Back to Journals » Journal of Pain Research » Volume 13
Peripheral Opioid Receptor Antagonists for Opioid-Induced Constipation: A Primer on Pharmacokinetic Variabilities with a Focus on Drug Interactions
Received 26 June 2019
Accepted for publication 9 January 2020
Published 25 February 2020 Volume 2020:13 Pages 447—456
DOI https://doi.org/10.2147/JPR.S220859
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Jeffrey Gudin, 1, 2 Jeffrey Fudin 3–6
1Department of Anesthesiology, Rutgers New Jersey Medical School, Newark, NJ, USA; 2Department of Anesthesiology, Englewood Hospital and Medical Center, Englewood, NJ, USA; 3Albany College of Pharmacy and Health Sciences, Albany, NY, USA; 4Western New England University College of Pharmacy, Springfield, MA, USA; 5Remitigate, LLC, Delmar, NY, USA; 6Stratton Veterans Affairs Medical Center, Albany, NY, USA
Correspondence: Jeffrey Gudin
Department of Anesthesiology, Englewood Hospital and Medical Center, 350 Engle Street, Englewood, NJ 07631, USA
Email [email protected]
Abstract: Opioid analgesics remain a treatment option for refractory acute and chronic pain, despite their potential risk for abuse and adverse events (AEs). Opioids are associated with several common AEs, but the most bothersome is opioid-induced constipation (OIC). OIC is often overlooked but has the potential to affect patient quality of life, increase associated symptom burden, and impede long-term opioid compliance. The peripherally acting μ-receptor antagonists (PAMORAs) are a class of drugs that include methylnaltrexone, naloxegol, and naldemedine. Collectively, each is approved for the treatment of OIC. PAMORAs work peripherally in the gastrointestinal tract, without impacting the central analgesic effects of opioids. However, each has unique pharmacokinetic properties that may be impacted by coadministered drugs or food. This review focuses on important metabolic and pharmacokinetic principals that are pertinent to drug interactions involving μ-opioid receptor antagonists prescribed for OIC. It highlights subtle differences among the PAMORAs that may have clinical significance. For example, unlike naloxegol or naldemedine, methylnaltrexone is not a substrate for CYP3A4 or p-glycoprotein; therefore, its plasma concentration is not altered when coadministered with concomitant medications that are CYP3A4 or p-glycoprotein inducers or inhibitors. With a better understanding of pharmacokinetic nuances of each PAMORA, clinicians will be better equipped to identify potential safety and efficacy considerations that may arise when PAMORAs are coadministered with other medications.
Keywords: drug-related side effects and adverse reactions, opioid or opiate mu (μ)-receptor antagonists, opioid analgesics, pharmacokinetics; opioid-induced constipation
Introduction
Clinicians choose opioids for the management of both acute and chronic pain as part of multimodal treatment plans.1 While most are familiar with the toxicities associated with opioid use, many overlook more common adverse events (AEs). Opioid-induced constipation (OIC) and other side effects such as nausea, vomiting, and somnolence are common and bothersome AEs that may be associated with increased symptom burden and limit long-term compliance with opioid therapy.1,2
Four drugs are currently approved by the US Food and Drug Administration (FDA) for the treatment of OIC. Lubiprostone, a chloride channel-2 agonist, increases fluid content in the gastrointestinal (GI) tract without known pharmacologic activity at opioid receptors.3 Three peripherally acting μ-opioid receptor antagonists (PAMORAs) are currently available for the treatment of OIC: methylnaltrexone, naloxegol, and naldemedine (Table 1). Each has demonstrated efficacy for OIC in patients taking opioid medication for chronic pain.4–6 PAMORAs bind to opioid receptors in the periphery, potentially blocking their activation by exogenous opioid exposure within the GI tract to prevent or minimize constipation. PAMORAs have specific properties such as low lipid solubility, large structure, and strong polarity that allow them to resist diffusion across the blood-brain barrier (BBB) at therapeutic doses;7–9 therefore, opioid withdrawal typically does not occur and central opioid analgesic effects are maintained.10
 |
Table 1 Comparison of Peripherally Acting µ-Receptor Antagonists Approved for the Treatment of Opioid-Induced Constipation |
Drug–drug, drug–food, and drug–disease interactions are common when treating both pain and analgesic side effects, especially in patients with comorbidities requiring polypharmacy. While not all drug interactions are clinically meaningful, some are therapeutically significant and can affect the safety and efficacy profiles of concomitantly used drugs. Moreover, adverse drug interactions may have a significant economic impact, including more doctor visits, additional treatments, and hospitalizations,11,12 which may contribute to increased morbidity and even mortality.
Each of the three PAMORAs approved for OIC has subtle pharmacokinetic differences that clinicians should consider. The objective of this review is to provide a primer of metabolic and pharmacokinetic principles that impact drug interactions involving µ-opioid receptor antagonists prescribed for OIC.
Overview of Pharmacokinetic Metabolism Important to Drug Interactions
Phase I and Phase II Metabolism
The major site of drug metabolism is the liver, and a process known as the “first-pass effect” attempts to keep a drug from reaching the systemic circulation immediately after enteric absorption by its rapid uptake and metabolism into inactive compounds by the liver. Not all drugs are metabolized on first pass through the liver, and some are excreted from the liver unchanged. Hepatic metabolism is composed of two phases. In Phase I, the drug may be functionalized or inactivated, and in Phase II, the drug is conjugated (Figure 1) to a more readily excretable form.13,14
 |
Figure 1 Phase I and Phase II metabolism. Note: Copyright © 2014. Springer. Reproduced from Taxak N, Bharatam PV. Drug metabolism: a fascinating link between chemistry and biology. Resonance. 2014;19(3):259–282.13 Abbreviation: CYP450, cytochrome P450. |
Most Phase I reactions are catalyzed by the cytochrome P450 system (CYP450) (see overview of CYP450 below). Common chemical reactions during Phase I include hydroxylation, dealkylation, oxidation, reduction, and hydrolysis.14 These reactions convert a parent drug to more polar metabolites by unmasking or inserting a polar functional group (eg, −OH, −SH, −NH2). The purpose of these reactions is to convert a highly lipophilic drug to a hydrophilic metabolite to prepare the drug for Phase II conjugation.13,15 Many drugs have more than one metabolite, any of which may be more or less pharmacologically active than the parent compound.
In Phase II, the products derived from the Phase I reactions are conjugated by a variety of transferase enzymes. During this process, a suitable moiety (eg, glucuronic acid, glutathione, sulphate, glycine) is conjugated to the Phase II metabolite.13 Common Phase II reactions include glucuronidation, sulfation, amino acid conjugation, acetylation, methylation, and glutathione conjugation, which enhance water solubility for secretion into the bile or urine.14
Cytochrome P450
The CYP450 system of heme proteins is integral to drug metabolism.16 Approximately 75% of medications are metabolized by CYP450 enzymes,17 and of these drugs, more than 80% are metabolized specifically by CYP450 3A4 and 3A5.17 In addition to the liver, CYP450 enzymes also are present in the small intestine, which may impact drug absorption during first-pass metabolism.16
To facilitate an understanding of this metabolic system, it is helpful for clinicians to think about medications as substrates, inducers, auto-inducers or inhibitors of CYP450 enzymes. Many medications are substrates, which can be generally defined as substances that are metabolized by one or more CYP enzymes to a more hydrophilic form for excretion.17,18 Substrates may also be inhibitors or inducers as described below.
Inducers are drugs that can increase the synthesis of one or more CYP450 enzymes.18 Drugs that induce CYP450 enzymes may result in lower plasma concentrations of the substrate (drug), potentially lowering their blood levels to subtherapeutic concentrations.16,17 Enzyme induction occurs within 3 weeks, and its effects decrease gradually as the inducer is discontinued.17 When an inducer is discontinued, it may take 2 to 4 weeks for its effects to wane, after which the substrate levels could increase significantly.17 Some drugs, including carbamazepine, are auto-inducers, meaning that they can induce their own CYP450-mediated metabolism because they are both a substrate and an inducer for the same enzyme.18 In this case, carbamazepine could have therapeutic serum levels for 2 to 3 weeks, and then become subtherapeutic as the induced CYP3A4 enzymes enhance its metabolism.
Inhibitors are drugs that can decrease the synthesis of CYP450 enzymes.18 Inhibition of CYP450 enzymes may increase plasma concentrations of a substrate via Phase I metabolism, because the enzyme responsible for metabolism is reduced by the inhibitor. There are warnings associated with many medications about strong inhibitors, as they can be a significant cause of drug toxicity.16,17 To add to the complexity, in some cases, the metabolite may be active, so inhibiting the metabolic conversion could reduce toxicity and efficacy. An example is the conversion of codeine to morphine by CYP2D6. Enzyme inhibition occurs within approximately 48 hrs, and its effect is rapidly diminished once the inhibitor is discontinued.17
The extent to which CYP450 enzymes alter drug concentrations is highly variable and is population dependent by phenotype. The clinical impact is determined by many factors, including the therapeutic index, co-administered drugs that also affect CYP450 enzymes, drug potency, variable and combined metabolic pathways for a single drug, drug dosage form (eg, intravenous [IV] vs oral), renal or hepatic function, patient age, and genetic polymorphisms.11,16 The degree of inhibition or induction varies and is defined by the FDA as strong, moderate, or weak (Table 2).19 Inasmuch, it is often difficult to predict the pharmacologic impact when drug concentrations are altered by inhibitors and inducers.
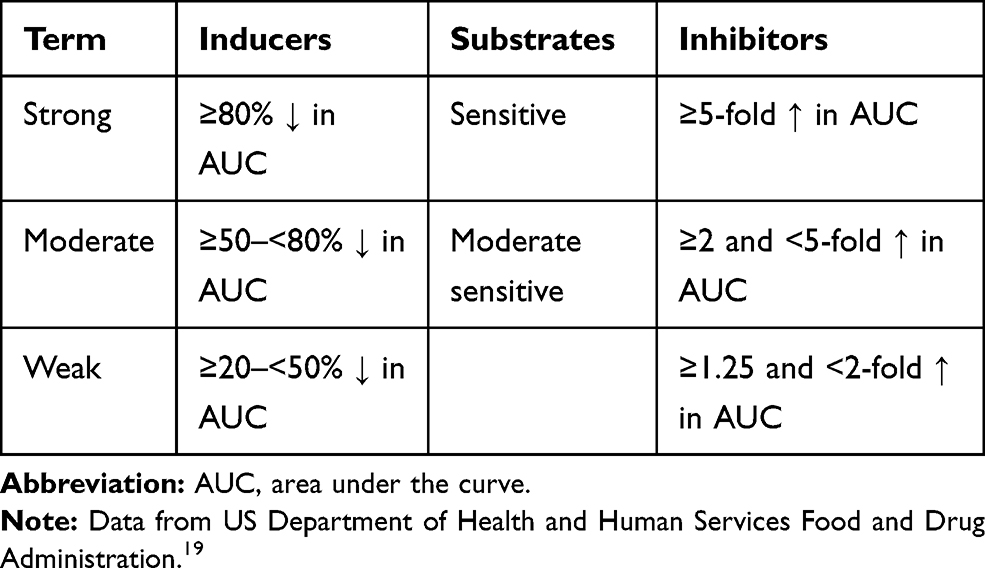 |
Table 2 FDA Definitions of Strong, Moderate, and Weak Inducers and Inhibitors. |
P-Glycoprotein
P-glycoprotein (P-gp) is a protein that actively transports drugs and toxins out of cells. It plays a significant role in drug absorption by functioning as a biological passage inhibitor.20 P-gp is an adenine triphosphate-dependent efflux transport protein for many substrates, including various analgesics.11,21–23 The primary function of P-gp is to prohibit substrates from crossing biological barriers. P-gp is found in the small intestine, kidney, and liver.24,25 It is a major transporter that protects passage through the BBB.22,23,25 P-gp has roles in drug absorption in the small intestine via the luminal facing epithelia (Figure 2)26 and in drug metabolism in the liver through the bile-facing canaliculi.11,22 P-gp also is expressed in the proximal tubules of the kidney, which aids in drug elimination.11
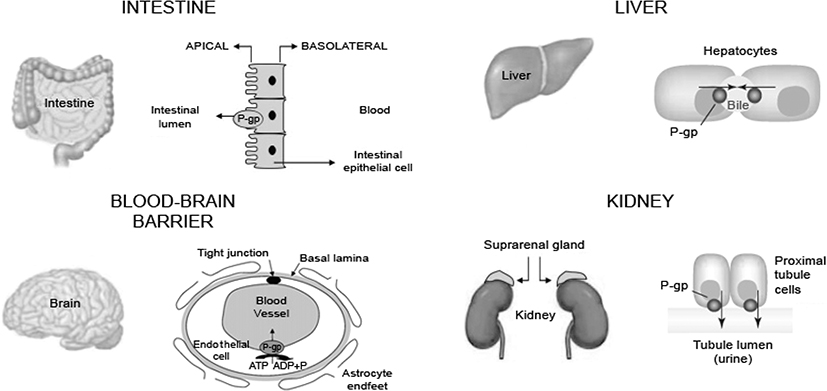 |
Figure 2 Efflux direction of P-gp transport in the key organs. Abbreviation: P-gp, p-glycoprotein.Note: Reproduced from Fortuna A, Alves G, Falcao A. In vitro and in vivo relevance of the P-glycoprotein probe substrates in drug discovery and development: focus on rhodamine 123, digoxin and talinolol. J Bioequiv Availab. 2011(suppl 2). Creative Commons license and disclaimer available from: http://creativecommons.org/licenses/by/4.0/legalcode.26 |
In vitro and in vivo studies have demonstrated that morphine, methadone, loperamide, meperidine, oxycodone, and fentanyl are P-gp substrates.27 Much like the CYP system, some of these substrates as well as other drugs may also serve as inducers or inhibitors of P-gp.
If P-gp is induced by a drug, it will prevent the passage of a P-gp substrate drug because there is more P-gp to pump the substrate drug back into the gut, thereby diminishing absorption. P-gp induction may influence the central nervous system (CNS) influx availability and potential efficacy of opioid agonists,21 as many opioids (eg, morphine, oxycodone) have a high P-gp substrate activity,28 and chronic administration may result in lower brain concentrations.29 Examples of P-gp inducers include carbamazepine, dexamethasone, doxorubicin, nefazodone, rifampin, Saint John’s Wort, trazadone, vinblastine, tipranavir, rifampin, and others.30,31
If P-gp is inhibited by a drug, it could result in elevated absorption of a P-gp substrate drug that otherwise relies on this protective mechanism to curtail absorption. Examples of P-gp inhibitors include clarithromycin, cyclosporine, diltiazem, itraconazole, ritonavir, progesterone, propranolol, quinidine, and verapamil, among others.31 Inhibitors of P-gp, such as verapamil or quinidine, may change the absorption of coadministered P-gp substrates, such as digoxin or loperamide.25 For example, when the P-gp substrate loperamide is coadministered with the P-gp inhibitor quinidine, the transport of loperamide across the BBB increases, which can lead to the CNS clinical effect of respiratory depression.25
Induction and inhibition by P-gp may lead to clinically significant drug interactions,25 and these effects may be dose-dependent.11 To add to the complexity of metabolism, CYP3A4 substrates also may be substrates or inhibitors of P-gp. Because of the overlapping substrate specificity between CYP3A4 and P-gp, and because of similarities in P-gp and CYP3A4 inhibitors and inducers, many drug interactions involve both P-gp and CYP3A4.20,32
Drug–Food Interactions
Foods are substances that can affect the activity of certain drugs. These interactions also include effects of herbal medicine and dietary supplements. Food effects can impact gastric emptying (ie, absorption) and alter pH, which in turn, changes the lipophilicity of certain drugs. In addition, various foods and nutraceuticals may be inducers, inhibitors, and/or substrates for CYP and P-gp discussed above. For example, grapefruit inhibits CYP3A4, CYP1A2, and P-gp;33 St. John’s Wort induces CYP3A4, CYP2B6, and P-gp;34 turmeric inhibits CYP1A2, CYP2C9, CYP3A4, CYP2C19 and 2D6;35 and oranges inhibit CYP3A4 and P-gp.33 Food can modulate metabolic enzymes and influx and efflux transporters.36 Some PAMORAs have food effects, each of which are described below (Table 3).37–39
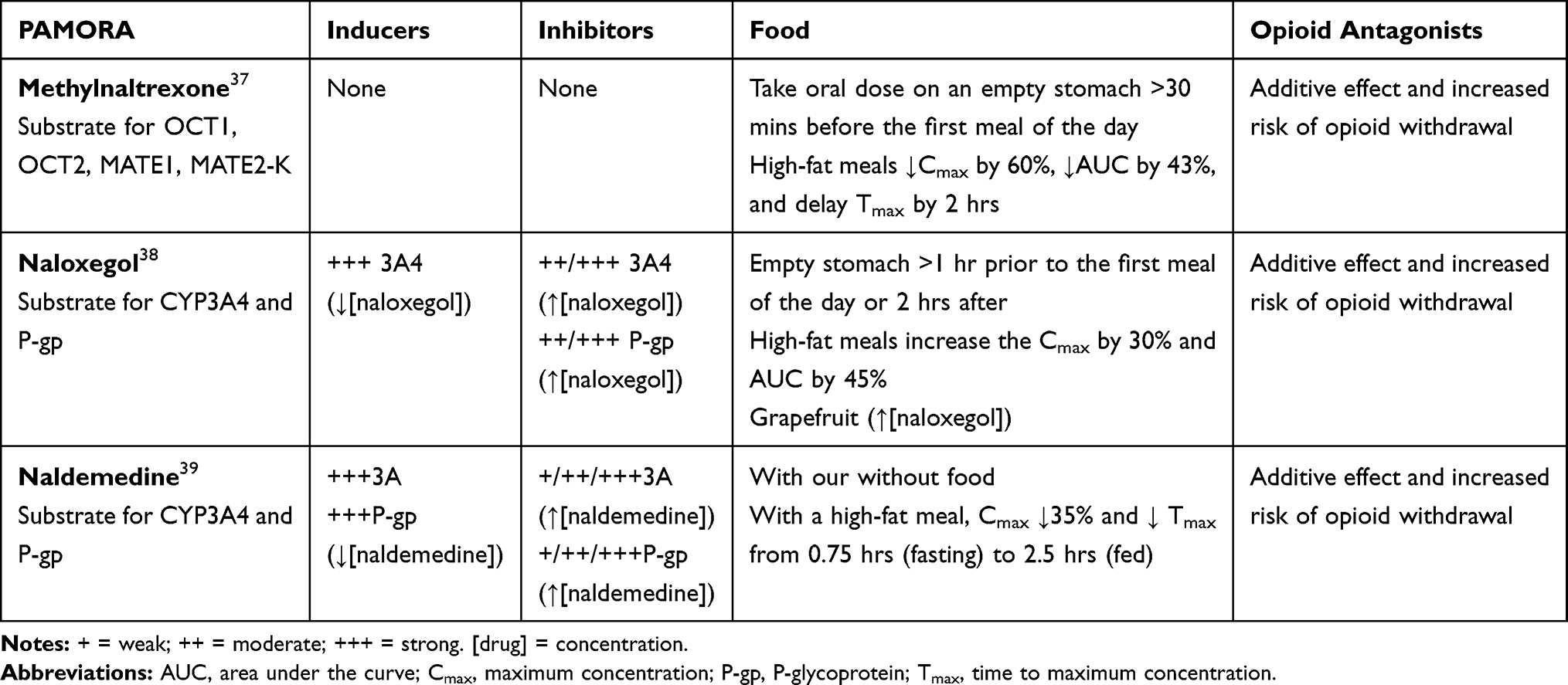 |
Table 3 Drug–Drug Interactions for PAMORAs: Effect of CYP450 Isoenzymes, P-Glycoprotein, Food, and Opioid Antagonists |
Drug Interaction Overview of PAMORAS Approved for OIC
Methylnaltrexone
Methylnaltrexone is available as an oral tablet or subcutaneous (SC) injection and is approved to treat OIC in adults with chronic noncancer pain, including patients with chronic pain related to prior cancer or its treatment who do not require frequent opioid dose escalation. The SC injection is also approved to treat OIC in adults with advanced illness or pain caused by active cancer who require opioid dose escalation for palliative care.37
In vitro studies have demonstrated that methylnaltrexone does not significantly inhibit or induce many CYP450 enzymes, including CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, or CYP3A4 (Table 3).37 In a Phase I, randomized, open-label, active- and placebo-controlled trial of healthy volunteers, methylnaltrexone (0.3 mg/kg, SC) did not affect the metabolism of dextromethorphan, a sensitive CYP2D6 substrate.37,40 In another Phase I, open-label study of healthy adults, a single dose of methylnaltrexone (24 mg IV, 20 min) was administered before and with the last dose of cimetidine, a moderate CYP3A4 inhibitor.37,41 The mean maximal plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC) of methylnaltrexone increased by only 10%.37 These examples suggest that among drugs known to induce or inhibit CYP450 enzymes, the plasma concentration of methylnaltrexone is not impacted in a clinically meaningful way.
The effect of food was assessed in a randomized, open-label, two-way crossover study, where healthy volunteers were given oral methylnaltrexone (450 mg) with a high-fat breakfast. Cmax of methylnaltrexone decreased by 60%, AUC decreased by 43%, and time to Cmax (Tmax) was delayed by 2 hrs.37 Thus, patients with chronic noncancer pain and OIC who take oral methylnaltrexone are advised to do so with water on an empty stomach ≥30 mins before the first meal of the day (Table 3).37
Pharmacokinetic differences based on gender have not been reported. Similarly, there are no pharmacokinetic studies in the pediatric population.37 However, case reports of methylnaltrexone use in terminally ill children who received approved doses showed that methylnaltrexone was safe and effective in this population.42 In a small study of a single intravenous infusion of methylnaltrexone in healthy adults showed that older patients (>65 years) had a mean clearance that was 20% lower and the AUC was 26% higher than in younger patients.37 However, in the clinical trial program of oral or injectable methylnaltrexone, no differences between younger and older patients were observed with respect to effectiveness or safety. Therefore, no age-specific dosage adjustments are recommended.37
Naloxegol
Naloxegol is a PAMORA used to treat OIC in patients with noncancer pain.38 Naloxegol is metabolized predominantly by CYP3A and is a substrate of the P-gp transporter.43 Weak inhibitors of CYP450 3A4 may have a minimal clinical effect on naloxegol,44 but strong or moderate inhibitors and strong inducers alter the plasma concentration of naloxegol.43 Elevated serum levels of naloxegol have been shown in clinical studies to increase AEs, particularly gastrointestinal events, and cause premature study discontinuation due to treatment-emergent AEs.45 The P-gp effect is primarily on absorption in the intestinal tract, however, because naloxegol is readily absorbed and highly soluble, the P-gp effect is likely negligible.44
Drug–drug interactions between naloxegol and strong CYP3A4 inhibitors have been reported (Table 3). Multiple doses of ketoconazole, a strong CYP3A inhibitor and P-gp inhibitor, increased Cmax and AUC of naloxegol by 9.58-fold (90% confidence interval, 8.10, 11.33) and 12.85-fold (90% confidence interval, 11.31, 14.61), respectively.38,44 Thus, the combination of naloxegol with strong CYP3A4 inhibitors is contraindicated because of the possibility of significantly increased exposure to naloxegol, the effects of which have not been established.38,46 Likewise, the combination of naloxegol with moderate CYP3A4 inhibitors should be avoided because of the potential for increased naloxegol concentrations. The product label advises that the dosage should be reduced to 12.5 mg once daily, and patients who require concomitant use should be monitored for AEs prior to increasing the dose.38 For example, multiple doses of diltiazem 240 mg extended-release formulation, a strong CYP3A4 inhibitor and P-gp inhibitor, increased Cmax and AUC of naloxegol by 2.86-fold (90% confidence interval, 2.59, 3.15) and 3.41-fold (90% confidence interval, 3.16, 3.69), respectively.38
Drug–drug interactions between naloxegol and CYP3A4 inhibitors or P-gp inhibitors also have been reported. Single doses of oral quinidine (600 mg), a strong CYP3A4 inhibitor and P-gp inhibitor, increased Cmax and AUC of oral naloxegol (25 mg) by 2.47-fold (90% confidence interval, 2.19, 2.78) and 1.39-fold (90% confidence interval, 1.31, 1.46), respectively, but did not antagonize morphine-related miosis.38,47 Similar changes for Cmax and AUC of naloxegol (2.44 and 1.38, respectively) were reported in another study.44
By contrast, the impact of drug–drug interactions between naloxegol and strong CYP3A4 or P-gp inducers are not as robust. Studies have shown that multiple doses of rifampin 600 mg, a strong CYP3A4 inducer, decreased Cmax and AUC of naloxegol by 0.24-fold (90% confidence interval, 0.20, 0.31) and 0.11-fold (90% confidence interval, 0.10, 0.12), respectively.38,44 Although the concomitant use of strong CYP3A4 inducers with naloxegol may slightly decrease the concentration of naloxegol, the clinical significance of this potential interaction is not known.38,46
PK modeling simulations suggest that coadministration of a single dose of oral naloxegol (25 mg) with efavirenz (400 mg once daily), a moderate CYP3A inducer, results in naloxegol exposure that is similar to that after a 12.5-mg dose of naloxegol alone.38,44
Naloxegol does not affect the systemic exposure of morphine and its circulating metabolites. When healthy subjects were given IV morphine (5 mg/70 kg) and a single dose of naloxegol (8 mg–1000 mg), morphine exposure did not increase or decrease with increasing naloxegol dose compared with morphine alone.38 A single dose of IV morphine (5 mg/70 kg) increased Cmax and AUC of naloxegol by 0.96-fold (90% confidence interval 0.78, 1.19) and 0.94-fold (90% confidence interval, 0.84, 1.07), respectively.38,47 However, in a partially double-blind, crossover, randomized trial of healthy subjects given a single IV dose of morphine (5 mg/70 kg) plus oral naloxegol (25 mg), morphine did not alter the pharmacokinetic properties of naloxegol and vice versa.47
Drug–drug interactions between naloxegol and drugs that alter gastric pH (eg, antacids and proton-pump inhibitors) have not been studied;38 however, food–drug interactions have been reported (Table 3). In a Phase I, open-label, randomized study in healthy volunteers, the extent and rate of naloxegol (25 mg) absorption was increased with a high-fat meal; Cmax was increased by 30% and AUC by 45%.38,48 According to the product label, naloxegol is to be taken on an empty stomach at least 1 hr prior to the first meal of the day (dosing used in clinical trials) or 2 hrs after the meal.38
Patients taking methadone for pain may have greater risk of GI AEs that may have been related to opioid withdrawal than patients taking other opioids.38 While the mechanism has not been established, the authors suspect it could be a function of methadone’s long tissue half-life compared with other opioids.
There does not appear to be an effect of gender or age on the pharmacokinetics of naloxegol. However, in a small pharmacokinetic study of elderly Japanese patients receiving multiple doses of naloxegol 25 mg daily, the mean Cmax and AUC values were 45% and 54%, respectively, greater than that observed in young healthy patients. The clinical significance of this effect has not been established.38
Naldemedine
Naldemedine is a PAMORA approved for the treatment of OIC in adult patients with chronic noncancer pain.39 Although naldemedine is a substrate of CYP 3A4, it does not inhibit or induce major CYP450 enzymes and does not inhibit P-gp.49 Concomitant use of naldemedine and strong CYP3A inducers, such as rifampin (Table 3), should be avoided because rifampin markedly decreases the Cmax and AUC of naldemedine, which may affect efficacy. A compensatory increase in naldemedine dose is not advised due to the risk of increased GI AEs.39,49 Likewise, concomitant use of naldemedine and itraconazole, a strong CYP3A4 inhibitor produced increases in Cmax and AUC of naldemedine,39,49 and concomitant use of naldemedine and fluconazole, a moderate CYP3A4 inhibitor, produced a modest increase in Cmax and AUC of naldemedine.39,49 The labeling for naldemedine advises monitoring for adverse reactions when it is used with moderate and strong CYP3A4 inhibitors because of an increase in plasma concentration, but dose adjustment is not needed.39
Naldemedine is a sensitive P-gp substrate and drug interactions between naldemedine and cyclosporine, a P-gp inhibitor (Table 3), have been reported. Concomitant use moderately increased the Cmax and AUC.39,49 Patients should be monitored for adverse reactions, but no dose adjustment is needed.39
No drug interaction studies of naldemedine and antacids, proton-pump inhibitors, or other drugs that alter gastric pH have been conducted;39 however, the results of drug–food interaction studies suggest that naldemedine may be taken with or without food (Table 3).39 In a Phase I, open-label, randomized study, healthy subjects consumed a high-fat breakfast ≤30 mins before dosing, after a 10 hr overnight fast.39,49 The rate of naldemedine absorption was noted to decrease (35% decrease in Cmax; Tmax increase from 0.75 h in the fasted state to 2.5 h in the fed state).
Like naloxegol, no studies have been conducted that show a pharmacokinetic effect based on gender or age.39
Naloxone and Opioid/Naloxone Combinations
Naloxone, a potent, competitive μ-opioid receptor antagonist, has been prescribed for OIC. Naloxone undergoes extensive first-pass metabolism and has a narrow therapeutic index and low systemic bioavailability (≤2%), which is highly variable between patients.50 Naloxone has not been shown to be a substrate for P-gp.21
Oral naloxone is available in the fixed-dose oral combinations of oxycodone/naloxone prolonged-release formulation,51 buprenorphine/naloxone,52 and pentazocine/naloxone.53 Oxycodone/naloxone was approved but not marketed in the United States; this combination is currently marketed in Europe, where its indication for severe pain is similar to other ER opioid analgesics.54 An analysis of oxycodone prolonged-release with naltrexone showed antagonism of peripheral μ-opioid receptors in the GI tract and reduction in OIC with minimal impact on centrally acting opioid analgesia.54–56 Buprenorphine/naloxone is indicated in various transmucosal dosage forms for the treatment of opioid use disorder, but it is not indicated for OIC.57
Pharmacodynamic Class Effects
There are a few pharmacodynamic interactions of PAMORAs that should be noted since they typically are of clinical importance. For all PAMORAs, the product labels each warn of opioid withdrawal risk alone or when coadministered with opioid antagonists.37–39 Concomitant use of the opioid antagonists naltrexone and nalmefene with buprenorphine/naloxone, for example, may precipitate opioid withdrawal symptoms and should be avoided.58,59 Concomitant use of mixed partial agonists (eg, pentazocine), partial agonist/antagonist opioid analgesics (eg, buprenorphine) or combinations of opioid agonists combined with an antagonist (eg, morphine/naltrexone) in patients taking any full agonist opioid (eg, oxycodone) may reduce the analgesic effect or precipitate withdrawal symptoms.51
Conclusions
OIC is a common adverse effect in patients receiving opioids for chronic pain. The PAMORAs methylnaltrexone, naloxegol, and naldemedine are currently approved for the treatment of OIC. It is important to understand the influence of CYP450 enzymes and P-gp induction and inhibition on the plasma concentration of each PAMORA and to identify PAMORAs such as methylnaltrexone, for example, that do not influence common CYP450 enzymes or P-gp. Lack of pharmacokinetic influence may lead to fewer drug–drug or drug–food interactions and may influence the selection of coadministered medications commonly prescribed for comorbidities associated with OIC.
Acknowledgments
Technical editorial and medical writing assistance was provided under the direction of the authors by Dana A. Franznick, PharmD, of Echelon Brand Communications, LLC, an OPEN Health company, Parsippany, NJ, USA. Funding for this assistance was provided by Salix Pharmaceuticals.
Disclosure
Jeffrey Gudin is part of the Advisory Board and/or Speakers Bureau for AcelRx Pharmaceuticals, AstraZeneca, BioDelivery Sciences International, Daiichi Sankyo, Scilex Pharmaceuticals, Salix Pharmaceuticals, and GlaxoSmithKline. He is a consultant for Mallinckrodt, Nektar, and Quest Diagnostics. He is also an advisor for Purdue. Jeffrey Fudin is part of Advisory Board and/or Speakers Bureau for AcelRx Pharmaceuticals, AstraZeneca, Daiichi Sankyo, GlaxoSmithKline, Quest Diagnostics, Scilex Pharmaceuticals and Salix Pharmaceuticals. He is also a speaker for Acutis Diagnostics, Inc and a consultant for Bio Delivery Sciences International and Firstox Laboratories. The authors report no other conflicts of interest in this work.
References
1. Morlion B, Clemens KE, Dunlop W. Quality of life and healthcare resource in patients receiving opioids for chronic pain: a review of the place of oxycodone/naloxone. Clin Drug Investig. 2015;35(1):1–11. doi:10.1007/s40261-014-0254-6
2. Morley KI, Ferris JA, Winstock AR, Lynskey MT. Polysubstance use and misuse or abuse of prescription opioid analgesics: a multi-level analysis of international data. Pain. 2017;158(6):1138–1144. doi:10.1097/j.pain.0000000000000892
3. Spierings ELH, Brewer RP, Rauck RL, Losch-Beridon T, Mareya SM. Lubiprostone for opioid-induced constipation does not interfere with opioid analgesia in patients with chronic noncancer pain. Pain Pract. 2017;17(3):312–319. doi:10.1111/papr.12444
4. Thomas J, Karver S, Cooney GA, et al. Methylnaltrexone for opioid-induced constipation in advanced illness. N Engl J Med. 2008;328(22):2332–2343. doi:10.1056/NEJMoa0707377
5. Chey WD, Webster L, Sostek M, Lappalainen J, Barker PN, Tack J. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med. 2014;370(25):2387–2396. doi:10.1056/NEJMoa1310246
6. Hale M, Wild J, Reddy J, Yamada T, Arjona Ferreira JC. Naldemedine versus placebo for opioid-induced constipation (COMPOSE-1 and COMPOSE-2): two multicentre, Phase 3, double-blind, randomised, parallel-group trials. Lancet Gastroenterol Hepatol. 2017;2(8):555–564. doi:10.1016/S2468-1253(17)30105-X
7. Pardridge WM. Transport of small molecules through the blood-brain barrier: biology and methodology. Adv Drug Deliv Rev. 1995;15(1–3):5–36. doi:10.1016/0169-409X(95)00003-P
8. Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2(1):3–14. doi:10.1602/neurorx.2.1.3
9. Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32(11):1959–1972. doi:10.1038/jcbfm.2012.126
10. Camilleri M, Drossman DA, Becker G, Webster LR, Davies AN, Mawe GM. Emerging treatments in neurogastroenterology: a multidisciplinary working group consensus statement on opioid-induced constipation. Neurogastroenterol Motil. 2014;26(10):1386–1395. doi:10.1111/nmo.12417
11. Lund M, Petersen TS, Dalhoff KP. Clinical implications of P-glycoprotein modulation in drug–drug interactions. Drugs. 2017;77(8):859–883. doi:10.1007/s40265-017-0729-x
12. Arnold RJG, Tang J, Schrecker J, Hild C. Impact of definitive drug–drug interaction testing on medication management and patient care. Drugs Real World Outcomes. 2018;5(4):217–224. doi:10.1007/s40801-018-0143-z
13. Taxak N, Bharatam PV. Drug metabolism: a fascinating link between chemistry and biology. Resonance. 2014;19(3):259–282. doi:10.1007/s12045-014-0031-0
14. Gunaratna C. Drug metabolism & pharmacokinetics in drug discovery: a primer for bioanalytical chemists, part I. Curr Sep. 2000;19(1):17–23.
15. Nassar AF, Hollenberg PF, Scatina J. Drug Metabolism Handbook: Concepts and Applications. Hoboken, NJ: John Wiley & Sons, Inc.; 2009.
16. McDonnell AM, Dang CH. Basic review of the cytochrome P450 system. J Adv Pract Oncol. 2013;4(4):263–268. doi:10.6004/jadpro.2013.4.4.7
17. Shapiro K, Brown SA, Garrett SD. Drug interactions. In: RxPrep Course Book: A Comprehensive Course for the NAPLEX and CPJE. El Segundo, CA: RxPrep; 2015:190–202.
18. Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 2008;9(4):310–322. doi:10.2174/138920008784220664
19. Guidance for industry: clinical drug interaction studies – study design, data analysis, and clinical implications; 2017. Available from: https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf.
20. Lin JH, Yamazaki M. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet. 2003;42(1):59–98. doi:10.2165/00003088-200342010-00003
21. Metcalf MD, Rosicky AD, Hassan HE, et al. Opioids and efflux transporters. Part 4: influence of N-substitution on P-glycoprotein substrate activity of noroxymorphone analogues. Bioorg Med Chem Lett. 2014;24(15):3592–3595. doi:10.1016/j.bmcl.2014.05.033
22. Fromm MF. P-glycoprotein: a defense mechanism limiting oral bioavailability and CNS accumulation of drugs. Int J Clin Pharmacol Ther. 2000;38(2):69–74. doi:10.5414/CPP38069
23. Chaves C, Remiao F, Cisternino S, Decleves X. Opioids and the blood-brain barrier: a dynamic interaction with consequences on drug disposition in brain. Curr Neuropharmacol. 2017;15(8):1156–1173. doi:10.2174/1570159X15666170504095823
24. Kolbow J, Modess C, Wegner D, et al. Extended-release but not immediate-release and subcutaneous methylnaltrexone antagonizes the loperamide-induced delay of whole-gut transit time in healthy subjects. J Clin Pharmacol. 2016;56(2):239–245. doi:10.1002/jcph.v56.2
25. Glaeser H. Importance of P-glycoprotein for drug–drug interactions. Handb Exp Pharmacol. 2011;(201):285–297.
26. Fortuna A, Alves G, Falcao A. In vitro and in vivo relevance of the P-glycoprotein probe substrates in drug discovery and development: focus on rhodamine 123, digoxin and talinolol. J Bioequiv Availab. 2011;(suppl 2). doi:10.4172/jbb.S2-001
27. Mercer SL, Coop A. Opioid analgesics and P-glycoprotein efflux transporters: a potential systems-level contribution to analgesic tolerance. Curr Top Med Chem. 2011;11(9):1157–1164. doi:10.2174/156802611795371288
28. Cunningham CW, Mercer SL, Hassan HE, Traynor JR, Eddington ND, Coop A. Opioids and efflux transporters. Part 2: P-glycoprotein substrate activity of 3- and 6-substituted morphine analogs. J Med Chem. 2008;51(7):2316–2320. doi:10.1021/jm701457j
29. Mercer SL, Hassan HE, Cunningham CW, Eddington ND, Coop A. Opioids and efflux transporters. Part 1: P-glycoprotein substrate activity of N-substituted analogs of meperidine. Bioorg Med Chem Lett. 2007;17(5):1160–1162. doi:10.1016/j.bmcl.2006.12.042
30. Horn JR, Hansten P. Drug transporters: the final frontier for drug interactions. Pharm Times. 2008;2:33.
31. Inhibitors and inducers of CYP enzymes and P-glycoprotein. Med Lett Drugs Ther. 2017;59(1517):e56.
32. Lin JH. Drug–drug interaction mediated by inhibition and induction of P-glycoprotein. Adv Drug Deliv Rev. 2003;55(1):53–81. doi:10.1016/S0169-409X(02)00171-0
33. Ased S, Wells J, Morrow LE, Malesker MA. Clinically significant food–drug interactions. Consult Pharm. 2018;33(11):649–657. doi:10.4140/TCP.n.2018.649.
34. Zhou S, Chan E, Pan SQ, Huang M, Lee EJ. Pharmacokinetic interactions of drugs with St John’s wort. J Psychopharmacol. 2004;18(2):262–276. doi:10.1177/0269881104042632
35. Sasaki T, Sato Y, Kumagai T, Yoshinari K, Nagata K. Effect of health foods on cytochrome P450-mediated drug metabolism. J Pharm Health Care Sci. 2017;3:14. doi:10.1186/s40780-017-0083-x
36. Mouly S, Lloret-Linares C, Sellier PO, Sene D, Bergmann JF. Is the clinical relevance of drug–food and drug–herb interactions limited to grapefruit juice and Saint-John’s Wort? Pharmacol Res. 2017;118:82–92. doi:10.1016/j.phrs.2016.09.038
37. Relistor [package insert]. Bridgewater, NJ: Salix Pharmaceuticals; 2018.
38. Movantik [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals; 2018.
39. Symproic [package insert]. Florham Park, NJ: Shionogi Inc.; 2018.
40. Effects of MNTX on CYP450 2D6 in metabolizers of dextromethorphan NCT01367535; 2011. Available from: https://clinicaltrials.gov/ct2/show/NCT01367535.
41. Effect of cimetidine on the single-dose PK of IV- administered MNTX NCT01366378; 2011.Available from: https://clinicaltrials.gov/ct2/show/NCT01366378.
42. Flerlage JE, Baker JN. Methylnaltrexone for opioid-induced constipation in children and adolescents and young adults with progressive incurable cancer at the end of life. J Palliat Med. 2015;18(7):631–633. doi:10.1089/jpm.2014.0364
43. Bui K, Zhou D, Sostek M, She F, Al-Huniti N. Effects of CYP3A modulators on the pharmacokinetics of naloxegol. J Clin Pharmacol. 2016;56(8):1019–1027. doi:10.1002/jcph.v56.8
44. Zhou D, Bui K, Sostek M, Al-Huniti N. Simulation and prediction of the drug–drug interaction potential of naloxegol by physiologically based pharmacokinetic modeling. CPT Pharmacometrics Syst Pharmacol. 2016;5(5):250–257. doi:10.1002/psp4.12070
45. Webster L, Dhar S, Eldon M, Masuoka L, Lappalainen J, Sostek M. A phase 2, double-blind, randomized, placebo-controlled, dose-escalation study to evaluate the efficacy, safety, and tolerability of naloxegol in patients with opioid-induced constipation. Pain. 2013;154(9):1542–1550. doi:10.1016/j.pain.2013.04.024
46. Yu J, Ritchie TK, Zhou Z, Ragueneau-Majlessi I. Key findings from preclinical and clinical drug interaction studies presented in new drug and biological license applications approved by the food and drug administration in 2014. Drug Metab Dispos. 2016;44(1):83–101. doi:10.1124/dmd.115.066720
47. Bui K, She F, Zhou D, Butler K, Al-Huniti N, Sostek M. The effect of quinidine, a strong P-glycoprotein inhibitor, on the pharmacokinetics and central nervous system distribution of naloxegol. J Clin Pharmacol. 2016;56(4):497–505. doi:10.1002/jcph.v56.4
48. US Food and Drug Administration and Center for Drug Evaluation and Research. Naloxegol medical review; 2013. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/204760Orig1s000MedR.pdf.
49. US Food and Drug Administration and Center for Drug Evaluation and Research. Naldemedine clinical pharmacology and biopharmaceutics review; 2008. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2008/021964s000_PharmR_P1.pdf.
50. Liu JC, Ma JD, Morello CM, Atayee RS, Best BM. Naltrexone metabolism and concomitant drug concentrations in chronic pain patients. J Anal Toxicol. 2014;38(4):212–217. doi:10.1093/jat/bku019
51. Targiniq ER [package insert]. Stamford, CT: Purdue Pharma; 2014.
52. Zubsolv [package insert]. Morristown, NJ: Orexo US; 2018.
53. Talwin Nx [package insert]. Bridgewater, NJ: Sanofi-aventis U.S.; 2011.
54. European Medicines Agency. Assessment report: oxynal-Targin and associated names 2014. Available from: https://www.ema.europa.eu/documents/referral/oxynal-targin-article-13-referral-assessment-report_en.pdf.
55. Smith K, Hopp M, Mundin G, et al. Naloxone as part of a prolonged release oxycodone/naloxone combination reduces oxycodone-induced slowing of gastrointestinal transit in healthy volunteers. Expert Opin Investig Drugs. 2011;20(4):427–439. doi:10.1517/13543784.2011.563236
56. Lowenstein O, Leyendecker P, Hopp M, et al. Combined prolonged-release oxycodone and naloxone improves bowel function in patients receiving opioids for moderate-to-severe non-malignant chronic pain: a randomised controlled trial. Expert Opin Pharmacother. 2009;10(4):531–543. doi:10.1517/14656560902796798
57. Suboxone Sublingual Film [package insert]. North Chesterfield, VA: Indivior Inc.; 2018.
58. Suboxone [summary of product characteristics]. Dublin, Ireland: Indivior Europe Limited; 2018.
59. Zubsolv [summary of product characteristics]. Cambridge, United Kingdom: Mundipharma Corporation Limited; 2018.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.