Back to Journals » Infection and Drug Resistance » Volume 19
Performance Evaluation of Targeted Nanopore Sequencing in Non-Tuberculous Mycobacteria Identification: A Comparative Study in Shenzhen, China
Authors Gui J ![]() , Long C, Fu Y
, Long C, Fu Y ![]() , He H, Li J, Wang F
, He H, Li J, Wang F ![]()
Received 21 October 2025
Accepted for publication 9 February 2026
Published 16 February 2026 Volume 2026:19 572430
DOI https://doi.org/10.2147/IDR.S572430
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Hazrat Bilal
Jing Gui,1,* Chan Long,2,* Yu Fu,1,* Huishan He,1 Jinli Li,1 Feng Wang1
1Department of Pathogenic Laboratory, Shenzhen Center for Chronic Disease Control, Shenzhen, People’s Republic of China; 2Department of Hematology, Huizhou First People’s Hospital, Huizhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Feng Wang, Department of Pathogenic Laboratory, Shenzhen Center for Chronic Disease Control, Shenzhen, People’s Republic of China, Email [email protected] Jing Gui, Department of Pathogenic Laboratory, Shenzhen Center for Chronic Disease Control, Shenzhen, People’s Republic of China, Email [email protected]
Objective: This study aims to analyze the performance differences between targeted nanopore sequencing, Sanger sequencing, and metagenomic sequencing in comparatively identifying non-tuberculous mycobacteria (NTM) species. Additionally, it explores the clinical application potential of targeted nanopore sequencing for identifying NTM clinical isolates in the Shenzhen region.
Methods: This retrospective study collected a total of 50 suspected NTM isolates from drug-resistant tuberculosis surveillance across 10 districts in Shenzhen, China, between December 2024 and June 2025. The species of the NTM isolates were initially identified using fluorescence PCR probe melting curve analysis. Genomic DNA was extracted from all 50 isolates, and species identification was performed using targeted nanopore sequencing (tNS), metagenomic sequencing (mNGS), and Sanger sequencing. The Jaccard similarity index, Kappa coefficient for classification consistency, and F1 score for model performance were calculated to evaluate the concordance among the three sequencing methods and assess the detection performance of targeted nanopore sequencing in NTM species identification.
Results: The most frequently detected NTM species by tNS, mNGS, and Sanger sequencing were M.abscessus and M.fortuitum, while M. tuberculosis was predominantly identified through mNGS results. Among the 50 suspected NTM samples, 18 (36%) showed complete concordance between tNS, mNGS, and Sanger sequencing, with the highest agreement observed between mNGS and tNS (28 samples, 56%). The final species identification reference results for the 50 samples were confirmed through a comprehensive evaluation using the Jaccard similarity coefficient, precision, and recall. Based on reference results, the F1 scores for tNS, mNGS, and Sanger sequencing were 0.927, 0.896, and 0.543, respectively. The tNS exhibited the highest concordance with the reference results, outperforming the other two methods.
Conclusion: tNS represents a preferred auxiliary methodology for clinical identification of NTM isolates in Shenzhen, China, with identification results optimally validated through integration with mNGS findings. This study provides strong support for the application of tNS technology for NTM species identification.
Keywords: non-tuberculous mycobacteria, targeted nanopore sequencing, species identification, evaluation
Introduction
Non-tuberculous mycobacteria (NTM) represent a significant and diverse group of mycobacteria that excludes both the Mycobacterium tuberculosis complex and Mycobacterium leprae. To date, more than 190 distinct NTM species have been identified.1 NTM are capable of infecting various human tissues, including the lungs, soft tissues, and lymph nodes, with pulmonary infections constituting the most common clinical manifestation and often presenting with symptoms that closely mimic those of tuberculosis.2 A comprehensive systematic review3 encompassing 18 countries, 47 independent studies, and 285,681 NTM isolates demonstrates that 82% of included studies document an increasing trend in NTM infection rates, while 68% report a corresponding rise in the incidence of NTM-related diseases. The estimated annual variation rates for global NTM infections and associated diseases are 4.0% and 4.1%, respectively. Notably, Mycobacterium avium complex (MAC) exhibits the most pronounced upward trajectory, with 78.9% and 83.3% of studies confirming escalating infection rates for this complex.3,4
High-risk populations for NTM infections include individuals with compromised immune function (such as those living with HIV, organ transplant recipients, and patients receiving long-term immunosuppressive therapy), individuals with underlying structural lung damage (including those with bronchiectasis or cystic fibrosis), elderly populations, and chronic smokers. As the global demographic continues to age and the prevalence of immunocompromised individuals increases, the pool of susceptible populations is projected to expand substantially. Furthermore, the management of NTM infections necessitates prolonged treatment regimens (frequently exceeding 12 months), involves multiple antimicrobial agents, carries a high risk of adverse drug reactions, and often requires prohibitively expensive novel therapeutic agents. Consequently, treatment costs far exceed those associated with typical pulmonary infections, thereby imposing a substantial economic burden on healthcare systems worldwide. This challenge is particularly acute in low- and middle-income countries, where the diagnostic and therapeutic expenses associated with NTM infections represent a critical constraint on medical resource allocation and contribute significantly to escalating healthcare expenditures.2–4
Currently, NTM infections have emerged as a major global public health threat, with their clinical impact demonstrating distinct regional epidemic characteristics across all continents, including China.3,4 For instance, in North America, MAC represents the predominant etiological agent of NTM pulmonary disease, accounting for 51–85% of all NTM isolates. In Europe, MAC and M. kansasii constitute the primary causative agents of NTM pulmonary disease, with MAC comprising 22–82% of cases. Asia, particularly East Asia (encompassing South Korea, Japan, mainland China, and the Taiwan region of China), has emerged as a high-incidence region for NTM infections. Beyond MAC (representing 34–76% of isolates) as the dominant species in this region, the prevalence of Mycobacterium abscessus complex (MAB) is notably higher than in other continents. In certain areas, such as southern Taiwan, MAB has become the predominant pathogenic species. As the nation bearing the third-highest tuberculosis burden globally, China has witnessed a dramatic escalation in NTM isolation rates, rising from 4.3% in 1979 to 11.1% in 2000, and further increasing to 22.9% in 2010, with continued upward trends observed throughout the past decade.5 A comprehensive systematic review and meta-analysis spanning 2013−20245 revealed that the co-infection rate of NTM among suspected tuberculosis patients in China was 11.27%, with marked regional heterogeneity. The prevalence was highest in Northeast China (24.18%), followed by southeastern coastal regions (12.83%), and lowest in Southwest China (2.30%). Regarding species distribution, slow-growing mycobacteria account for 68.07% of NTM isolates in China, predominantly MAC (particularly M. intracellulare), while rapidly growing mycobacteria comprise 26.57%, primarily M. abscessus, with higher prevalence observed in southern provinces. Additionally, the Taiwan region demonstrates relatively elevated NTM infection rates, with MAB as the dominant species, paralleling epidemic patterns observed in southern regions of mainland China.
Evidence indicates5 that cities along China’s southern coastal regions, such as Shenzhen, face heightened risk of NTM infections due to warm, humid climatic conditions and substantial population mobility. MAC and M. abscessus constitute the principal pathogenic species in these geographic areas. Given the continuous expansion of high-risk populations for NTM infections, coupled with elevated resistance rates among rapidly growing mycobacteria such as M. abscessus, therapeutic management has become progressively more challenging. Moreover, the persistent high rate of misdiagnosis for NTM infections underscores the critical importance of enhancing differential diagnostic accuracy to optimize clinical management. Species-level identification constitutes a fundamental component of this differential diagnostic process. Conventional methods for distinguishing NTM are labor-intensive and time-consuming, frequently requiring molecular biological techniques to achieve precise species-level characterization. Advances in contemporary sequencing methodologies (encompassing both second- and third-generation platforms) have created novel opportunities for NTM species identification. Among these approaches, targeted nanopore sequencing (tNS), which integrates the advantages of targeted amplification with nanopore sequencing technology, demonstrates rapid detection capability, high analytical sensitivity, and robust specificity. This platform has exhibited excellent performance in the detection of pulmonary infectious diseases.5–7
Based on this context, the present study focuses on the Shenzhen metropolitan area (a representative region along China’s southeastern coast) to comparatively analyze the performance differences among tNS, metagenomic sequencing (mNGS), and Sanger sequencing in NTM species identification. The objectives are to explore the clinical application potential of tNS, provide technical support for rapid and accurate diagnosis of NTM infections in China, enhance the diagnostic and therapeutic standards for NTM infections nationwide, and ultimately mitigate the burden of these infections on public health systems and healthcare infrastructure.
Materials and Methods
Strain Collection
This study retrospectively collected a total of 50 strains of suspected NTM isolated from drug-resistant tuberculosis surveillance conducted in 10 districts of Shenzhen, China, between December 2024 and June 2025. The species identification was initially performed using fluorescent PCR probe melting curve analysis, which provided preliminary identification of the NTM species. The identified strains included M. intracellular (3 strains), M. lentiflavum (2 isolates), a mixed M. tuberculosis and unidentified NTM strain (1 isolate), Mycobacterium kansasii (1 isolate), Mycobacterium abscessus (13 isolates), Mycobacterium fortuitum (6 isolates), and 24 isolates identified as unclassified NTM. This study is a secondary analysis of retrospective samples and data from the China Shenzhen MTBC Drug-Resistant Surveillance (DRS) program, which was routinely conducted without collecting additional data or samples. This trial has been approved by the Ethics Committee of the Shenzhen Chronic Disease Prevention Center.
Sample Preparation
The retrospective samples were cultured on neutral Roche solid media, and once colonies appeared, total genomic DNA was extracted using a Cetyltrimethy -lammonium Bromide(CTAB)-based method. Briefly, 200 µL of Tris-EDTA buffer was added to the samples, which were then heat-treated at 85°C to lyse the bacteria. After incubation at −20°C for 15 minutes, 40 µL of lysozyme (20 mg/mL) was added. Following a 1-hour incubation at 37°C, the bacterial cell membranes and proteins were digested using 1% sodium dodecyl sulfate (SDS) and proteinase K (final concentration 250 µg/mL). To separate the aqueous and organic phases, 1% N-acetyl-N, N, N-trimethylammonium bromide reagent and NaCl were added, followed by DNA precipitation with ethanol. The extracted DNA was stored at −20°C for subsequent analyses.
Targeted Nanopore Sequencing
Genomic DNA was extracted from NTM cultures using the R9 Mycobacterium Identification and Antimicrobial Resistance Detection Kit (Invision BioTech, Zhejiang, China) according to the manufacturer’s protocol. The extracted DNA was sent to Guangzhou Dian Medical Laboratory (Guangzhou, China) for targeted amplification of the hsp65 gene fragment as the basis for species identification. In short, an equivalent of 100 ng of the prepared library was loaded onto the GridION (Mk1) platform for sequencing. MinKNOW23.07.5 (focal) software was used to collect raw electronic signal data. The raw sequencing reads were filtered out for quality, removing sequences shorter than 200 bp and eliminating host DNA reads by aligning them to the human reference genome (GRCh38). Then, the remaining filtered reads were aligned to the mycobacteria and drug resistance gene databases (TBDReaMDB, MUBII-TB-DB, and RESEQTB.ORG), generating the analysis results (analysis platform Nano TNGS V1.0, database NanoTarget DB V1.0).6
Metagenomic Sequencing
Genomic DNA was fragmented randomly using a Covaris ultrasonicator and then subjected to end repair, A-tailing, adapter ligation, purification, and PCR amplification to complete the library preparation. After library construction, preliminary quantification was performed using the Qubit 2.0 fluorometer, followed by library dilution. The insert size distribution was assessed using the Agilent 2100 Bioanalyzer. Once the insert size met the required specifications, the library concentration was adjusted for sequencing, and high-throughput sequencing was carried out on the Illumina NovaSeq 6000 platform. Species identification was performed using Kraken27–11 and NTM-profiler (https://bioinformatics.lshtm.ac.uk/ntm-profiler). NTM-profiler utilizes the ANI (Average Nucleotide Identity) algorithm from GTDB-Tk12 to calculate the ANI between the assembled genome and reference sequences from the NTM-DB database, outputting NTM species with an ANI value greater than 0.90. The initial species determination was based on the Kraken2 results to confirm whether Mycobacterium species were the dominant species (with the highest proportion) or present at a level greater than 5%. If Mycobacterium was identified, species identification was further refined using the ANI values output by NTM-profiler. When Kraken2 indicated Mycobacterium as the predominant species, NTM species with an ANI greater than 0.95 were considered detected. A more relaxed ANI threshold (≥ 0.90) was applied if Mycobacterium was not the predominant species but still present.
Sanger Sequencing
The 16S rRNA gene was amplified using specific primers designed for its conserved regions (Primers: 16S rRNA-F: AGAGTTTGATCMTGGCTCAG; 16S rRNA-R: CCGTCAATTCMT TTRAGTTT; Fragment length: 1536 bp). The amplification products were sent to Jingnuo Biotechnology Co., Ltd. (Shanghai, China) for Sanger sequencing. The resulting sequences were compared against the NTM-DB database (https://github.com/pathogen-profiler/ntm-db) to confirm the species identification.
Statistical Analysis
The analysis pipeline was implemented using Python 3.6, with additional performance metrics, including Jaccard index, kappa coefficient, and F1 score, computed using scikit-learn 0.24.2, and mean values were calculated on a per-sample basis. A paired χ2-test was employed to assess the differences in detection rates between the two diagnostic methods. Wilson’s method was used to compute 95% confidence intervals. All statistical analyses were conducted using SPSS 23.0, and a two-tailed P< 0.05 was considered statistically significant.
Results
Distribution of Primary Experimental Species Identified by Three Sequencing Methods
Among the 50 samples subjected to analysis, one specimen failed amplification due to suboptimal nucleic acid quality, precluding Sanger sequencing, whereas all remaining samples were successfully sequenced and satisfied quality control criteria. The distribution of primary experimental species identified by the three sequencing methodologies is summarized in Table 1. The most frequently detected species was M.abscessus, followed by M.fortuitum, both of which represent rapidly growing NTM species. Notably, tNS detected M. tuberculosis in 4 initial screening samples of NTM, while mNGS identified M. tuberculosis in 12 initial screening samples of NTM.
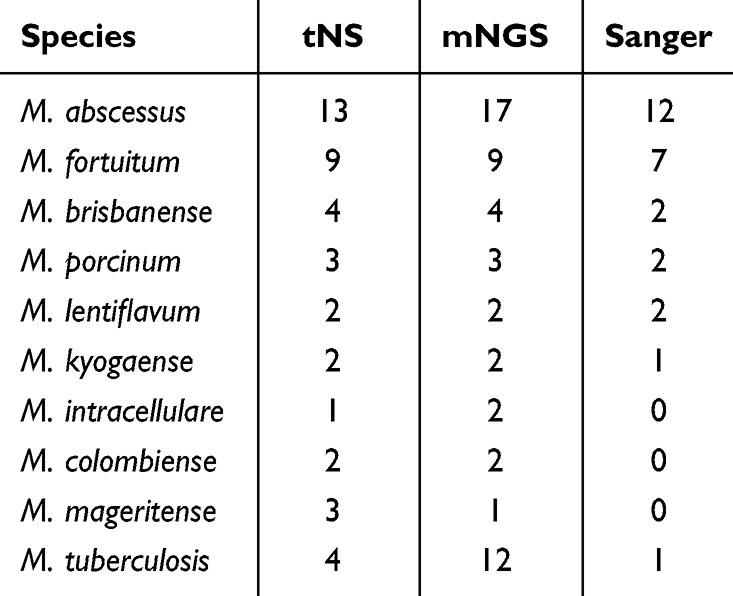 |
Table 1 Distribution of Primary Experimental Species Identified by Three Sequencing Methods |
Consistency Analysis of NTM Species Identification by Three Sequencing Methods
Among the 50 suspected NTM samples analyzed, complete concordance across all three sequencing methods (tNS, mNGS, and Sanger sequencing) for full species identification was observed in 18 samples (36%). The highest degree of consistency was demonstrated between tNS and mNGS, with both methods yielding identical species identification results in 28 samples (56%) (see Figure 1 and Supplemental Table S1). For NTM samples exhibiting discordant results, high-throughput sequencing methods (tNS and mNGS) demonstrated superior reproducibility in species identification. In contrast, Sanger sequencing failed to accurately identify 13 samples (26%) due to complications arising from overlapping sequence peaks. In cases involving mixed-species NTM samples, tNS identified 9 samples, whereas mNGS detected 14 samples. The identification results exhibited some variation; however, this difference was not statistically significant (χ2 = 1.08, P > 0.05) (see Supplemental Table S1).
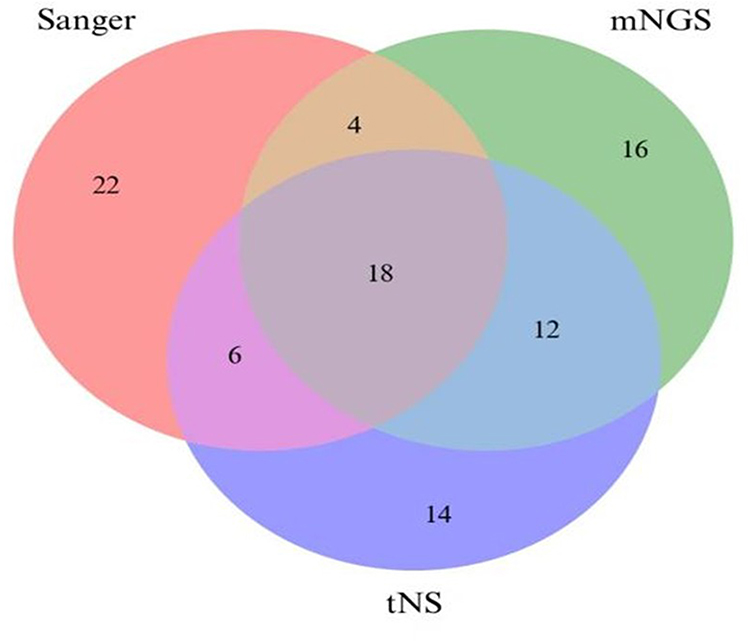 |
Figure 1 Comparison of Full Species Identification Results by Three Sequencing Methods. |
Given that NTM species function as opportunistic pathogens that are abundantly present in environmental reservoirs and demonstrate substantial species diversity, the clinical significance of low-abundance, diverse NTM species detected in clinical specimens remains uncertain. Therefore, we prioritized comparison of identification results for the predominant NTM species present in the samples. This focused analysis revealed that 28 samples (56%) demonstrated consistent results across all three sequencing methods for the primary species, representing a higher concordance rate than that observed in the full-species analysis (18 samples, 36%). However, this difference did not reach statistical significance (χ2 = 2.32, P > 0.05). Notably, tNS and mNGS yielded concordant results for 46 samples, representing 92% of the total sample set. These findings indicate a high level of agreement between high-throughput sequencing methodologies in identifying dominant NTM species (see Figure 2).
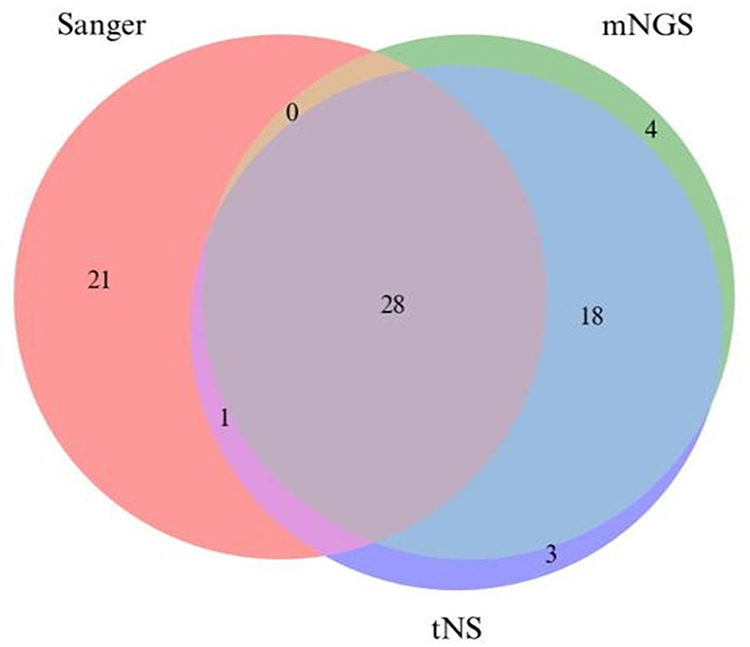 |
Figure 2 Comparison of Main Species Identification Results by Three Sequencing Methods. |
Performance Analysis of Three Sequencing Methods in NTM Species Identification
A comprehensive evaluation of sequencing results was conducted to establish definitive species identification outcomes for the 50 suspected strains, which served as reference standards for this comparative investigation (see Supplemental Table S1). Among these isolates, the species identification results and reference results for 32 strains with controversial detection outcomes are detailed in Supplemental Table S2. Comparisons between the total species identification results and major species identification results obtained by the three sequencing methods against reference standards were performed, with relevant statistical parameters presented in Table 2 and Table 3. Given that the primary objective of this investigation was to evaluate the NTM species differentiation capacity of distinct sequencing methodologies, statistical metrics unaffected by true negative results were selected for analysis. These included Precision, Recall, the F1-score (a composite metric derived from Precision and Recall), and the Jaccard coefficient (commonly employed in multi-label classification tasks to quantify similarity between detected and actual results).
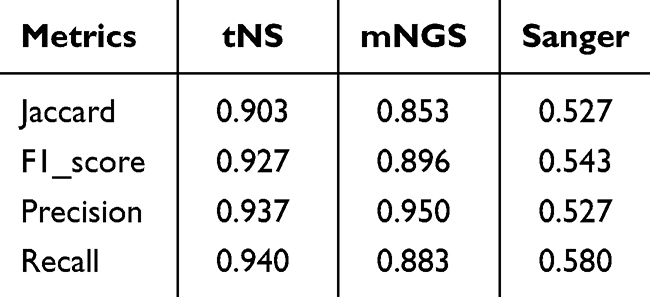 |
Table 2 Consistency Metrics of Full Species Identification Results by Three Sequencing Methods Compared with Reference Results |
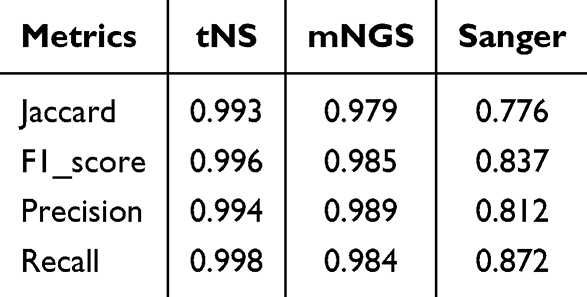 |
Table 3 Consistency Metrics of Main Species Identification Results by Three Sequencing Methods Compared with Reference Results |
Overall, tNS exhibited optimal performance across all evaluated metrics. Its Precision for total species identification was 0.937, marginally lower than that of mNGS (0.950), while achieving superior performance in all remaining indicators. Sanger sequencing failed to identify mixed species throughout this investigation and remained compromised by identification failures resulting from overlapping peaks caused by species mixtures, thereby demonstrating suboptimal performance in NTM species identification. The overall performance of mNGS was slightly inferior to that of tNS, which was primarily attributable to detection sensitivity associated with analytical algorithms—specifically, differential weighting assigned to two parameters (sequence proportion and sequence consistency) could yield divergent detection outcomes. However, the performance differential between these two detection methodologies did not achieve statistical significance in this study.
Additionally, we conducted further analysis of the sensitivity, specificity, accuracy (ACC), and kappa values of the three sequencing methods for the predominant bacterial species (comprising 9 distinct NTM species). Detailed statistical parameters are presented in Table 4. Detection performance among the three bacterial species with relatively elevated detection frequencies (M. abscessus, M. fortuitum, and M. tuberculosis) demonstrated significant variations. For M. abscessus, tNS achieved a sensitivity of 92.86% (95% CI: 68.53–98.73), specificity of 100.00% (95% CI: 90.36–100.00), ACC of 98.00% (95% CI: 89.50–99.65), and a kappa value of 0.949. The kappa values for mNGS and Sanger sequencing were 0.860 and 0.896, respectively, with all three detection methods demonstrating high identification concordance. For M. fortuitum, both tNS and mNGS achieved 100.00% sensitivity, specificity, and ACC, with kappa values of 1.000, indicating excellent identification consistency. However, Sanger sequencing demonstrated a sensitivity of only 66.67% (95% CI: 35.42–87.94) and a kappa value of 0.703, reflecting relatively low detection efficiency. For M.tuberculosis, all three detection methods exhibited low kappa values. mNGS achieved the highest sensitivity (71.43%, 95% CI: 35.89–91.78) but demonstrated limited specificity of only 83.72% (95% CI: 70.03–91.88) and a kappa value of 0.425. tNS achieved a sensitivity of 57.14% (95% CI: 25.05–84.18), specificity of 100.00%, and a kappa value of 0.696. Sanger sequencing yielded the lowest sensitivity (14.29%, 95% CI: 2.57–51.31) and a kappa value of merely 0.223, with all three detection methods demonstrating poor identification concordance.
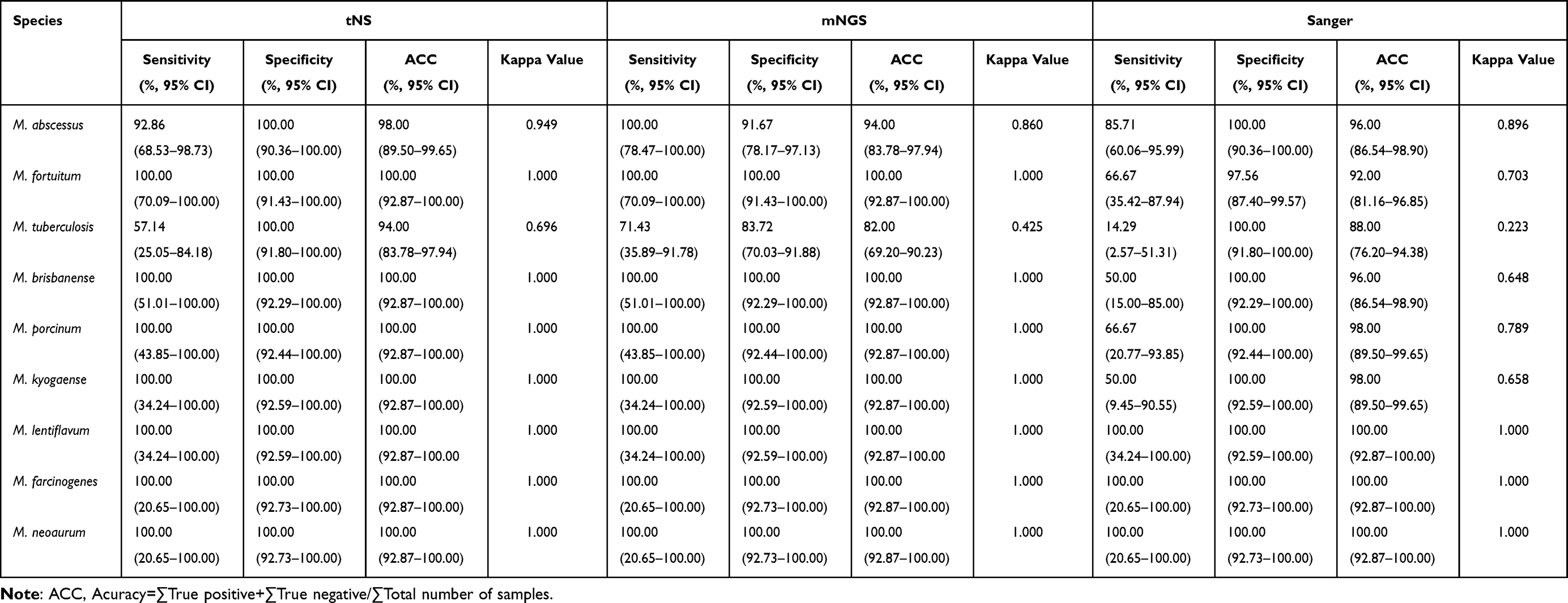 |
Table 4 Performance Metrics Analysis of Three Molecular Detection Methods Compared with Reference Results |
Discussion
In China, the distribution of NTM exhibits pronounced geographical heterogeneity. For instance, coastal provinces such as Guangdong and Zhejiang demonstrate higher NTM species diversity, whereas inland regions exhibit comparatively lower diversity. This regional variation may be attributable to factors including frequent exposure to surface water sources and elevated urbanization levels in southeastern coastal areas, which collectively facilitate the transmission and clinical detection of multiple NTM species.4,5
In the present investigation, 50 suspected NTM isolates (comprising 26 known species and 24 uncharacterized strains) were selected for methodological evaluation utilizing three distinct molecular sequencing technologies. The sample size conforms to established clinical research specifications for methodological evaluation of NTM molecular detection platforms (comparative studies of species identification methodologies typically incorporate 30–200 isolates),13–17 thereby enabling calculation of robust statistical performance metrics including kappa values and F1-scores. Moreover, the composition of the included strains takes both known and unknown NTM species into account (each accounting for approximately 50%), which is considered to effectively evaluate the discriminatory ability of the three sequencing technologies. Specifically, it can test the identification performance of tNS and mNGS for unknown species, avoiding result bias caused by evaluating only known species.
Among the 50 suspected NTM samples examined in this study, full species identification results revealed that only 18 samples (36%) demonstrated complete concordance across all three sequencing methods. Upon re-evaluation and comparison of the predominant NTM species present in the samples, 28 samples (56%) exhibited consistent identification results across all three sequencing methods, representing a modest improvement over the concordance rate observed in full-species analysis; however, this difference did not achieve statistical significance (χ2 = 2.32, P > 0.05). While the overall concordance among the three sequencing methods remained relatively modest, tNS demonstrated superior performance across all evaluation metrics (Precision, Recall, F1-score, and Jaccard coefficient). We attribute these findings primarily to inherent differences in detection principles and analytical algorithms among the three sequencing methodologies.6,13–18 As a conventional first-generation sequencing technology, Sanger sequencing operates on the principle of dideoxy chain termination and is restricted to sequencing individual gene fragments (the 16S rRNA gene in this investigation), rendering it highly susceptible to interference from overlapping chromatogram peaks. When samples contain mixed species or exhibit gene fragment polymorphisms, this approach is prone to generating ambiguous peak patterns and alignment failures (the failure rate for Sanger sequencing in this study reached 26%), resulting in discordant results compared with tNS and mNGS. These findings align with conclusions from related studies14–16 demonstrating that Sanger sequencing exhibits limited efficacy in identifying mixed species and low-abundance organisms. As a second-generation high-throughput sequencing technology, mNGS performs untargeted sequencing of the entire genomic complement within samples. During analysis, Kraken2 is initially employed to identify the most abundant mycobacterial species, followed by confirmation of specific NTM taxa through reference to average nucleotide identity (ANI) values calculated by NTM-profiler. While this approach enables detection of mixed species, it remains susceptible to influences from sequencing depth, sequence alignment algorithms, and sequence proportion weighting parameters, potentially leading to misidentification of low-abundance mycobacteria as target strains or generating discrepancies with tNS identification results due to variations in ANI consistency threshold settings.16–19 As a technology integrating third-generation nanopore sequencing with targeted amplification, tNS exclusively performs targeted amplification and sequencing of the hsp65 gene (a primary discriminatory locus for NTM species identification). Its analytical algorithm relies solely on sequence proportion as the species determination criterion. Although this approach enhances specificity, it neglects consideration of sequence consistency, and the inherently lower per-read accuracy characteristic of nanopore sequencing may facilitate errors in gene fragment alignment. These observations are consistent with findings from related references15–20 indicating that tNS identification results are substantially influenced by target gene selection and single-read accuracy.
Furthermore, this investigation revealed poor concordance between tNS and mNGS in identifying M. tuberculosis (with kappa values of 0.696 and 0.425, respectively). The primary explanation lies in divergent identification criteria between mNGS and tNS. Specifically, mNGS integrates both sequence proportion and sequence consistency with greater weighting assigned to the latter parameter, whereas tNS utilizes sequence proportion as the sole determinant. Given that this investigation focused on NTM species, the sequence data for Mycobacterium tuberculosis in the reference database employed were relatively limited.17–19 Additionally, the lower single-read sequencing accuracy inherent to tNS, combined with reliance on a single gene fragment for strain identification, increases susceptibility to alignment errors, thereby reducing the success rate of sequence alignment for Mycobacterium tuberculosis.19–21 Conversely, for the two other NTM species exhibiting higher detection frequencies, the kappa values for identification results obtained by second-generation and third-generation sequencing methods both exceeded 0.86, indicating that these sequencing approaches demonstrate high accuracy in identifying common NTM species.16–20
Based on the design and findings of this investigation, we acknowledge several limitations inherent to this research. First, this study exclusively selected 50 suspected NTM isolates obtained from drug-resistant tuberculosis surveillance programs across 10 districts of Shenzhen, China. The sample sources are restricted to populations associated with drug-resistant tuberculosis and do not encompass isolates from other high-risk groups for NTM infections, such as patients with common pulmonary infections and immunocompromised individuals. The relatively limited sample size may compromise the generalizability of research findings, potentially hindering extrapolation to NTM strain identification in other geographic regions or population cohorts nationwide. Second, the extremely low proportion of rare NTM species resulted in insufficient statistical power for evaluation of these uncommon taxa. Additionally, all analyses were conducted using pure cultures rather than direct clinical specimens. Factors including host DNA interference and low pathogen burden in actual clinical samples may substantially influence sequencing performance. Finally, this investigation focused exclusively on evaluations addressing the core objective of species identification and did not analyze the efficacy of the three sequencing methods in detecting NTM resistance genes. In clinical practice, NTM sequencing technology is required not only for species identification but also to provide guidance for antimicrobial resistance-informed treatment strategies. However, this study did not incorporate comparative analysis of resistance gene detection; therefore, the research scope remains incomplete. Future studies should encompass multicenter, large-sample prospective investigations to further validate the predictive efficacy of the three sequencing methods for diverse NTM species and resistance gene detection, while comprehensively exploring the advantages, limitations, and clinical application value of each approach.20,21
In summary, tNS represents a preferred auxiliary methodology for clinical identification of NTM isolates in Shenzhen, China, with identification results optimally validated through integration with mNGS findings. This study provides strong support for the application of tNS technology for NTM species identification.
Ethics Approval Statement
This study was approved by the Ethics Committee of Shenzhen Chronic Disease Prevention Center (SZCCC-2025-004-01-PJ). In addition, the Declaration of Helsinki was followed during our research. The necessary standards and legislation were followed in the execution of all procedures, including the declarations in “Declarations” section.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Clinical Research Center for Infectious and the Key Projects of Huizhou City’s Science and Technology Program (2023CZ01000 8).
Disclosure
The authors have declared that they have no conflicts of interest in this work.
References
1. Chinese Medical Association, Tuberculosis and Respiratory Diseases Branch. Guidelines for the diagnosis and treatment of non-tuberculous mycobacterial diseases (2020 Edition). Chinese Journal of Tuberculosis and Respiratory Diseases. 2020;43:918–11. doi:10.3760/cma.j.cn112147-20200508-00570
2. Tan Y, Deng Y, Yan X, et al. Nontuberculous mycobacterial pulmonary disease and associated risk factors in China: a prospective surveillance study. J Infect. 2021;83:46–53. doi:10.1016/j.jinf.2021.05.019
3. Dahl VN, Molhave M, Floe A, et al. Global trends of pulmonary infections with nontuberculous mycobacteria: a systematic review. Int J Infect Dis. 2022;125:120–131. doi:10.1016/j.ijid.2022.10.013
4. Hamed KA, Tillotson G. A narrative review of nontuberculous mycobacterial pulmonary disease: microbiology, epidemiology, diagnosis, and management challenges. Expert Rev Respir Med 2023;. 17(11):973–988. doi:10.1080/17476348.2023.2283135
5. Xu X, Lei Y, Zheng L. Non-tuberculous mycobacterial infections in mainland China and Taiwan: a systematic review and meta-analysis of epidemiology, species distribution, and drug resistance (2013–2024). Front Public Health. 2025;13:1676715. eCollection 2025. doi:10.3389/fpubh.2025.1676715
6. ping CL, Wang L, Fei WY, et al. Nanopore-targeted sequencing: a new and effective technique for the diagnosis of non-tuberculous mycobacteria pulmonary disease. Int J Med Microbiol. 2025:320. doi:10.1016/j.ijmm.2025.151663
7. Sun L, Zhang K, Liu Y, et al. Metagenomic next-generation sequencing targeted and metagenomic next-generation sequencing for pulmonary infection in HIV-infected and non-HIV-infected individuals. Front Cell Infect Microbiol. 2024;14:1–9. doi:10.3389/fcimb.2024.1438982
8. Schenk JJ, Becklund LE, Carey SJ, Fabre PP. What is the “modified” CTAB protocol? Characterizing modifications to the CTAB DNA extraction protocol. Appl Plant Sci. 2023;11:1–11. doi:10.1002/aps3.11517
9. Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90. doi:10.1093/bioinformati-cs/bty560
10. Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. Using SPAdes De Novo Assembler. Curr Protoc Bioinforma. 2020. doi:10.1002/cpbi.102
11. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:1–13. doi:10.1186/s13059-019-1891-0
12. Parks DH, Chuvochina M, Rinke C, Mussig AJ, Chaumeil P-A, Hugenholtz P. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022;50:D785–94. doi:10.1093/nar/gkab776
13. De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34:2666–2669. doi:10.1093/bioinformatics/bty149
14. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi:10.1093/bioinformatics/bty191
15. Zhang H, Tang M, Li D, Xu M, Ao Y, Lin L. Applications and advances in molecular diagnostics: revolutionizing non-tuberculous mycobacteria species and subspecies identification. Front Public Heal. 2024;12. doi:10.3389/fpubh.2024.1410672
16. Matsumoto Y, Kinjo T, Motooka D, et al. Comprehensive subspecies identification of 175 nontuberculous mycobacteria species based on 7547 genomic profiles. Emerg Microbes Infect. 2019;8:1043–1053. doi:10.1080/22221751.2019.1637702
17. Zheng J, Zhang H, Banerjee S, et al. A comprehensive assessment of Next-Generation Sequencing variants validation using a secondary technology. Mol Genet Genomic Med. 2019;7:1–7. doi:10.1002/mgg3.748
18. Wang S, Xing L. Metagenomic next-generation sequencing assistance in identifying non-tuberculous mycobacterial infections. Front Cell Infect Microbiol. 2023;13:1–11. doi:10.3389/fcimb.2023.1253020
19. Yang C, Wang T, Guo Y, Zeng Y, Gao W. Nanopore-targeted sequencing (NTS) for intracranial tuberculosis: a promising and reliable approach. Ann Clin Microbiol Antimicrob. 2024. doi:10.1186/s12941-024-00751-x
20. Dhasmana DJ, Whitaker P, van der Laan R, Frost F. A practical guide to the diagnosis and management of suspected non-tuberculous mycobacterial pulmonary disease (NTM-PD) in the United Kingdom. Npj Prim Care Respir Med. 2024;34. doi:10.1038/s41533-024-00403-9
21. Daley CL, Iaccarino JM, Lange C, et al. Treatment of nontuberculous mycobacterial pulmonary disease: an official ATS/ERS/ESCMID /IDSA clinical practice guideline. Eur Respir J. 2020;56. doi:10.1183/13993003.00535-2020
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Evaluation of the MeltPro Myco Assay for the Identification of Non-Tuberculous Mycobacteria
Lin J, Zhao Y, Wei S, Dai Z, Lin S
Infection and Drug Resistance 2022, 15:3287-3293
Published Date: 22 June 2022