Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Personalization of biologic therapy in patients with rheumatoid arthritis: less frequently accounted choice-driving variables
Authors Niccoli L, Nannini C, Blandizzi C, Mantarro S, Mosca M, Di Munno O ![]() , Goletti D
, Goletti D ![]() , Benucci M
, Benucci M ![]() , Li Gobbi F
, Li Gobbi F ![]() , Cassarà E, Kaloudi O, Cantini F
, Cassarà E, Kaloudi O, Cantini F ![]()
Received 30 May 2018
Accepted for publication 9 September 2018
Published 24 October 2018 Volume 2018:14 Pages 2097—2111
DOI https://doi.org/10.2147/TCRM.S175772
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Laura Niccoli,1 Carlotta Nannini,1 Corrado Blandizzi,2 Stefania Mantarro,2 Marta Mosca,3 Ombretta Di Munno,3 Delia Goletti,4 Maurizio Benucci,5 Francesca Li Gobbi,5 Emanuele Cassarà,1 Olga Kaloudi,1 Fabrizio Cantini1
1Department of Rheumatology, Hospital of Prato, Prato, Italy; 2Section of Pharmacology and Pharmacovigilance, Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy; 3Division of Rheumatology, Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy; 4Translational Research Unit, Department of Epidemiology and Preclinical Research, National Institute for Infectious Diseases, Rome, Italy; 5Rheumatology Unit, Hospital S. Giovanni di Dio, Florence, Italy
Objective: To propose appropriate statements that drive the choice of biologic therapies in patients with rheumatoid arthritis (RA), factoring in their impact on the following issues: anti-drug antibody (ADAb) formation, suspicion and management of infections, lupus-like syndrome (LLS), effects on bone mass and sexual sphere, and relationship between RA and periodontal disease (PD).
Methods: An overview of existing evidence was undertaken by an expert panel on behalf of the Italian board for the TAilored BIOlogic therapy (ITABIO). Data were extracted from controlled trials, national registries, national health care databases, post-marketing surveys, and, when required by the paucity of controlled studies, from open-label clinical series. Anti-tumor necrosis factor (anti-TNF) and non-anti-TNF-targeted biologics approved for RA were investigated.
Results: ADAb formation is chiefly associated with anti-TNFs, and it is reduced by combination therapy with methotrexate. To date, ADAb titration is not advisable for clinical practice, and, in case of anti-TNF secondary failure, a non-anti-TNF biologic is indicated. LLS is observed in anti-TNF receivers and, in most cases, resolves without anti-TNF withdrawal. A non-anti-TNF biologic is advisable in patients experiencing LLS. Non-anti-TNFs demonstrated a low or absent infection risk and are preferable in patients with comorbidities. Due to their positive effects on bone mass, anti-TNFs are indicated in women at osteoporosis risk, whereas non-anti-TNF have been poorly investigated. The emerging evidence of the relationship between RA and PD and the effects on anti-TNF efficacy should lead clinicians to consider the periodontal status in RA patients. Anti-TNFs may exert a positive effect on fertility and sexuality, and clinicians should explore these aspects in RA patients.
Conclusion: The optimization of biologic therapies by taking into proper account the above issues would improve patient outcomes.
Keywords: biologics, immunogenicity, infections, lupus-like syndrome, osteoporosis, periodontal disease, sexuality
Introduction
The results of randomized clinical trials (RCTs) and real-life reports, with special reference to national registries, have clarified many of the aspects of biologic disease-modifying anti-rheumatic drugs (bDMARD) in patients with inflammatory rheumatic disorders, including the mechanism of action, efficacy and safety, availability of response predictors, and possibility of optimizing their use in single patients.1 However, some biologic-related issues that may influence therapeutic choice are often disregarded, or still need to be fully addressed, by clinicians; these include the immunogenicity, suspicion and management of infections, anti-tumor necrosis factor (TNF)-related lupus-like syndrome (LLS), skeletal effects of biologics, relationship between periodontal disease (PD) and rheumatoid arthritis (RA), and effects of biologics on both fertility and sexuality. Indeed, rheumatologists are very careful in evaluating the disease activity and clinical course of RA, but an overshadowing of several features for a complete patient management frequently occurs. By contrast, the abovementioned topics may play a pivotal role for the best outcome, not only in terms of articular response but also for the improvement of the overall quality of care and the prevention of complications.
With these considerations in mind, the present article was intended to formulate practical indications on the impact of immunogenicity, infections, and LLS; effects of biologics on the skeleton; role of coexisting PD in patients with RA; and influence of RA and related therapies on fertility and sexuality.
Methods
As previously reported,1 a multidisciplinary expert panel on behalf of the Italian board for the TAilored BIOlogic therapy (ITABIO) reviewed the literature on the existing evidence on the immunogenicity of biologics, suspicion and management of infections, LLS, biologic skeletal effects, association of RA with PD and its impact on both disorders and on biologic therapy, impairment of fertility and sexual sphere, and effects of biologics. Taking in account the emerging evidence on these topics, appropriate statements useful to therapy tailoring were formulated.
Literature search
The literature review was conducted by using the PubMed database to identify English-language articles related to the abovementioned topics. Data were extracted from RCTs, national registries of biologics, national health care databases, and post-marketing surveys. When these source data were not available for specific topics, the evidence was retrieved from open-label studies on variable sample-size clinical series.
The following drugs were investigated: infliximab (IFX), etanercept (ETN), adalimumab (ADA), golimumab (GOL), certolizumab pegol (CZP), tocilizumab (TCZ), abatacept (ABA), and rituximab (RTX). The research was conducted by crossing each drug name with the following key terms: RA, early RA (ERA), efficacy, safety, immunogenicity, anti-drug antibody, infection, opportunistic infection, tuberculosis, osteoporosis, bone mass, skeletal manifestations, periodontal disease, periodontitis, fertility, and sexuality. The literature review was extended to literature published up to April 30, 2018.
Mechanism of immunogenicity and fallout on clinical practice
It has long been recognized that biologics used for treatment of RA can give rise to the development of antibodies targeting the respective biodrug, designated as “antidrug antibodies” (ADAbs).2–4 ADAb induction is commonly a polyclonal response, resulting in the formation of antibodies that may differ widely in terms of serum titer, antigenic affinity, and biologic actions. Accordingly, immunogenic responses against biologics give rise to pools of antibodies that comprise different subtypes (commonly, immunoglobulin M [IgM] and IgG, but, in a minority of cases, IgE as well) and isotypes (eg, IgG1, IgG2, IgG3), targeting different epitopes of the same biodrug and displaying different binding patterns.2 Based on the targeted epitopes and their intrinsic biological properties, ADAbs are currently classified as non-neutralizing and neutralizing antibodies. The former, usually comprising IgM, IgG, and IgE, bind epitopes located in molecular domains of the biodrug that are not critical for the interaction with its molecular target, which usually accelerates the biodrug clearance (clearing antibodies), enhances the bioavailability (sustaining antibodies), or elicits hypersensitivity adverse reactions.5,6
By contrast, neutralizing antibodies (usually IgG) bind epitopes that are critical for the interaction of the biodrug with its molecular target, thereby impairing its therapeutic efficacy, or eliciting adverse reactions through suppression of the biological actions of an endogenous mediator. Furthermore, similarly as non-neutralizing ADAbs, neutralizing ADAbs may affect the biodrug pharmacokinetic profile, mainly preventing its absorption from the injection site, and/or can elicit hypersensitivity reactions. In addition, as compared to neutralizing ADAbs, non-neutralizing ADAbs are usually characterized by higher titer, early induction, and prolonged persistence throughout the treatment period.5,6
The immune-pathogenesis of ADAbs relies basically on two main mechanisms. Most commonly, the biodrug is processed by antigen-presenting cells with subsequent presentation of its antigenic moieties to competent T lymphocytes, which undergo activation and in turn activate B lymphocytes, followed by clonal expansion and differentiation into plasma cells, which initiate ADAb production. The second mechanism, which is T-cell-independent, occurs usually when a number of biodrug molecules cluster to form macromolecular aggregates that behave as polyvalent antigenic structures. Such aggregates have the potential of promoting B-cell-receptor clustering on the membrane surface of B lymphocytes, thus stimulating their direct activation, which is followed by clonal expansion, differentiation into plasma cells, and initiation of ADAb production.7 In some instances, the immunogenic response triggered by the biodrug amplifies (epitope spreading) with a first phase of ADAb production through the abovementioned clustering mechanism and a second phase where ADAb–biodrug complexes are taken up by naïve B lymphocytes which then behave as antigen-presenting cells to process the biodrug and present its antigenic moieties to competent T lymphocytes; this results in the production of more potent ADAbs targeted against different epitopes than those that were involved in the first phase.5,8
In patients developing ADAbs by the mechanism of direct B-cell activation, the possible presence of biologic impurities – in combination with the disease-related inflammatory background – can behave as danger signals, leading to the stimulation of toll-like receptors (TLRs); this is followed by a further amplification of activated B lymphocytes and results in a switch from low-affinity IgM to high-affinity IgG ADAbs, with a consequent increased risk of detrimental clinical consequences.5,9
ADAb titration
Several attempts have been made to set up assay methods suitable for testing both the presence and titer of ADAbs as well as to follow-up their variations over the time in the sera of patients. Ideally, to fulfill the purpose of generating clinically useful results, laboratory methods for the assessment of ADAbs should be suitable for selectively detecting antibodies of any isotype that may develop against the different epitopes of the biodrug, and they should be sensitive enough for detecting both low- and high-affinity ADAbs.4 Unfortunately, ADAb titration assays have yielded highly variable results to such an extent that the actual significance of ADAb testing in clinical practice remains a matter of debate. This extreme variability depends largely on severe methodological limitations affecting the available assays, as extensively discussed in previous reviews.3,4,10 Thus, given the unique technical features and peculiar drawbacks of each assay method, comparative analyses of immunogenicity results obtained by means of different methods are inappropriate, likely to lead to biased conclusions, and should therefore be avoided.4 This statement is reinforced by the wide percentages of ADAb positivity associated with each biodrug, ranging from 0% to 83% for IFX, 0% to 54% for ADA, 3% to 37% for CZP, 0% to 19% for GLM, and 0% to 13% for ETN.11
At least five additional major factors can account for the heterogeneity of results on ADAb assessments, and these include: 1) sampling procedures (sampling time and sample handling); 2) patients’ characteristics in terms of intrinsic propensity to develop immunogenic responses; RA disease activity and/or duration; and presence of comorbidities; 3) intrinsic biodrug molecular properties as well as product composition and formulation, including different immunogenic molecular domains, presence of murine moieties, different glycosylation patterns, and presence of impurities or aggregates in the formulation; 4) differences in administration routes and treatment regimens, with greater immunogenicity promotion by subcutaneous administration and intermittent treatment; and 5) combination therapy with methotrexate (MTX), although not completely explained, reduces the immunogenicity of anti-TNFs.2,12–14 However, in the real-life world, approximately 30% of RA patients receive anti-TNF monotherapy,1 and, at least in part, the lower retention rate of these biologics, as observed with such a therapeutic regimen, is related to ADAbs.
Immunogenicity of individual biologics
Focusing on the immunogenicity of anti-TNFs, ADAbs can target their Fab or Fc fragment. With specific regard for IFX, ADA, GOL, and CZP, the majority of ADAbs target their Fab fragment and are endowed with neutralizing activity. Such a pattern of immunogenic response likely results from anti-idiotype reactions, and this may explain why the development of humanized therapeutic antibodies has decreased – although not suppressed – the risk of ADAb induction. ADAbs targeting the Fc fragment (particularly, the hinge region) do not exert neutralizing activity, but can significantly decrease their circulating levels by enhancing the biodrug pharmacokinetic clearance. On the other hand, receptor fusion proteins, such as ETN or ABA, can promote the induction of ADAbs directed against the receptor moiety (mainly exerting neutralizing activity) or the antibody Fc fragment (mainly, the hinge region). However, current evidence on ETN suggests only the development of non-neutralizing ADAbs.6,13,14
According to three systematic reviews,14–16 the majority of clinical studies agree on at least three points of evidence: serum ADAb positivity appears to be a risk factor for a loss of therapeutic efficacy; the development of ADAbs increases the risk of immune adverse reactions, which include infusion reactions, injection-site reactions, and, more occasionally, serum sickness and Arthus reactions; and concomitant treatment with MTX or azathioprine appears to decrease the risk of ADAb formation against anti-TNFs. Several authors have indeed reported that ADAbs can be responsible for decrease in biodrug concentration, leading to secondary treatment failure,14–16 and similar data have been yielded by real-world clinical practice non-interventional studies.17
With regard to non-anti-TNF-targeted biologics, the risk of ADAb development seems consistently reduced. In a recent review, ADAb positivity was recorded in 1.5% of RA patients receiving intravenous or subcutaneous TCZ, with no differences between TCZ monotherapy or combination therapy with conventional synthetic DMARDs.18 Similar results have been achieved in studies on RA patients that were designed specifically to correlate serum ADAb titers and TCZ levels with disease activity.19,20 Likewise, low ADAb positivity percentages have been reported with ABA (2%–20%) and RTX (0%–21%).11
Infection suspicion and diagnosis in patients taking biologics
RA itself is associated with an increased in the risk of infections,21 and the risk is increased in patients with severe disease requiring biologics, especially anti-TNFs.22
Anti-TNFs increase the risk of infection by different mechanisms: 1) impairment of the maintenance of granulomas,23,24 leading to increased risk of new tuberculosis infection, reactivation of latent tuberculosis,25 and other granulomatous infections, such as Histoplasma capsulatum26 and 2) impairment of phagosome formation and clearance of intracellular pathogens;24 biologic-induced neutropenia predisposing to infections such as Candida or Aspergillus,27 or interference with immune response against viral infections and consequent enhanced complications in patients infected with hepatitis B virus (HBV)28 or varicella zoster virus (VZV).29,30
However, more frequently, the serious infections associated with biologics are bloodstream infections, septic arthritis, osteomyelitis, pneumonia, urinary tract infections with urosepsis and pyelonephritis, soft-tissue cellulitis, gastroenteritis, and intra-abdominal infections. The risk of serious infections is highest in the first 6 months of biologic treatment, plateauing off by 24–36 months.31 The results of most analyses show that, among anti-TNFs, IFX is associated with a higher risk of serious infections than ETN. Concerning the non-anti-TNF-targeted biologics, the results of the head-to-head RCT of ADA and ABA (AMPLE study) showed a comparable occurrence of infections in the two treatment arms.32 However, it should be noted that patients treated in real-world practice differ from those, highly selected subjects enrolled in RCTs,33,34 and data from registries and observational studies demonstrated a low or absent infection risk in patients receiving non-anti-TNF-targeted biologics, including TCZ, ABA, and RTX.35
Clinical presentation and diagnostic approach to severe infections
Table 1 reports the clinical presentation and the major diagnostic approaches useful to diagnose severe infections in patients treated with biologics. Herein, we describe mainly the bloodstream infection, which is a life-threatening condition that arises when the body’s response to infection injures its own tissues.1,36–41 There may be symptoms related to a specific infection, such as a cough with pneumonia or painful urination with a kidney infection. In people with a weakened immune system and in the very young or old people, there may be no symptoms of a specific infection and the body temperature may be low or normal. Severe sepsis causes poor organ function or insufficient blood flow, leading to low blood pressure, high blood lactate, or low urine output and, finally, septic shock.
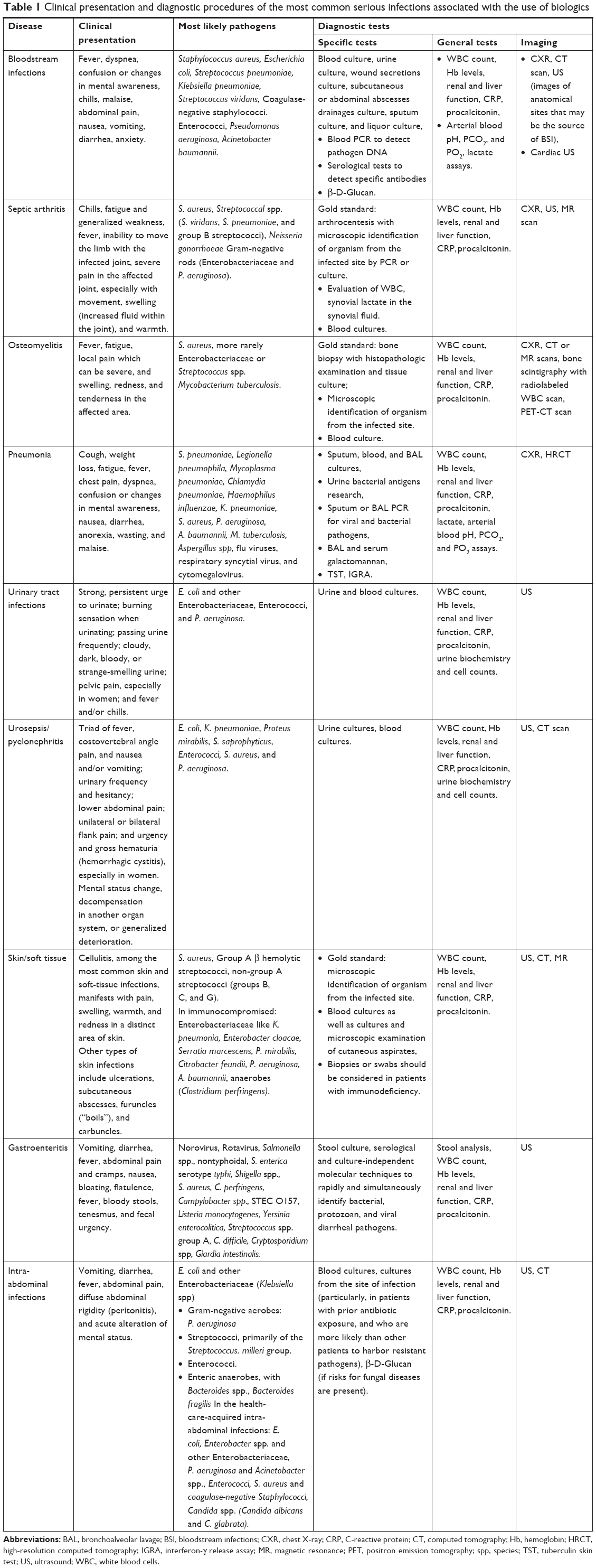 | Table 1 Clinical presentation and diagnostic procedures of the most common serious infections associated with the use of biologics |
Furthermore, a small but significant risk of serious opportunistic infection is associated with anti-TNF-α therapies.30 Opportunistic infection is defined as a usually serious and progressive infection by an organism that, under normal circumstances, possesses little or no pathologic capabilities. However, predisposing factors such as underlying disease or medical treatment can reduce the patient’s immune response, permitting the organism to cause an infectious disease.21 Beyond the role of biologics, in patients with RA, several adjunctive risks may lead to decreased innate immune function and increased risk of opportunistic infections, such as aging, diabetes, alcoholism, concomitant immunosuppressive therapies, malnutrition, and micronutrient deficiency.21,30,39,41
Among those considered as opportunistic infections, we should consider bacterial, fungal, viral, and parasitic infections.30 When possible, patients should be adequately vaccinated and closely monitored for early signs of infection.42
Anti-TNF and LLS
Since the introduction of anti-TNF therapy, there have been reports on autoantibody production and of lupus-like manifestations associated with these therapies.43–51
The appearance of antinuclear antibodies (ANA) during anti-TNF treatment has been described in the initial IFX trials and also confirmed after exposure to other anti-TNF drugs such as ETN and ADA.43–45 The development of ANA or increase of ANA titer in previously positive patients when on anti-TNFs has been reported in percentages ranging from 20% up to 70%. In addition, the appearance of anti-double-strand DNA (anti-dsDNA) antibodies and anticardiolipin antibodies (usually IgM) has been reported.
Drug-induced lupus erythematosus (DILE) has long been recognized as a condition associated with a number of different medications,45 but anti-TNF-induced-LLS has specific clinical and serological characteristics and differs from the classical DILE in many aspects.46–49
The true incidence of LLS is difficult to be established but it appears low, being reported in 0.18%–0.41% of patients treated with different anti-TNFs.46–49 The mean latency until the first manifestation is in the range from 2 to 40 weeks and the female:male ratio is 5:1. LLS has been reported in anti-TNF-treated patients with RA, psoriatic arthritis, and inflammatory bowel disease.46
The clinical picture of LLS is characterized by fever and constitutional symptoms (70%), high frequency of cutaneous manifestations (malar rash, discoid rash in up to 70%), arthritis (50%), myalgias (50%), and serositis (18%), whereas the renal system and CNS are affected more rarely (<10% of patients).48,49 Serologically, LLS is characterized by the positivity of anti-dsDNA antibodies and lower positivity of anti-histone antibodies (50%) and hypo-complementemia (60%).47–51
In the majority of cases, LLS resolves with anti-TNF withdrawal; however, up to 40% of patients require corticosteroids and a smaller percentage (approximately 10%) require immunosuppressive therapy.47
Potential mechanisms for LLS development
TNF plays a double role in systemic lupus erythematosus (SLE). Some studies, in fact, have shown an increased expression of TNF in kidney biopsies and in skin of patients with SLE, thus suggesting a possible pathogenic role of this cytokine.
Indeed, there are data in the literature showing a positive effect of short-duration anti-TNF therapy in the treatment of class V glomerulonephritis and joint involvement in SLE. No major side effects have been reported and there were no disease flares, although a transient increase of anti-dsDNA antibodies titers and anticardiolipin antibodies has been described in these patients during therapy.52,53 However, in animal models, a reduction of TNF predisposes to autoimmunity and antibody development. This might be due to the suppression of Th1 cytokines and a subsequent increase of Th2 cytokines, leading to increased autoantibodies production. Alternatively, the reduction of TNF may interfere with apoptosis thereby leading to a reduced clearance of nuclear antigens, consequently leading again to increased antibody production.54
Is ANA assessment to be recommended prior to anti-TNF therapy initiation?
No data in the literature thus far suggest that ANA positivity prior to therapy initiation has a prognostic role for LLS development, and there are no data supporting the need for antibody testing at baseline.54,55 However, it has been hypothesized that patients developing LLS may have a latent or subclinical form of SLE and, therefore, a careful history aimed at assessing the clinical manifestations is suggested.
Biologic agents and skeletal effects in RA
Systemic osteoporosis (OP) and increased fracture rates are well-known comorbidities in RA.56,57 Improved knowledge of the immune/inflammatory pathways which characterize the pathophysiology of RA provided the link between inflammation and bone loss via a complex network of bone cells; T and B cells; proinflammatory cytokines such as TNF-α, IL-1, IL-6, IL-17, and IL 23; co-stimulator molecules; signaling pathways, including both the RANK ligands – RANK and osteoprogeterin (RANKL/RANK/OPG); and Wnt inhibitor dickkopf-1 Wnt (DKK-1) signaling.58–60
As a consequence, it has been postulated that the novel targeted biologic therapies, which are characterized by powerful anti-inflammatory activity, would reduce both systemic bone loss and fracture incidence.58,61,62
Several cross-sectional and prospective studies evaluated the effect of biologics on bone mineral density (BMD) and/or bone turnover markers (BTMs) in RA with conflicting results. Most of these studies have been conducted with anti-TNFs, whereas there are very few studies with non-anti-TNF-targeted TCZ, RTX, and ABA.
Anti-TNFs and bone mass
TNF-α plays a central role in the physiopathology of bone loss by decreasing osteoblast activity and survival and increasing osteoclast differentiation, recruitment, and activity.58–60 Consequently, these findings suggest a beneficial effect of anti-TNFs also in preventing systemic bone loss.
Krieckaert et al63 documented that ADA treatment over 4 years arrested bone loss in the lumbar spine in 184 patients with severe RA, whereas hip BMD continued to decrease. A study evaluating the effect of synthetic DMARDs and biologics on systemic bone loss in RA patients who were followed for 10 years showed that a more aggressive strategy, including anti-TNFs, was effective in reducing the rate of bone loss.64 By contrast, a review by Kawai et al65 concluded that anti-TNFs were not effective in preventing bone loss in RA. Interestingly, data deriving from a recent meta-analysis on the effects of anti-TNFs on BMD in RA and AS trials66 showed a significant improvement of both lumbar spine and hip BMD only in AS patients. Recent data deriving from clinical studies between 2005 and 2014 added evidence that TNF-α inhibitors are able to arrest systemic bone loss as assessed by BMD and BTMs in RA.67 The beneficial effects of anti-TNFs against bone loss have been confirmed in other reviews.62,68
IL-6 blockade
The IL-6 pathway is one of the major contributing factors to the disruption of bone metabolism in immuno-mediated inflammatory diseases.59–61 IL-6 is a pleiotropic cytokine strongly connected to osteoclast physiology, as it interacts synergistically with IL-1 and TNF-α to promote the differentiation of osteoclast precursors (OCPs), to prolong the survival and increase the activity of mature osteoclasts. Moreover, this cytokine plays a critical role in the generation of proinflammatory TH17 cells, thereby enhancing IL-17 levels.60
The effect of TCZ on BTMs has been investigated in some prospective studies of RA patients. A rapid and significant increase of bone-formation markers (BFMs) associated with a significant decrease of bone-resorption markers (BRMs) followed the initiation of TCZ combination therapy with MTX in 416 patients with active RA.69 In addition, in the RADIATE study, TCZ–MTX treatment significantly decreased BRMs and increased BFMs, although not significantly, in RA patients.70 A critical role of TCZ on bone metabolism was also suggested by a pilot study which, in a smaller sample of RA patients (22 women), documented that the IL-6 blockade increased the OPG/RANKL ratio and decreased DKK-1 levels after 2 months of treatment.71 More recently, the effect of TCZ has been evaluated on BMD in patients with MTX-resistant active RA.72 Interestingly, TCZ stably maintained the BMD in patients with normal baseline BMD and increased BMD in osteopenic patients at baseline. In contrast, a 1-year open prospective study in patients with active RA receiving TCZ and MTX did not show significant changes in BMD.73 However, a significant decrease of serum levels of DKK-1 and a significant increase of BFMs were associated with TCZ–MTX treatment.
Inhibition of B and T cells
Together with activated T cells, B cells highly express RANKL and IL-6, thereby contributing to osteoclastogenesis.59 Moreover, B cells differentiate into plasma cells, which in turn inhibit bone formation through the expression of the DKK-1.59
Clinical studies evaluating the effect of RTX on systemic bone loss are not available. Nevertheless, as a consequence of the critical role played by B cells on systemic bone homeostasis, it can be speculated that RTX can positively influence bone mass.54 Boumans et al74 showed that 12-month treatment with RTX was associated with an increased OPG/RANKL ratio in the serum of 28 RA patients. In contrast, in a prospective study with a follow-up of 15 months that evaluated 13 RA patients, a non-significant decrease of RANKL was observed after RTX treatment.75 Wheater et al76 documented a significant decrease of BRMs associated with a significant increase of BFMs after 6 months of treatment with RTX in 46 RA patients.
So far, there are no clinical studies with BMD and/or BTs as primary outcomes in RA patients treated with ABA. CTLA-4 is a T-cell-associated antigen which competes with CD28 to bind to CD80/CD86, which is a potent costimulatory signal for T cells and OCPs.58,61 In a model of TNF-α-induced arthritis, CTLA-4 dose-dependently and directly inhibited osteoclast differentiation and maturation by binding to their precursor cells.77 Recently, Bozec et al78 reported that the binding of CTLA-4 to CD80/86 in OCPs induced their apoptosis. Thus, these data may suggest that ABA could exert a beneficial effect on systemic bone loss.60,67
Biologics and fractures
To date, the effect of biologics on fracture risk remains uncertain. Data from the CORRONA registry, which evaluated 11,429 RA patients treated with synthetic DMARDs or TNF-α inhibitors as monotherapy or synthetic DMARDs in combination with TNF-α inhibitors, showed a decrease in overall fracture risk associated with anti-TNF treatment when compared with other treatments.79 In contrast, Kim et al80 found a similar risk of non-vertebral fractures in RA patients treated with anti-TNFs as monotherapy or in combination with a synthetic DMARD, MTX alone, or another synthetic DMARD alone. Similar findings resulted from a nested case–control study of 1,515 RA patients treated with biologics, mostly anti-TNFs, and 6,023 controls.81 Furthermore, the risk of combined non-vertebral fractures did not differ between initiators of a synthetic agent and an anti-TNF in a large retrospective cohort of patients with RA and spondyloarthritis.82 Recently, three reviews which evaluated the anti-fracture activity of biologics, chiefly anti-TNFs, failed to demonstrate a clinically meaningful effect.65,67,68
Biologics and PD
The relationship between autoimmune rheumatic disorders and PD has been chiefly investigated in patients with RA with a focus on the epidemiology, pathogenic interplay, effect of coexisting PD on the clinical course of RA and response to biologic therapies, and the effects of biologics on PD.
Confirming previous reports,83–89 in a case–control study of 287 patients with established RA, there was a higher frequency of PD as compared with 330 matched controls (35%; unadjusted OR 1.49; 95% CI 1.06, 2.11; P=0.02). The difference was more pronounced in anti-citrullinated protein antibodies (ACPA)-positive cases (37%; unadjusted OR 1.65; 95% CI 1.15, 2.36; P=0.006). The association persisted significantly and more frequently after multivariate analysis adjustment in ACPA-positive patients (OR 1.59; 95% CI 1.01, 2.49; P=0.043).90 Two large population-based studies confirmed these findings,91,92 whereas another large-scale epidemiologic research study did not.93
These conflicting results were observed in patients with longstanding RA, where two studies of new-onset RA confirmed the association with PD.88,94 The relationship between RA and PD seems to be strengthened by the circumstance that the two inflammatory conditions share increased TNF production.95
Although controversial, these epidemiologic results suggest common pathogenic pathways between RA and PD; moreover, the role of the oral microbiome as a triggering factor for RA has been especially emphasized.96,97 In particular, a component of the oral microbiome – Porphyromonas gingivalis – seems to play an important pathogenic role in PD, ACPA production, and RA development.98,99
Mutual effects of biologic therapy and PD
The possible connection between the two inflammatory conditions seems to be confirmed by the negative influx on biologic therapy effectiveness exerted by coexistent PD in patients with RA. In a limited series of 18 RA patients with PD, a significant lower efficacy of anti-TNF therapy was observed in RA patients with PD;100 furthermore, a significant association between the coexistence of PD and the risk of ETN discontinuation resulted in a large population-based cohort of 3,359 patients (HR 1.27; 95% CI 1.01, 1.60).101 Conversely, several studies reported the beneficial effect of non-surgical PD treatment on RA severity.102–105
The effects of biologics on PD have been chiefly investigated in patients with RA receiving anti-TNFs. Overall, the majority of the studies evidenced a reduction of gingival inflammation with improvement of parameters of PD severity.106–110 Furthermore, similar results were observed in patients with ankylosing spondylitis.111 However, the strength of this evidence is limited by the open-label design of the studies and by the limited number of patients evaluated.
Fertility and sexuality in RA patients treated with biologics
Dealing with these topics implies a consideration of several aspects, including the role exerted by RA itself, the effects of biologic-combined therapies, and the direct effects of biologics. Although often less regarded, the fallout on patients’ psychological status may be detrimental, especially if the RA-induced depressive condition in at least one third of the patients is taken in account.112
With regard to fertility in RA as reported in other reports,113 a recent study observed unexplained subfertility and anovulation in 48% and 28%, respectively, of 178 women.114 Moreover, RA itself impairs the sexual function in up to 70% of women,115 with a direct correlation with the disease activity.116 Both fertility status and sexual function in males have been poorly investigated, with inconclusive results. However, reduced levels of testosterone have been detected in a low number of cohorts of males with RA.117,118 Even data on sexual function in males with RA are extremely limited, with erectile dysfunction being reported in ten (33%) out of 31 males in only one study.117 Among the traditional immunosuppressants employed for the treatment of RA, sulfasalazine has been proven to impair fertility,118 whereas data on MTX are unavailable.119,120 No impact on fertility has been reported for cyclosporine and azathioprine.119
Anti-TNFs have been extensively investigated for use in pregnancy; however, to the best of our knowledge, studies to assess their effects on fertility and sexual function in patients with RA are lacking. Data from patients with ankylosing spondylitis indicate that anti-TNFs do not impair sperm quality and fertility121 and may improve sexual dysfunction in males.122 To date, no data on fertility and sexual dysfunction are available for non-anti-TNF-targeted biologics.
Discussion
Available recommendations for the management of patients with RA provide important indications for the appropriate use of biologics, their efficacy and safety, and correct patient follow-up.123,124 Similarly, systematic reviews and meta-analyses have been chiefly focused on the same topics.125 However, the fallout on real-life clinical practice of the issues discussed in the present paper and the consequent management of patients have been rarely evaluated. In most cases, rheumatologists carefully address and measure the clinical response, but often neglect other patient characteristics that, in turn, may reduce or complicate the effects of therapeutic interventions.
As shown in Table 2, the ITABIO task force has formulated several evidence-based statements useful for improving the outcome of patients with RA.
 | Table 2 Frequently disregarded issues in the management of patients with rheumatoid arthritis: evidence-based ITABIO statements useful for the rheumatology clinical practice |
Focusing on immunogenicity, a consistent number of studies have evaluated the immune mechanisms and the effects on efficacy of anti-TNFs; however, in most cases, no suggestions for clinical practice were formulated.122 In this regard, when anti-TNFs are indicated, Statement 1 suggests combination therapy with MTX; otherwise, non-anti-TNFs are preferable. In the case of anti-TNF secondary failure, switching to a non-anti-TNF is advisable.127,128
Statement 2 provides indications for the suspect and diagnosis of infections in patients receiving biologics, with the suggestion to treat patients with high infection risk with a non-anti-TNF biologic.
As indicated in statement 3, although LLS is infrequent, rheumatologists should be aware of the possible occurrence of this syndrome in a patient taking anti-TNFs who develops fever and skin and articular complaints associated with a positive ANA titer. After anti-TNF interruption, at LLS recovery, a non-anti-TNF-targeted biologic is advisable.
Statement 4 reflects the existing evidence, and the key message for clinicians is to drive the biologic choice toward an anti-TNF in the case of a female patient with adjunctive risk factors for osteoporosis.
As pointed out in Statement 5, the connection between PD and RA requires further investigation; nevertheless, rheumatologists should consider the periodontal status of the patient and motivate him/her to undergo appropriate dental care.
Finally, Statement 6 explores a delicate issue that, often, is omitted by the patient due to reserve or shame in discussing it. However, to provide complete assistance, rheumatologists should ask the patient about his/her limitations and counsel psychiatric support.
Conclusion
In a correct and complete evaluation of the patient with RA, the discussed issues are often disregarded in clinical practice; however, an “in toto” evaluation of all patient features may drive the choice of biologic therapy and improve the outcome. We believe that information summarized in this article would be useful to clinicians for a more comprehensive patient management.
Disclosure
The authors report no conflicts of interest in this work.
References
Cantini F, Niccoli L, Nannini C. Italian board for the TAilored BIOlogic therapy (ITABIO). Tailored first-line biologic therapy in patients with rheumatoid arthritis, spondyloarthritis, and psoriatic arthritis. Semin Arthritis Rheum. 2016;45:519–532. | ||
Mok CC, Tsai WC, Chen DY, Wei JCC. Immunogenicity of anti-TNF biologic agents in the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2016;16(2):201–211. | ||
Meroni PL, Valentini G, Ayala F, Cattaneo A, Valesini G. New strategies to address the pharmacodynamics and pharmacokinetics of tumor necrosis factor (TNF) inhibitors: A systematic analysis. Autoimmun Rev. 2015;14(9):812–829. | ||
Gunn GR, Sealey DCF, Jamali F, Meibohm B, Ghosh S, Shankar G. From the bench to clinical practice: understanding the challenges and uncertainties in immunogenicity testing for biopharmaceuticals. Clin Exp Immunol. 2016;184(2):137–146. | ||
Sethu S, Govindappa K, Alhaidari M, Pirmohamed M, Park K, Sathish J. Immunogenicity to biologics: mechanisms, prediction and reduction. Arch Immunol Ther Exp. 2012;60(5):331–344. | ||
Jullien D, Prinz JC, Nestle FO. Immunogenicity of biotherapy used in psoriasis: the science behind the scenes. J Invest Dermatol. 2015;135(1):31–38. | ||
Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of therapeutic proteins: Influence of aggregation. J Immunotoxicol. 2014;11(2):99–109. | ||
Goins CL, Chappell CP, Shashidharamurthy R, Selvaraj P, Jacob J. Immune complex-mediated enhancement of secondary antibody responses. J Immunol. 2010;184(11):6293–6298. | ||
Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nat Rev Immunol. 2009;9(1):15–27. | ||
Wadhwa M, Thorpe R. Strategies and Assays for the Assessment of Unwanted Immunogenicity. J Immunotoxicol. 2006;3(3):115–121. | ||
Strand V, Balsa A, Al-Saleh J, et al. Immunogenicity of Biologics in Chronic Inflammatory Diseases: A Systematic Review. BioDrugs. 2017;31(4):299–316. | ||
Krieckaert C, Rispens T, Wolbink G. Immunogenicity of biological therapeutics: from assay to patient. Curr Opin Rheumatol. 2012;24:306–311. | ||
Schaeverbeke T, Truchetet M-E, Kostine M, Barnetche T, Bannwarth B, Richez C. Immunogenicity of biologic agents in rheumatoid arthritis patients: lessons for clinical practice. Rheumatology. 2016;55(2):210–220. | ||
Garcês S, Demengeot J, Benito-Garcia E. The immunogenicity of anti-TNF therapy in immune-mediated inflammatory diseases: a systematic review of the literature with a meta-analysis. Ann Rheum Dis. 2013;72(12):1947–1955. | ||
Maneiro JR, Salgado E, Gomez-Reino JJ. Immunogenicity of Monoclonal Antibodies Against Tumor Necrosis Factor Used in Chronic Immune-Mediated Inflammatory Conditions. JAMA Intern Med. 2013;173(15):1416–1428. | ||
Thomas SS, Borazan N, Barroso N, et al. Comparative Immunogenicity of TNF Inhibitors: Impact on Clinical Efficacy and Tolerability in the Management of Autoimmune Diseases. A Systematic Review and Meta-Analysis. BioDrugs. 2015;29(4):241–258. | ||
Moots RJ, Xavier RM, Mok CC, et al. The impact of anti-drug antibodies on drug concentrations and clinical outcomes in rheumatoid arthritis patients treated with adalimumab, etanercept, or infliximab: Results from a multinational, real-world clinical practice, non-interventional study. PLoS One. 2017;12(4):e0175207. | ||
Burmester GR, Choy E, Kivitz A, et al. Low immunogenicity of tocilizumab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76(6):1078–1085. | ||
Benucci M, Meacci F, Grossi V, et al. Correlations between immunogenicity, drug levels, and disease activity in an Italian cohort of rheumatoid arthritis patients treated with tocilizumab. Biologics. 2016;10:53–58. | ||
Sigaux J, Hamze M, Daien C, et al. Immunogenicity of tocilizumab in patients with rheumatoid arthritis. Joint Bone Spine. 2017;84(1):39–45. | ||
Listing J, Gerhold K, Zink A. The risk of infections associated with rheumatoid arthritis, with its comorbidity and treatment. Rheumatology. 2013;52(1):53–61. | ||
Minozzi S, Bonovas S, Lytras T, et al. Risk of infections using anti-TNF agents in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: a systematic review and meta-analysis. Expert Opin Drug Saf. 2016;15(sup1):11–34. | ||
Roach DR, Bean AGD, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol. 2002;168(9):4620–4627. | ||
Koo S, Marty FM, Baden LR. Infectious Complications Associated with Immunomodulating Biologic Agents. Infect Dis Clin North Am. 2010;24(2):285–306. | ||
Cantini F, Niccoli L, Goletti D. Tuberculosis Risk in Patients Treated with Non-Anti-Tumor Necrosis Factor- (TNF-) Targeted Biologics and Recently Licensed TNF-Inhibitors: Data from Clinical Trials and National Registries. J Rheumatol Suppl. 2014;91:56–64. | ||
Wallis RS, Broder M, Wong J, Lee A, Hoq L. Reactivation of latent granulomatous infections by infliximab. Clin Infect Dis. 2005;41(Supplement_3):S194–S198. | ||
Toruner M, Loftus EV, Harmsen WS, et al. Risk factors for opportunistic infections in patients with inflammatory bowel disease. Gastroenterology. 2008;134(4):929–936. | ||
Cantini F, Boccia S, Goletti D, et al. HBV Reactivation in Patients Treated with Antitumor Necrosis Factor-Alpha (TNF-α) Agents for Rheumatic and Dermatologic Conditions: A Systematic Review and Meta-Analysis. Int J Rheumatol. 2014;2014(2):926836. | ||
Strangfeld A, Listing J, Herzer P, et al. Risk of Herpes Zoster in Patients With Rheumatoid Arthritis Treated With Anti–TNF-α Agents. JAMA. 2009;301(7):737–744. | ||
Ali T, Kaitha S, Mahmood S, Ftesi A, Stone J, Bronze MS. Clinical use of anti-TNF therapy and increased risk of infections. Drug Health Patient Saf. 2013;5:79–99. | ||
Askling J, Fored CM, Brandt L, et al. Time-dependent increase in risk of hospitalisation with infection among Swedish RA patients treated with TNF antagonists. Ann Rheum Dis. 2007;66(10):1339–1344. | ||
Weinblatt ME, Schiff M, Valente R, et al. Head-to-head comparison of subcutaneous abatacept versus adalimumab for rheumatoid arthritis: Findings of a phase IIIb, multinational, prospective, randomized study. Arthritis Rheum. 2013;65(1):28–38. | ||
Zink A, Strangfeld A, Schneider M, et al. Effectiveness of tumor necrosis factor inhibitors in rheumatoid arthritis in an observational cohort study: Comparison of patients according to their eligibility for major randomized clinical trials. Arthritis Rheum. 2006;54(11):3399–3407. | ||
Aaltonen KJ, Ylikylä S, Tuulikki Joensuu J, et al. Efficacy and effectiveness of tumour necrosis factor inhibitors in the treatment of rheumatoid arthritis in randomized controlled trials and routine clinical practice. Rheumatology. 2017;56(5):725–735. | ||
Singh JA. Infections With Biologics in Rheumatoid Arthritis and Related Conditions: a Scoping Review of Serious or Hospitalized Infections in Observational Studies. Curr Rheumatol Rep. 2016;18(10):61. | ||
Rhodes A, Evans LE, Alhazzani W, et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med. 2017;43(3):304–377. | ||
Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–810. | ||
Seymour CW, Liu VX, Iwashyna TJ, et al. Assessment of Clinical Criteria for Sepsis: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):762–774. | ||
Laupland KB. Incidence of bloodstream infection: a review of population-based studies. Clin Microbiol Infect. 2013;19(6):492–500. | ||
Buetti N, Atkinson A, Marschall J, Kronenberg A. Swiss Centre for Antibiotic Resistance (ANRESIS). Incidence of bloodstream infections: a nationwide surveillance of acute care hospitals in Switzerland 2008–2014. BMJ Open. 2017;7:e013665. | ||
Chandra RK. Nutrition, immunity and infection: From basic knowledge of dietary manipulation of immune responses to practical application of ameliorating suffering and improving survival. Proc Natl Acad Sci U S A. 1996;93(25):14304–14307. | ||
Fernandez-Martinez S, Cortes X, Borrás-Blasco J, Gracia-Pérez A, Casterá MDE. Effectiveness of a systematic vaccination program in patients with autoimmune inflammatory disease treated with anti-TNF alpha drugs. Expert Opin Biol Ther. 2016;16(11):1317–1322. | ||
Williams EL, Gadola S, Edwards CJ. Anti-TNF-induced lupus. Rheumatology. 2009;48(7):716–720. | ||
Pereira Vaz JL, Fernandes V, Nogueira F, Arnobio AM, Levy RA. Infliximab induced autoantibodies. A multicenter study. Clin Rheumatol. 2016;35:325–332. | ||
Chimenti MS, Spinelli FR, Giunta A, et al. Emergence of antinuclear antibodies in psoriatic patients treated with Infliximab: personal experience and literature review. Drug Dev Res. 2014;75(Suppl 1):S61–S63. | ||
Ramos-Casals M, Brito-Zerón P, Soto M-J, Cuadrado M-J, Khamashta MA. Autoimmune diseases induced by TNF-targeted therapies. Best Pract Res Clin Rheumatol. 2008;22(5):847–861. | ||
Katz U, Zandman-Goddard G. Drug-induced lupus: An update. Autoimmun Rev. 2010;10(1):46–50. | ||
De Bandt M, Sibilia J, Le Loët X, et al. Systemic lupus erythematosus induced by anti-tumour necrosis factor alpha therapy: a French national survey. Arthritis Res Ther. 2005;7(3):R545–R551. | ||
Wetter DA, Davis MDP. Lupus-Like Syndrome Attributable to Anti – Tumor Necrosis Factor α Therapy in 14 Patients During an 8-Year Period at Mayo Clinic. Mayo Clin Proc. 2009;84(11):979–984. | ||
Costa MF, Said NR, Zimmermann B. Drug-Induced Lupus due to Anti-Tumor Necrosis Factor α Agents. Semin Arthritis Rheum. 2008;37(6):381–387. | ||
Vannucchi V, Grazzini M, Pieralli F, Giannotta M, Biagioni C, Nozzoli C. Adalimumab-induced lupus erythematosus with central nervous system involvement in a patient with Crohn’s disease. J Gastrointestin Liver Dis. 2011;20(2):201–203. | ||
Aringer M, Smolen JS. Therapeutic blockade of TNF in patients with SLE – Promising or crazy? Autoimmun Rev. 2012;11(5):321–325. | ||
Mosca M, Tani C, Filice ME, et al. TNF-alpha inhibitors in Systemic Lupus Erythematosus. A case report and a systematic literature review. Mod Rheumatol. 2015;25(4):642–645. | ||
Emer JJ, Frankel A, Zeichner JA. A practical approach to monitoring patients on biological agents for the treatment of psoriasis. J Clin Aesthet Dermatol. 2010;3(8):20–26. | ||
Ding T, Ledingham J, Luqmani R, et al. BSR and BHPR rheumatoid arthritis guidelines on safety of anti-TNF therapies. Rheumatology. 2010;49(11):2217–2219. | ||
Sinigaglia L, Nervetti A, Mela Q, et al. A multicenter cross sectional study on bone mineral density in rheumatoid arthritis. Italian Study Group on Bone Mass in Rheumatoid Arthritis. J Rheumatol. 2000;27(11):2582–2589. | ||
Okano T, Inui K, Tada M, et al. High frequency of vertebral fracture and low bone quality in patients with rheumatoid arthritis – Results from TOMORROW study. Mod Rheumatol. 2017;27(3):398–404. | ||
Schett G, Stach C, Zwerina J, Voll R, Manger B. How antirheumatic drugs protect joints from damage in rheumatoid arthritis. Arthritis Rheum. 2008;58(10):2936–2948. | ||
Shaw AT, Gravallese EM. Mediators of inflammation and bone remodeling in rheumatic disease. Semin Cell Dev Biol. 2016;49:2–10. | ||
Amarasekara DS, Yu J, Rho J. Bone Loss Triggered by the Cytokine Network in Inflammatory Autoimmune Diseases. J Immunol Res. 2015;2015;2015:832127. | ||
Redlich K, Smolen JS. Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat Rev Drug Discov. 2012;11(3):234–250. | ||
Dimitroulas T, Nikas SN, Trontzas P, Kitas GD. Biologic therapies and systemic bone loss in rheumatoid arthritis. Autoimmun Rev. 2013;12(10):958–966. | ||
Krieckaert CLM, Nurmohamed MT, Wolbink G, Lems WF. Changes in bone mineral density during long-term treatment with adalimumab in patients with rheumatoid arthritis: a cohort study. Rheumatology. 2013;52(3):547–553. | ||
Haugeberg G, Helgetveit KB, Førre Ø, Garen T, Sommerseth H, Prøven A. Generalized bone loss in early rheumatoid arthritis patients followed for ten years in the biologic treatment era. BMC Musculoskelet Disord. 2014;15(1):289. | ||
Kawai VK, Stein CM, Perrien DS, Griffin MR. Effects of anti-tumor necrosis factor α agents on bone. Curr Opin Rheumatol. 2012;24(5):576–585. | ||
Siu S, Haraoui B, Bissonnette R, et al. Meta-analysis of tumor necrosis factor inhibitors and glucocorticoids on bone density in rheumatoid arthritis and ankylosing spondylitis trials. Arthritis Care Res. 2015;67(6):754–764. | ||
Manara M, Sinigaglia L. Bone and TNF in rheumatoid arthritis: clinical implications. RMD Open. 2015;1(Suppl 1):e000065. | ||
Zerbini CAF, Clark P, Mendez-Sanchez L, et al. Biologic therapies and bone loss in rheumatoid arthritis. Osteoporos Int. 2017;28(2):429–446. | ||
Garnero P, Thompson E, Woodworth T, Smolen JS. Rapid and sustained improvement in bone and cartilage turnover markers with the anti-interleukin-6 receptor inhibitor tocilizumab plus methotrexate in rheumatoid arthritis patients with an inadequate response to methotrexate: Results from a substudy of the multicenter double-blind, placebo-controlled trial of tocilizumab in inadequate responders to methotrexate alone. Arthritis Rheum. 2010;62(1):33–43. | ||
Karsdal MA, Schett G, Emery P, et al. IL-6 receptor inhibition positively modulates bone balance in rheumatoid arthritis patients with an inadequate response to anti-tumor necrosis factor therapy: biochemical marker analysis of bone metabolism in the tocilizumab RADIATE study (NCT00106522). Semin Arthritis Rheum. 2012;42(2):131–139. | ||
Terpos E, Fragiadaki K, Konsta M, Bratengeier C, Papatheodorou A, Sfikakis PP. Early effects of IL-6 receptor inhibition on bone homeostasis: a pilot study in women with rheumatoid arthritis. Clin Exp Rheumatol. 2011;29(6):921–925. | ||
Kume K, Amano K, Yamada S, et al. The effect of tocilizumab on bone mineral density in patients with methotrexate-resistant active rheumatoid arthritis. Rheumatology. 2014;53(5):900–903. | ||
Briot K, Rouanet S, Schaeverbeke T, et al. The effect of tocilizumab on bone mineral density, serum levels of Dickkopf-1 and bone remodeling markers in patients with rheumatoid arthritis. Joint Bone Spine. 2015;82(2):109–115. | ||
Boumans MJH, Thurlings RM, Yeo L, et al. Rituximab abrogates joint destruction in rheumatoid arthritis by inhibiting osteoclastogenesis. Ann Rheum Dis. 2012;71(1):108–113. | ||
Hein G, Eidner T, Oelzner P, et al. Influence of Rituximab on markers of bone remodeling in patients with rheumatoid arthritis: a prospective open-label pilot study. Rheumatol Int. 2011;31(2):269–272. | ||
Wheater G, Hogan VE, Teng YKO, et al. Suppression of bone turnover by B-cell depletion in patients with rheumatoid arthritis. Osteoporosis International. 2011;22(12):3067–3072. | ||
Axmann R, Herman S, Zaiss M, et al. CTLA-4 directly inhibits osteoclast formation. Ann Rheum Dis. 2008;67(11):1603–1609. | ||
Bozec A, Zaiss MM, Kagwiria R, et al. T cell costimulation molecules CD80/86 inhibit osteoclast differentiation by inducing the IDO/tryptophan pathway. Sci Transl Med. 2014;6(235):235ra60. | ||
Coulson KA, Reed G, Gilliam BE, Kremer JM, Pepmueller PH. Factors influencing fracture risk, T score, and management of osteoporosis in patients with rheumatoid arthritis in the Consortium of Rheumatology Researchers of North America (CORRONA) registry. J Clin Rheumatol. 2009;15(4):155–160. | ||
Kim SY, Schneeweiss S, Liu J, Solomon DH. Effects of disease-modifying antirheumatic drugs on nonvertebral fracture risk in rheumatoid arthritis: A population-based cohort study. J Bone Miner Res. 2012;27(4):789–796. | ||
Roussy J-P, Bessette L, Bernatsky S, Rahme E, Lachaine J. Biologic disease-modifying anti-rheumatic drugs and the risk of non-vertebral osteoporotic fractures in patients with rheumatoid arthritis aged 50 years and over. Osteoporos Int. 2013;24(9):2483–2492. | ||
Kawai VK, Grijalva CG, Arbogast PG, et al. Initiation of tumor necrosis factor α antagonists and risk of fractures in patients with selected rheumatic and autoimmune diseases. Arthritis Care Res. 2013;65(7):1085–1094. | ||
Kässer UR, Gleissner C, Dehne F, Michel A, Willershausen-Zönnchen B, Bolten WW. Risk for periodontal disease in patients with longstanding rheumatoid arthritis. Arthritis Rheum. 1997;40(12):2248–2251. | ||
Mercado FB, Marshall RI, Klestov AC, Bartold PM. Relationship Between Rheumatoid Arthritis and Periodontitis. J Periodontol. 2001;72(6):779–787. | ||
Pischon N, Pischon T, Kröger J, et al. Association among rheumatoid arthritis, oral hygiene, and periodontitis. J Periodontol. 2008;79(6):979–986. | ||
Garib BT, Qaradaxi SS. Temporomandibular joint problems and periodontal condition in rheumatoid arthritis patients in relation to their rheumatologic status. J Oral Maxillofac Surg. 2011;69(12):2971–2978. | ||
Smit M, Westra J, Vissink A, Doornbos-van der Meer B, Brouwer E, van Winkelhoff A. Periodontitis in established rheumatoid arthritis patients: a cross-sectional clinical, microbiological and serological study. Arthritis Res Ther. 2012;14(5):R222. | ||
Scher JU, Ubeda C, Equinda M, et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum. 2012;64(10):3083–3094. | ||
Potikuri D, Dannana KC, Kanchinadam S, et al. Periodontal disease is significantly higher in non-smoking treatment-naive rheumatoid arthritis patients: results from a case-control study. Ann Rheum Dis. 2012;71(9):1541–1544. | ||
Joseph R, Rajappan S, Nath SG, Paul BJ. Association between chronic periodontitis and rheumatoid arthritis: a hospital-based case–control study. Rheumatol Int. 2013;33(1):103–109. | ||
Mikuls TR, Payne JB, Yu F, et al. Periodontitis and Porphyromonas gingivalis in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2014;66(5):1090–1100. | ||
Arkema EV, Karlson EW, Costenbader KH. A prospective study of periodontal disease and risk of rheumatoid arthritis. J Rheumatol. 2010;37(9):1800–1804. | ||
Demmer RT, Molitor JA, Jacobs DR, Michalowicz BS. Periodontal disease, tooth loss and incident rheumatoid arthritis: results from the First National Health and Nutrition Examination Survey and its epidemiological follow-up study. J Clin Periodontol. 2011;38(11):998–1006. | ||
Eriksson K, Nise L, Kats A, et al. Prevalence of Periodontitis in Patients with Established Rheumatoid Arthritis: A Swedish Population Based Case-Control Study. PLoS One. 2016;11(5):e0155956. | ||
Wolff B, Berger T, Frese C, et al. Oral status in patients with early rheumatoid arthritis: a prospective, case-control study. Rheumatology. 2014;53(3):526–531. | ||
Javed F, Ahmed HB, Mikami T, Almas K, Romanos GE, Al-Hezaimi K. Cytokine profile in the gingival crevicular fluid of rheumatoid arthritis patients with chronic periodontitis. J Investig Clin Dent. 2014;5(1):1–8. | ||
Scher JU, Bretz WA, Abramson SB. Periodontal disease and subgingival microbiota as contributors for rheumatoid arthritis pathogenesis. Curr Opin Rheumatol. 2014;26(4):424–429. | ||
Brusca SB, Abramson SB, Scher JU. Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr Opin Rheumatol. 2014;26(1):101–107. | ||
Algood HM, Lin PL, Yankura D, Jones A, Chan J, Flynn JL. TNF influences chemokine expression of macrophages in vitro and that of CD11b+ cells in vivo during Mycobacterium tuberculosis infection. J Immunol. 2004;172(11):6846–6857. | ||
Araújo VMA, Melo IM, Lima V. Relationship between Periodontitis and Rheumatoid Arthritis: Review of the Literature. Mediators Inflamm. 2015;2015:259074. | ||
Savioli C, Ribeiro AC, Fabri GM, et al. Persistent periodontal disease hampers anti-tumor necrosis factor treatment response in rheumatoid arthritis. J Clin Rheumatol. 2012;18(4):180–184. | ||
Chen HH, Chen DY, Lai KL, et al. Periodontitis and etanercept discontinuation risk in anti-tumor necrosis factor-naive rheumatoid arthritis patients: a nationwide population-based cohort study. J Clin Rheumatol. 2013;19(8):432–438. | ||
Ortiz P, Bissada NF, Palomo L, et al. Periodontal therapy reduces the severity of active rheumatoid arthritis in patients treated with or without tumor necrosis factor inhibitors. J Periodontol. 2009;80(4):535–540. | ||
Bıyıkoğlu B, Buduneli N, Aksu K, et al. Periodontal therapy in chronic periodontitis lowers gingival crevicular fluid interleukin-1beta and DAS28 in rheumatoid arthritis patients. Rheumatol Int. 2013;33(10):2607–2616. | ||
Erciyas K, Sezer U, Ustün K, et al. Effects of periodontal therapy on disease activity and systemic inflammation in rheumatoid arthritis patients. Oral Dis. 2013;19(4):394–400. | ||
Okada M, Kobayashi T, Ito S, et al. Periodontal treatment decreases levels of antibodies to Porphyromonas gingivalis and citrulline in patients with rheumatoid arthritis and periodontitis. J Periodontol. 2013;84(12):e74–e84. | ||
Mayer Y, Elimelech R, Balbir-Gurman A, Braun-Moscovici Y, Machtei EE. Periodontal condition of patients with autoimmune diseases and the effect of anti-tumor necrosis factor-α therapy. J Periodontol. 2013;84(2):136–142. | ||
Kadkhoda Z, Amirzargar A, Esmaili Z, Vojdanian M, Akbari S. Effect of TNF-α Blockade in Gingival Crevicular Fluid on Periodontal Condition of Patients with Rheumatoid Arthritis. Iran J Immunol. 2016;13(3):197–203. | ||
Kobayashi T, Yokoyama T, Ito S, et al. Periodontal and serum protein profiles in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitor adalimumab. J Periodontol. 2014;85(11):1480–1488. | ||
Han JY, Reynolds MA. Effect of anti-rheumatic agents on periodontal parameters and biomarkers of inflammation: a systematic review and meta-analysis. J Periodontal Implant Sci. 2012;42(1):3–12. | ||
Fabri GM, Pereira RM, Savioli C, et al. Periodontitis Response to Anti-TNF Therapy in Ankylosing Spondylitis. J Clin Rheumatol. 2015;21(7):341–345. | ||
Matcham F, Rayner L, Steer S, Hotopf M. The prevalence of depression in rheumatoid arthritis: a systematic review and meta-analysis. Rheumatology. 2013;52(12):2136–2148. | ||
Ostensen M. Rheumatoid arthritis: The effect of RA and medication on female fertility. Nat Rev Rheumatol. 2014;10(9):518–519. | ||
Brouwer J, Fleurbaaij R, Hazes JMW, Dolhain R, Laven JSE. Subfertility in Women With Rheumatoid Arthritis and the Outcome of Fertility Assessments. Arthritis Care Res. 2017;69(8):1142–1149. | ||
Costa TF, Silva CR, Muniz LF, Mota LM. Prevalence of sexual dysfunction among female patients followed in a Brasília Cohort of early rheumatoid arthritis. Rev Bras Reumatol. 2015;55(2):123–132. | ||
Brouwer J, Hazes JM, Laven JS, Dolhain RJ. Fertility in women with rheumatoid arthritis: influence of disease activity and medication. Ann Rheum Dis. 2015;74(10):1836–1841. | ||
Gordon D, Beastall GH, Thomson JA, Sturrock RD. Androgenic status and sexual function in males with rheumatoid arthritis and ankylosing spondylitis. Q J Med. 1986;60(231):671–679. | ||
Cutolo M, Balleari E, Giusti M, Monachesi M, Accardo S. Sex hormone status of male patients with rheumatoid arthritis: evidence of low serum concentrations of testosterone at baseline and after human chorionic gonadotropin stimulation. Sex hormone status of male patients with rheumatoid arthritis: evidence of low serum concentrations of testosterone at baseline and after human chorionic gonadotropin stimulation. Arthritis Rheum. 1988;31:1314–1317. | ||
Leroy C, Rigot JM, Leroy M, et al. Immunosuppressive drugs and fertility. Orphanet J Rare Dis. 2015;10:136. | ||
Gutierrez JC, Hwang K. The toxicity of methotrexate in male fertility and paternal teratogenicity. Expert Opin Drug Metab Toxicol. 2017;13(1):51–58. | ||
Ramonda R, Foresta C, Ortolan A, et al. Influence of tumor necrosis factor α inhibitors on testicular function and semen in spondyloarthritis patients. Fertil Steril. 2014;101(2):359–365. | ||
Oh JS, Heo HM, Kim YG, Lee SG, Lee CK, Yoo B. The effect of anti-tumor necrosis factor agents on sexual dysfunction in male patients with ankylosing spondylitis: a pilot study. Int J Impot Res. 2009;21(6):372–375. | ||
Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–977. | ||
Singh JA, Saag KG, Bridges SL, et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol. 2016;68(1):1–26. | ||
Tarp S, Eric Furst D, Boers M, et al. Risk of serious adverse effects of biological and targeted drugs in patients with rheumatoid arthritis: a systematic review meta-analysis. Rheumatology. 2017;56(3):417–425. | ||
Atkins D, Best D, Briss PA, et al. Grading quality of evidence and strength of recommendations. BMJ. 2004;328(7454):1490. | ||
Cantini F, Niccoli L, Nannini C, et al. Second-line biologic therapy optimization in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. Semin Arthritis Rheum. 2017;47(2):183–192. | ||
Favalli EG, Raimondo MG, Becciolini A, Crotti C, Biggioggero M, Caporali R. The management of first-line biologic therapy failures in rheumatoid arthritis: Current practice and future perspectives. Autoimmun Rev. 2017;16(12):1185–1195. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.