. | |
Teng R, Maya J, Butler K. Evaluation of the pharmacokinetics and pharmacodynamics of ticagrelor co-administered with aspirin in healthy volunteers. Platelets. 2013;24(8):615–624. | |
Husted SE, Storey RF, Bliden K, et al. Pharmacokinetics and pharmacodynamics of ticagrelor in patients with stable coronary artery disease: results from the ONSET-OFFSET and RESPOND studies. Clin Pharmacokinet. 2012;51(6):397–409. | |
Nylander S, Femia EA, Scavone M, et al. Ticagrelor inhibits human platelet aggregation via adenosine in addition to P2Y antagonism. J Thromb Haemost. 2013;11(10):1867–1876. | |
Bonello L, Pansieri M, Mancini J, et al. High on-treatment platelet reactivity after prasugrel loading dose and cardiovascular events after percutaneous coronary intervention in acute coronary syndromes. J Am Coll Cardiol. 2011;58(5):467–473. | |
Mega JL, Close SL, Wiviott SD, et al. Cytochrome P450 genetic polymorphisms and the response to prasugrel: relationship to pharmacokinetic, pharmacodynamic, and clinical outcomes. Circulation. 2009:119(19):2553–2560. | |
Franken CC, Kaiser AF, Krüger JC, Overbeck K, Mügge A, Neubauer H. Cytochrome P450 2B6 and 2C9 genotype polymorphism – a possible cause of prasugrel low responsiveness. Throm Haemost. 2013;110(1):131–140. | |
Cuisset T, Loosveld M, Morange PE, et al. CYP2C19*2 and *17 alleles have a significant impact on platelet response and bleeding risk in patients treated with prasugrel after acute coronary syndrome. JACC Cardiovasc Interv. 2012;5(12):1280–1287. | |
Xiang Q, Cui Y, Zhao X, Zhao N. Identification of PEAR1 SNPs and their influences on the variation in prasugrel pharmacodynamics. Pharmacogenomics. 2013;14(10):1179–1189. | |
Tantry US, Bliden KP, Wei C, et al. First analysis of the relation between CYP2C19 genotype and pharmacodynamics in patients treated with ticagrelor versus clopidogrel: the ONSET/OFFSET and RESPOND genotype studies. Circ Cardiovasc Genet. 2010;3(6):556–566. | |
Wallentin L, James S, Storey RF, et al; PLATO investigators. Effect of CYP2C19 and ABCB1 single nucleotide polymorphisms on outcomes of treatment with ticagrelor versus clopidogrel for acute coronary syndromes: a genetic substudy of the PLATO trial. Lancet. 2010;376(9749):1320–1328. | |
Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. New Engl J Med. 2009;361:1045–1057. | |
Varenhorst C, Eriksson N, Johansson Å, et al. Ticagrelor plasma levels but not clinical outcomes are associated with transporter and metabolism enzyme genetic polymorphisms. J Am Coll Cardiol. 2014;63(12_S). | |
International Warfarin Pharmacogenetics C, Klein TE, Altman RB, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med. 2009;360(8):753–764. | |
Budnitz DS, Lovegrove MC, Shehab N, Richards CL. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med. 2011;365(21):2002–2012. | |
Pirmohamed M, James S, Meakin S, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ. 2004;329(7456):15–19. | |
Fung E, Patsopoulos NA, Belknap SM, et al. Effect of genetic variants, especially CYP2C9 and VKORC1, on the pharmacology of warfarin. Semin Thromb Hemost. 2012;38(8):893–904. | |
Johnson JA, Cavallari LH, Beitelshees AL, Lewis JP, Shuldiner AR, Roden DM. Pharmacogenomics: application to the management of cardiovascular disease. Clin Pharmacol Ther. 2011;90(4):519–531. | |
Lee MT, Klein TE. Pharmacogenetics of warfarin: challenges and opportunities. J Hum Genet. 2013;58(6):334–338. | |
Jorgensen AL, FitzGerald RJ, Oyee J, Pirmohamed M, Williamson PR. Influence of CYP2C9 and VKORC1 on patient response to warfarin: a systematic review and meta-analysis. PloS One. 2012;7(8):e44064. | |
Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood. 2008;111(8):4106–4112. | |
Takeuchi F, McGinnis R, Bourgeois S, et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 2009;5(3):e1000433. | |
Pirmohamed M, Burnside G, Eriksson N, et al; EU-PACT Group. A randomized trial of genotype-guided dosing of warfarin. N Engl J Med. 2013;369(24):2294–2303. | |
Kimmel SE, French B, Kasner SE, et al; COAG Investigators. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N Engl J Med. 2013;369(24):2283–2293. | |
Verhoef TI, Ragia G, de Boer A, et al; EU-PACT Group. A randomized trial of genotype-guided dosing of acenocoumarol and phenprocoumon. N Engl J Med. 2013;369(24):2304–2312. | |
Kirchheiner J, Ufer M, Walter EC, et al. Effects of CYP2C9 polymorphisms on the pharmacokinetics of R- and S-phenprocoumon in healthy volunteers. Pharmacogenetics. 2004;14(1):19–26. | |
Schalekamp T, Oosterhof M, van Meegen E, et al. Effects of cytochrome P450 2C9 polymorphisms on phenprocoumon anticoagulation status. Clin Pharmacol Ther. 2004;76(5):409–417. | |
Teichert M, Eijgelsheim M, Uitterlinden AG, et al. Dependency of phenprocoumon dosage on polymorphisms in the VKORC1, CYP2C9, and CYP4F2 genes. Pharmacogenet Genomics. 2011;21(1):26–34. | |
Cavallari LH, Perera MA. The future of warfarin pharmacogenetics in under-represented minority groups. Future Cardiol. 2012;8(4):563–576. | |
Perera MA, Cavallari LH, Limdi NA, et al. Genetic variants associated with warfarin dose in African-American individuals: a genome-wide association study. Lancet. 2013;382(9894):790–796. | |
Paré G, Eriksson N, Lehr T, et al. Genetic determinants of dabigatran plasma levels and their relation to bleeding. Circulation. 2013;127(13):1404–1412. | |
Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139–1151. | |
Pirmohamed M, Burnside G, Eriksson N, et al; EU-PACT Group. A randomized trial of genotype-guided dosing of warfarin. New Engl J Med. 2013;369:2294–2303. | |
Kimmel SE, French B, Kasner SE, et al; COAG Investigators. A pharmacogenetic versus a clinical algorithm for warfarin dosing. New Engl J Med. 2013;369(24):2283–2293. | |
Roberts JD, Wells GA, Le May MR, et al. Point-of-care genetic testing for personalisation of antiplatelet treatment (RAPID GENE):a prospective, randomised, proof-of-concept trial. Lancet. 2012;379(9827):1705–1711. | |
Xie X, Ma YT, Yang YN, et al. Personalized antiplatelet therapy according to CYP2C19 genotype after percutaneous coronary intervention: a randomized control trial. Int J Cardiol. 2013;168(4):3736–3740. | |
Johnson JA, Gong L, Whirl-Carrillo M, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther. 2011; 90(4):625–629. | |
Kazi DS, Garber AM, Shah RU, et al. Cost-effectiveness of genotype-guided and dual antiplatelet therapies in acute coronary syndrome. Ann Intern Med. 2014;160(4):221–232. | |
Lala A, Berger JS, Sharma G, Hochman JS, Scott Braithwaite R, Ladapo JA. Genetic testing in patients with acute coronary syndrome undergoing percutaneous coronary intervention: a cost-effectiveness analysis. J Thromb Haemost. 2013;11(1):81–91. | |
Reese ES, Daniel Mullins C, Beitelshees AL, Onukwugha E. Cost-effectiveness of cytochrome P450 2C19 genotype screening for selection of antiplatelet therapy with clopidogrel or prasugrel. Pharmacotherapy. 2012;32(4):323–332. | |
Sorich MJ, Horowitz JD, Sorich W, Wiese MD, Pekarsky B, Karnon JD. Cost-effectiveness of using CYP2C19 genotype to guide selection of clopidogrel or ticagrelor in Australia. Pharmacogenomics. 2013;14(16):2013–2021. | |
Pink J, Pirmohamed M, Lane S, Hughes DA. Cost-effectiveness of pharmacogenetics-guided warfarin therapy vs alternative anticoagulation in atrial fibrillation. Clin Pharmacol Ther. 2014;95(2):199–207. | |
Verhoef TI, Redekop WK, Veenstra DL, et al; EU-PACT Group. Cost-effectiveness of pharmacogenetic-guided dosing of phenprocoumon in atrial fibrillation. Pharmacogenomics. 2013;14(8):869–883. | |
You JH. Pharmacogenetic-guided selection of warfarin versus novel oral anticoagulants for stroke prevention in patients with atrial fibrillation: a cost-effectiveness analysis. Pharmacogenet Genomics. 2014;24(1):6–14. | |
You JH, Tsui KK, Wong RS, Cheng G. Cost-effectiveness of dabigatran versus genotype-guided management of warfarin therapy for stroke prevention in patients with atrial fibrillation. PloS One. 2012;7(6):e39640. | |
Leey JA, McCabe S, Koch JA, Miles TP. Cost-effectiveness of genotype-guided warfarin therapy for anticoagulation in elderly patients with atrial fibrillation. Am J Geriatr Pharmacother. 2009;7(4):197–203. | |
Patrick AR, Avorn J, Choudhry NK. Cost-effectiveness of genotype-guided warfarin dosing for patients with atrial fibrillation. Circ Cardiovasc Qual Outcomes. 2009;2(5):429–436. | |
Eckman MH, Rosand J, Greenberg SM, Gage BF. Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with nonvalvular atrial fibrillation. Ann Intern Med. 2009;150(2):73–83. | |
You JH, Tsui KK, Wong RS, Cheng G. Potential clinical and economic outcomes of CYP2C9 and VKORC1 genotype-guided dosing in patients starting warfarin therapy. Clin Pharmacol Ther. 2009;86(5):540–547. | |
Sangkuhl K, Klein TE, Altman RB. Clopidogrel pathway. Pharmacogenetics and genomics [webpage on the Internet]. PharmGKB; 2010. Available from: http://www.pharmgkb.org/pathway/PA154424674. Accessed May 7, 2014. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.