Back to Journals » Drug Design, Development and Therapy » Volume 10
Per-residue energy decomposition pharmacophore model to enhance virtual screening in drug discovery: a study for identification of reverse transcriptase inhibitors as potential anti-HIV agents
Authors Cele N, Ramesh M, Soliman M
Received 1 September 2015
Accepted for publication 1 December 2015
Published 11 April 2016 Volume 2016:10 Pages 1365—1377
DOI https://doi.org/10.2147/DDDT.S95533
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Favourite N Cele, Muthusamy Ramesh, Mahmoud ES Soliman
Molecular Modelling and Drug Design Research Group, School of Health Sciences, University of KwaZulu-Natal, Durban, South Africa
Abstract: A novel virtual screening approach is implemented herein, which is a further improvement of our previously published “target-bound pharmacophore modeling approach”. The generated pharmacophore library is based only on highly contributing amino acid residues, instead of arbitrary pharmacophores, which are most commonly used in the conventional approaches in literature. Highly contributing amino acid residues were distinguished based on free binding energy contributions obtained from calculation from molecular dynamic (MD) simulations. To the best of our knowledge; this is the first attempt in the literature using such an approach; previous approaches have relied on the docking score to generate energy-based pharmacophore models. However, docking scores are reportedly unreliable. Thus, we present a model for a per-residue energy decomposition, constructed from MD simulation ensembles generating a more trustworthy pharmacophore model, which can be applied in drug discovery workflow. This work is aimed at introducing a more rational approach to the field of drug design, rather than comparing the validity of this approach against those previously reported. We recommend additional computational and experimental work to further validate this approach. This approach was used to screen for potential reverse transcriptase inhibitors using the pharmacophoric features of compound GSK952. The complex was subjected to docking, thereafter, MD simulation confirmed the stability of the system. Experimentally determined inhibitors with known HIV-reverse transcriptase inhibitory activity were used to validate the protocol. Two potential hits (ZINC46849657 and ZINC54359621) showed a significant potential with regard to free binding energy. Reported results obtained from this work confirm that this new approach is favorable in the future of the drug design industry.
Keywords: HIV-1, reverse transcriptase, GSK952, molecular dynamic simulations, pharmacophore model, molecular docking
Introduction
HIV infection is the leading cause of death across the globe.1 There are two strains of HIV, namely, HIV-1 and HIV-2. HIV-1 is the most infectious and prevalent globally.2 Worldwide statistics by the American foundation for AIDS Research reported that sub-Saharan Africa is the most affected region, with approximately 70% of adults and 91% of children HIV positive.3
HIV-1 reverse transcriptase (RT) is currently an essential target for US Food and Drug Administration (FDA) approved HIV-1 therapy4 and a prominent target of many approved anti-HIV drugs that are key components of highly active antiretroviral therapies.5 The HIV-1 RT enzyme catalyzes the conversion of viral RNA into cDNA, which enters the host nucleus and is incorporated into host chromosomal DNA of the host cell by the IN enzyme.6 It is the sole viral enzyme required for the catalytic formation of cDNA generated from viral RNA, hence playing a central role in HIV replication,7 making it a prime target for HIV-1 therapy. HIV-1 RT is a heterodimer consisting of two subunits; a p66 subunit (DNA polymerization site and RNase H active site), which is responsible for the replication of the single-stranded RNA genome found in virions into the double-stranded DNA; and a p51 subunit, which is responsible for the proper folding of p66 rather than enzymatic activities subunit (Figure 1).8
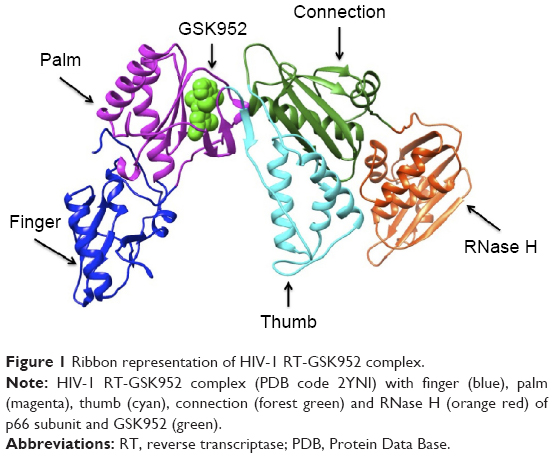 | Figure 1 Ribbon representation of HIV-1 RT-GSK952 complex. |
HIV-1 RT does not possess any proofreading activity. Thus, DNA synthesis prone to errors can be carried out by HIV-1 RT, resulting in a higher mutation rate and the production of multiple HIV variants.2 Etravirine and rilpivirine are the most recent non-nucleoside reverse transcriptase inhibitors (NNRTIs) to which mutated RT viruses are resistant.9 To date, NNRTIs’ resistance still poses a challenge with regard to NNRTI therapy. Thus, there is a clear need for the discovery of new drugs with greater resistance profiles capable of inhibiting HIV-I RT mutated viruses. Numerous studies have made significant attempts in the discovery of new potent NNRTIs using pharmacophore model approaches.10–12
The potential anti-HIV drug needs to be effective against resistant strains. The present study looked at GSK952, a newly discovered NNRTI. The crystal structure (Protein Data Base [PDB] 2YNI)8 of the catalytic domain of HIV-1 RT enzyme bound to GSK952 is available. GSK952 shows high potency against the mutated Y188L strain as well as the mutated Y188C and K103N strains. Previous work on this compound confirmed it has a good inhibitory activity against HIV-1 RT, including the mutated viral Y188L HIV-1 RT strains.8 It exhibited a high antiviral profile when compared to a group of compounds sharing a common imidazole-amide biaryl ether scaffold.8 It possesses a linear arrangement of hydrogen bond acceptors (C=O) and donors (NH), which are required for binding to HIV-1 RT and to form hydrogen bonds with residues in the active site, including K101, K103, and P236.13 GSK952 forms hydrogen bonds with NH and C=O groups (Figure S1), which are positioned along the protein backbone rather than the side chains. This could explain the inhibitory activity against the K103N mutation.14
In this mutation, the side chain of K103 is tilted away from the inhibitor due to the mutation of the wild type to Asn103.8 GSK952 (Figure 2) has revealed an extraordinary antiviral activity against a wide range of NNRTI-resistant viruses8 and forms favorable pharmacokinetic profiles across multiple species. We believe it has great potential and deserves further exploration. Thus, it is an ideal compound for the current study.
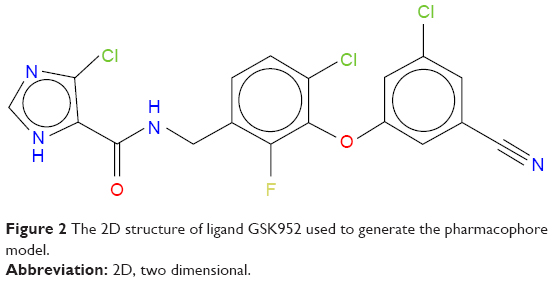 | Figure 2 The 2D structure of ligand GSK952 used to generate the pharmacophore model. |
Computational tools and capability escalation have led to virtual screening (VS) becoming a routine method in pharmaceutical drug discovery.15 Numerous computational screening tools are accessible for mining of inhibitors with properties of interest.16 VS is one of the most trusted and convenient tools in drug design. Literature confirms its reliability in the discovery of novel HIV-1 RT inhibitors through VS.17 VS is either ligand based or structure based.18 Structure-based VS uses the three-dimensional (3D) structure of the receptor to search for potential ligands.19 Ligand-based VS (LBVS) explores features or properties of known bioactive ligands and searches for compounds with similarities.20 In LBVS, also known as pharmacophore-based VS, favorable features of a known active site are used to build a pharmacophore model. Among other models, pharmacophore searches are best at discovering a range of chemical structures with feasible features, hence the principal method for the initial selection of compounds.21 Ligand-based pharmacophore approaches generate libraries based on a set of known ligands illustrative of crucial interactions between the ligands and a particular target;22 whereas structure-based pharmacophore models are based on the knowledge of the 3D structure of the target.23 Numerous studies have combined LBVS and structure-based VS with the aim of improving the VS process.24,25 A number of studies have attempted to improve VS and pharmacophore models.26–30 A previous study proposed a target-bound generated pharmacophore model to further improve pharmacophore-based VS. It has been confirmed that target-bound pharmacophore-based VS is a more rational approach.19 Yet to this date, general standards for VS with regard to method evaluation are insufficient. The aim of this work is to identify more potential NNRTIs by exploiting the structural features of GSK952 using a pharmacophore model.
We propose implementing an approach that aims to improve and refine the current pharmacophore approach. This approach is centered on the type of interactions witnessed at a molecular level, which includes hydrogen bonding, charge, and hydrophobic interactions (HPIs).31 This approach will search for compounds that interact with the highly contributing residues based on the free binding energy (FBE).
In this study, we performed molecular dynamic (MD) simulations, pharmacophore-based VS and per-residue energy decomposition (PRED) analysis for residues with the greatest FBE contributions. VS depends primarily on docking calculations, although results based exclusively on docking calculations are rather questionable.32 To test the validity of our proposed approach, we applied the same docking procedure to a set of experimentally determined inhibitors with known HIV-1 RT inhibition activity. In our approach, we intend to unshackle the limitations of previous approaches, such as using noncrucial residues, which retrieves a large number of hits with assumed activity. In an attempt to enhance the accuracy of the pharmacophore model, we only selected highly contributing amino acid residues (HCAAR). A target-bound ensemble was employed in the current study, in the hope of implementing a better approach. A pharmacophore model was created using HCAAR in the protein’s active site, whereas conventional methods use pharmacophore maps created without considering the energy contributions of interacting residues. Only potential pharmacophore moieties were considered; thus, the library of potential compounds generated is more concise and direct. In our approach, the energy-based pharmacophore map is generated using the FBE calculated from MD, which is a more reliable approach, compared to the conventional approach using the docking score. We believe this approach will be more effective than the conventional approach due to these key factors highlighted earlier. The refinement of the method proposed in this study could be implemented as a potential tool for medicinal chemists in the search for more potent HIV-1 RT inhibitors.
Computational methodology
The computational tools implemented in this study are represented in the workflow in Figure 3. A diagrammatic representation of the pharmacophore model used in this study is shown in Figure 4. The residues with the highest energy contributions are numbered and ranked from 1 to 6, with 1 being the highest FBE contributing residue (Figure 4). The highest ranked contributors were used to select the corresponding pharmacophore moieties on the ligand to generate a pharmacophore model used to screen a database for potential hit compounds.
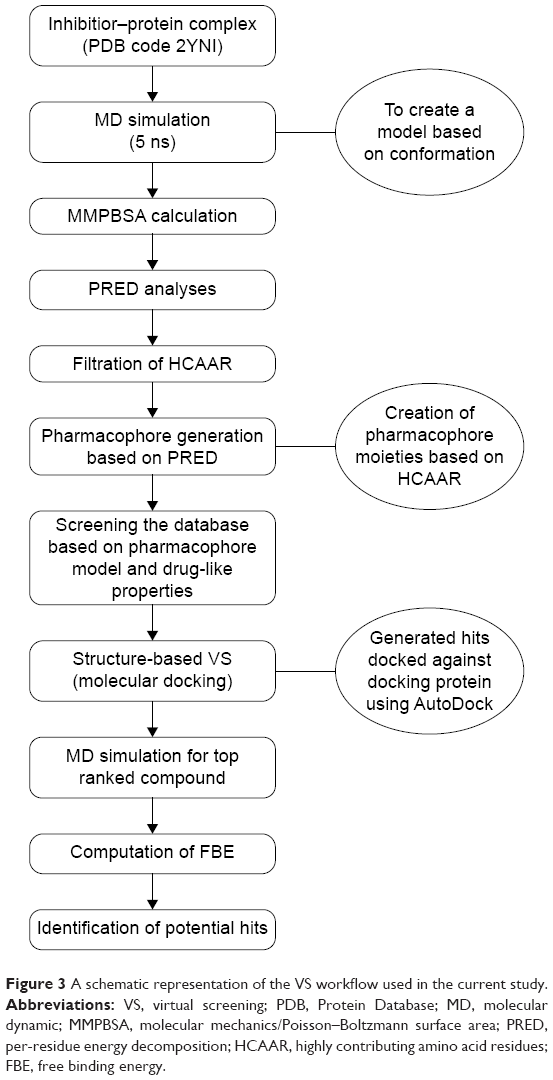 | Figure 3 A schematic representation of the VS workflow used in the current study. |
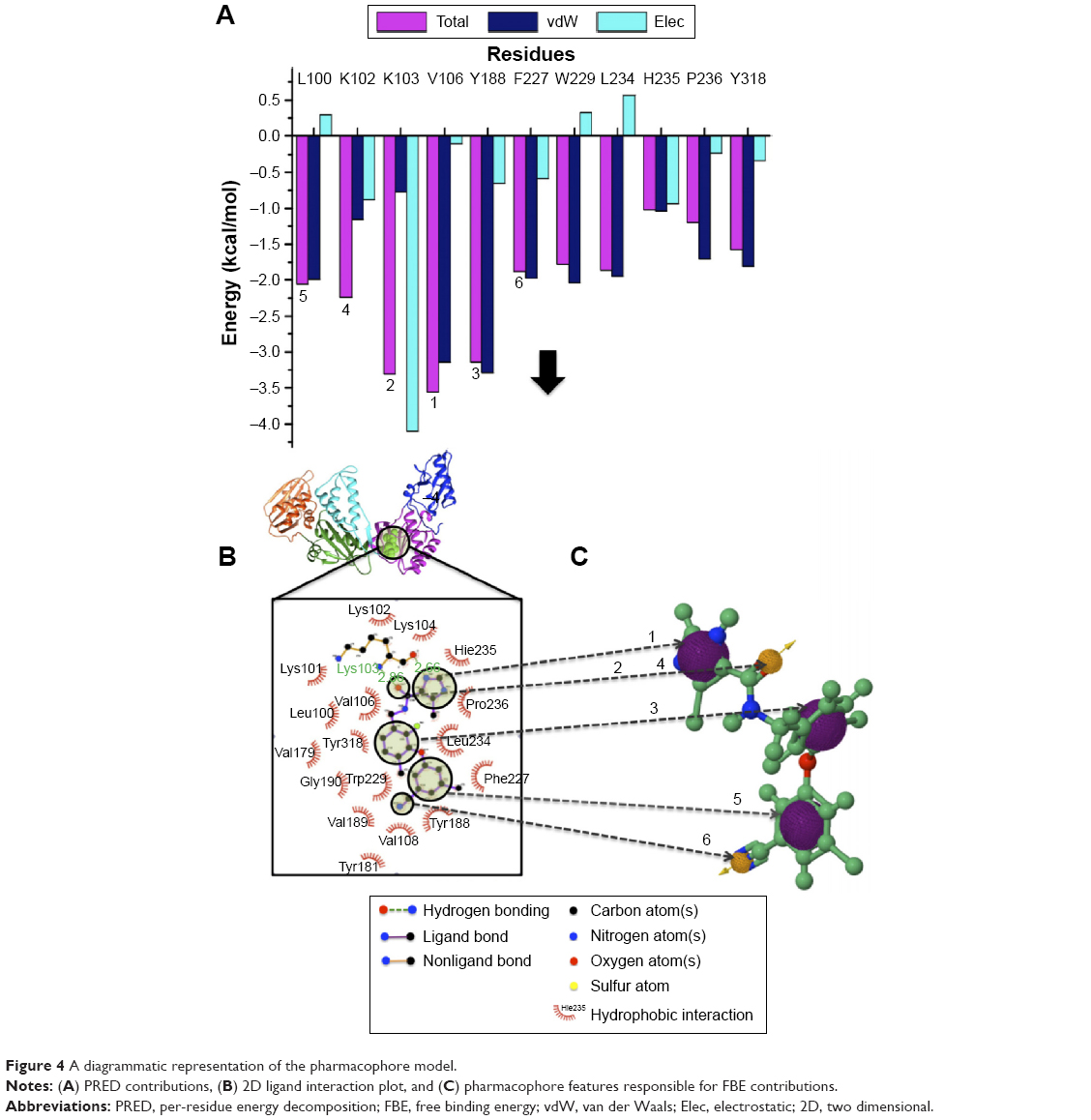 | Figure 4 A diagrammatic representation of the pharmacophore model. |
Ligand and receptor preparation
The crystal structure of HIV-1 RT-GSK952 complex was obtained from PDB (2YNI).8 The steepest decent method and MMFF94S force field in Avogadro software33 were used to minimize conformation of HIV-1 RT-GSK952 complex. The crystal structure was opened on UCSF chimera34 to delete chain B and solvents (H2O, MG [magnesium atoms] and TAR [tartaric acid]). GSK952 was selected and deleted. GSK952 was prepared by adding hydrogen. The ligand was prepared using Antechamber and tLeap of Amber1435 to optimize both HIV-1 RT (enzyme) and GSK952 (ligand), respectively, to ensure all parameters are present for MD simulations. Topology files generated were submitted for MD simulation.
MD simulations
Solvation of MD simulations36 was performed on HIV-1 RT-GSK952 using the graphics processing units version of the PMEMD (Particle Mesh Ewald Molecular Dynamics) engine integrated with Amber14.35 Leap module in Amber 1435 was used to add hydrogen atoms to the proteins. The standard Amber force field was employed to treat the receptor for bioorganic systems (ff99sb).37 A box with equilibrated TIP3P38 water molecules that were arranged around the receptor at a distance of 10 Å around the enzyme and Cl− ions was added to neutralize the systems. Cubic periodic boundary conditions were employed, and particle-mesh Ewald method39 applied in Amber12 was used to treat the long-range electrostatic interactions with nonbonding at 2fs integration step. The initial minimization of the system was carried out using the steepest descent method for 1,000 steps. A canonical ensemble (amount of substance volume temperature [NVT]) MD was carried out for 50 ps, during which the system was progressively heated up from 0 to 300 K by means of a Langevin thermostat.40 The system was then equilibrated at 300 K under 1 atm pressure while preserving the force constants on the restrained solute. Throughout all MD simulations, the SHAKE algorithm41 was employed on atoms covalently bonded to a hydrogen atom. A production run was achieved, with no restraints, for 5 ns in an isothermal isobaric (amount of substance, pressure, and temperature [NPT]). ensemble employing a Berendsen barostat.42 Trajectory exploration including the root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and radius of gyration (Rg) were carried out using PTRAJ (process trajectory) and CPPTRAJ modules applied in AMBER14.35
The trajectory was saved every 1 ps and examined every 1 ps using the PTRAJ module applied in Amber14.35 The structure was visualized using graphical user interface of the UCSF chimera package.34 Data were plotted using the GUI of Microcal Origin data analysis software Version 6 (www.originlab.com).
Molecular docking
AutoDock Vina was used for docking calculation.43 Geister partial charges were allocated during docking. AutoDock Graphical user interface provided by MGL tools were used to outline the AutoDock atom types.44 The docked conformations were obtained using the Lamarckian genetic algorithm.45 The magnitude of the grid box was x =14 Å, y =14 Å, z =18 Å, enclosing the anticipated active site residues including the highest contributing Leu100, Lys102, Lys103, Val106, Try188, and Phe227 residues. The classification of the compounds was in accordance with their docking score (DS) in a descending order.
FBE calculations
The FBE of the docked complexes was calculated to support the docking calculations and to predict the binding efficiencies of the HIV-1 RT against the targets. The FBE predictions are performed using molecular mechanics/Poisson–Boltzmann surface area (MMPBSA) that incorporates Equation 146 and molecular mechanics/generalized-Boltzmann surface area (MMGBSA) method that incorporates Equation 2.47
|
|
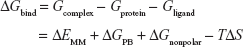
|
|
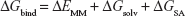
Here, ΔEMM is the difference between the minimized energies of the HIV-1 RT-GSK952 complex and the total energies of the HIV-1 RT and HIV-1 RT inhibitor including the electrostatic and the van der Waals energies, TΔS is the change in entropy of the ligand binding conformations, ΔGsolv is the difference in the P/GBSA solvation energies of the HIV-1 RT-GSK952 complex and the sum of the solvation energies of the HIV-1 RT and HIV-1 RT inhibitor, ΔGSA is the difference in the surface area energies for the HIV-RT enzymes and HIV-1 RT inhibitor. Both MMPBSA and MMGBSA methods have been realized to ensure the accurate ranking of inhibitors based on their FBE, and hence can serve as a powerful tool in drug design research.
Results and discussion
PRED pharmacophore model
The pharmacophore model exploits both the structural features of the proteins as well as the chemical features of ligands. To generate a PRED-based pharmacophore model, PRED decomposition was computed from MMPBSA calculations after 5 ns MD simulations of the (2YNI-GSK952) complex. Residues Leu100, Lys102, Lys103, Val106, Try188, and Phe227 were found to be the highest contributing residues that interact with the ligands (Table S1). The pharmacophoric features of the ligands’ HPI, hydrogen acceptor, and hydrogen bond interactions were found to interact with Leu100, Lys102, Val106, Try188, Lys103, Phe227, and Lys103, respectively. These ligand features were set as a query to generate a PRED-based pharmacophore model in ZINCpharmer.48 Furthermore, the PRED-based pharmacophore model (Figure S2) was used to screen the ZINC database49 for compounds with similar features to obtain the novel hits. Additionally, a further selection criterion was implemented when screening ZINCpharmer database. Seven hundred and eighty-eight hits were obtained from the ZINC database.
Molecular docking
All 788 hits were docked into the crystal structure (2YNI) to assess their chemical and physical feasibility. Thus, only ones with the correct pose and physical properties were selected for further consideration. This provided valuable insights into the nature of the binding site and the key ligand–protein interactions that are responsible for the molecular recognition and served as a validation step in the proposed workflow. A set of four compounds with experimentally determined activity (half maximal inhibitory concentration [IC50] values) was selected to further validate our findings. These four compounds were docked into the crystal structure of 2YNI as described earlier in the Molecular docking section. Calculated DS were correlated against the inhibitors’ experimentally determined IC50 values (Table 1). DS correlated (R2=0.62128) (Figure 5) with the IC50 values. The comparison by means of correlation serves as an additional validation step and adds robustness and validity to the docking protocol used in the current study. After the validation, molecular docking was carried out for all 788 hits.
 | Table 1 Validation of molecular docking approach |
 | Figure 5 Validation of molecular docking: docking score vs half maximal inhibitory concentration (IC50). |
The top ten compounds with the highest DS were selected from the library of 788 hits. The DS for the top ten compounds ranged from −11.5 to −12.4 kcal/mol (Table 2). It should be noted that there is not much difference in the binding energy of the top ten compounds with a rough range of 0.9 kcal/mol. Hit compounds were found to be more stable due to conservation of vital pharmacophoric properties when generating the pharmacophore model.
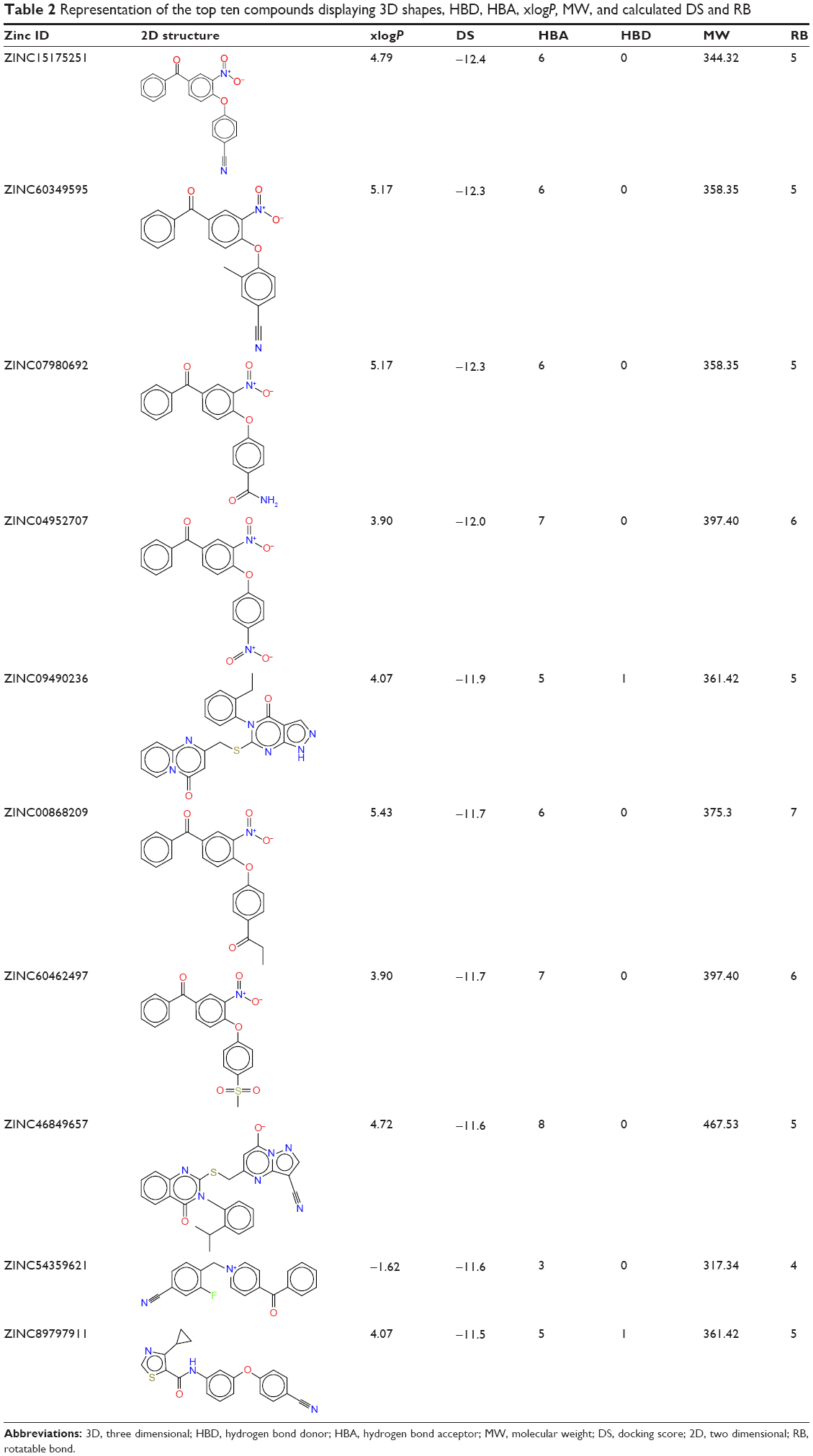 | Table 2 Representation of the top ten compounds displaying 3D shapes, HBD, HBA, xlogP, MW, and calculated DS and RB |
It is of high importance to consider residues that are directly accountable for the efficacy of HIV-1 RT when evaluating whether the molecules bind strongly with HIV-1 RT. This will ensure effective inhibition. Lig-plot analysis is best suited for displaying the two-dimensional interaction between the ligand and the residue contributing to the ligand binding. Lig-plot shows the interacting residues with Leu100, Lys102, Lys103, Val106, Tyr188, and Phe227 with the highest FBE contributing to the ligand binding. Leu100, Lys102, Val106, Phe227, Pro229, and Y188 have HPIs whereas Lys103 displays a hydrogen interaction with the inhibitor (Figure 4). The PRED calculations presented in this study are valuable tools that can be employed to highlight the most significant residues involved in the binding of the inhibitor.
As a result, this will serve as a guidance in the design of drug candidates that can strongly bind with such residues. The anti-HIV activity of the top ten hits is not evidenced, as the FDA-approved anti-HIV drugs were omitted throughout the entire screening process to guarantee the design of compound libraries grounded on novel structural scaffolds. Such findings verify the refined pharmacophore approach adopted in this work, rendering the suggested concept secure and dependable enough for the mining of novel drug candidates against the enzyme of interest. We believe the conveyed methodology in this current work can be applied to more biological drug targets with determined protein structures and binders that are recognized.
MD simulations and MMPBSA calculations
As previously mentioned, docking alone cannot provide reliable results. Hence, it is of high importance to correlate docking results with MD simulations. MD simulation (5 ns) was performed on the top ten screened hits from the molecular docking to confirm the change in mobility resulting from the binding of each hit to HIV-1 RT. Common MD-type force fields were employed to evaluate and rescore the docked complexes, this will lead to more accurate estimates of the binding affinities. All the free energy components are representative of average values over the 5 ns MD simulations calculated using the MMPBSA approach shown in Table 3. Among the top ten, ZINC54359621 and ZINC46849657 resulted in the highest FBE. The total calculated FBE (ΔGbind) of GSK952 against HIV-1 RT protein is −58.8 kcal/mol compared to −58.5 kcal/mol and −59.0 kcal/mol of ZINC46849657 and ZINC54359621, respectively. These findings are more reliable than the energy contributions obtained from the docking calculations. It was also observed that the ΔEvdW of ZINC54359621 and ZINC46849657 was higher (Table 3; Figure 6), which is advantageous and a contributing factor toward the high binding affinity.50 Electrostatic forces contribute to inhibitor molecules gaining binding energy.51 ZINC54359621 and ZINC46849657 electrostatic interactions are similar to that of GSK952, which explains their higher binding affinity and stability.
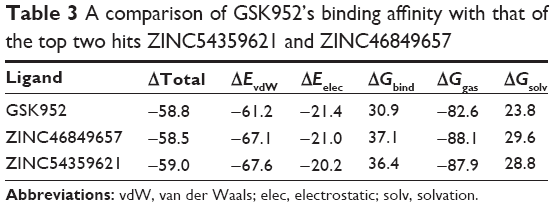 | Table 3 A comparison of GSK952’s binding affinity with that of the top two hits ZINC54359621 and ZINC46849657 |
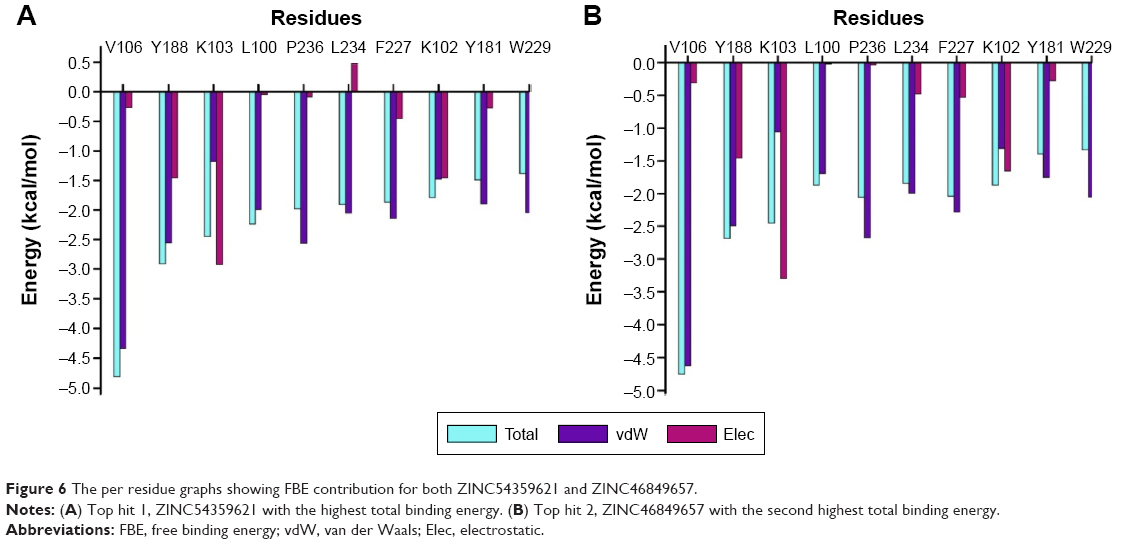 | Figure 6 The per residue graphs showing FBE contribution for both ZINC54359621 and ZINC46849657. |
PRED of the top two hits showed consistency compared to that of GSK952. The residues that contributed to the binding of GSK952 to HIV-1 RT protein also contributed to the binding of the top two hits (Figure 6).
It is noted that ZINC54359621 has a hydrogen bond between the oxygen atom of Pro236 and the NH group of the aromatic ring, NH group of Lys103 and the oxygen atom of the aromatic ring. ZINC46849657 has a hydrogen bond between the oxygen atom of Pro236 and the NH group of the aromatic ring, the NH group of residue Lys103 and the oxygen atom of the aromatic ring (Figure 7). These are one of the key interactions required for the binding to HIV-1 RT. This indicates the compounds’ suitability as an RT inhibitor.
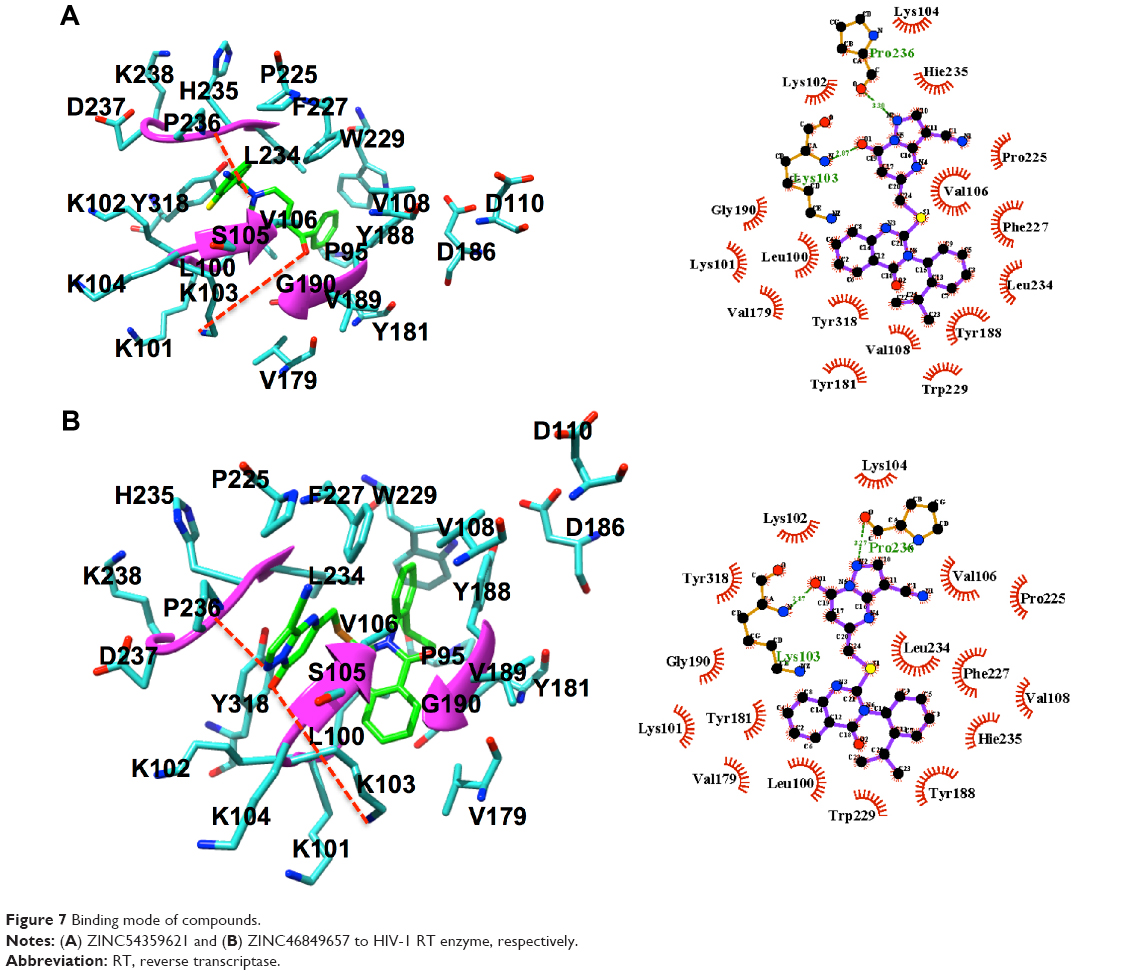 | Figure 7 Binding mode of compounds. |
HIV-1 RT is highly flexible in nature,52,53 and therefore the stability of the system during MD simulation was analyzed by computing 1) RMSD (Figure S3A) and 2) RMSF (Figure S3B). The average values of RMSD for ZINC54359621 and ZINC46849657 are 3.12 Å and 2.58 Å, respectively. Literature on HIV-1 RT with RMSD with a similar behavior as RMSD (Figure S3A) validates the system’s stability54,55 due to the high flexibility of HIV-1 RT. RMSF shows good stability of all residues interacting with the ligands from residues 100 to 236. The average values of RMSF for ZINC54359621 and ZINC4684965 are 1.87 Å and 1.53 Å, respectively. Further, the compactness of the system was analyzed by computing Rg of the systems. The average values of Rg for ZINC54359621 and ZINC46849657 are 51.2 Å and 51.2 Å, respectively. Results show that the target protein folded correctly and was able to retain a stable compact structure (Figure S3C).
Conclusion
VS was carried out to identify the potential inhibitors against mutated HIV-1 RT based on 1) PRED-based pharmacophore model, 2) molecular docking and, 3) MMPBSA approaches. The study identified two novel hits ZINC54359621 and ZINC46849657. These compounds may be representatives of a new series of NNRTIs possessing high resistance profiles against mutated HIV viruses. They have the potential to inhibit the transcription of the viral RNA by binding to the active site of the RT’s p66 subunit, thereby decreasing the replication rate of the virus. The pharmacophore approach was further refined, where the candidate molecules for structure-based VS were chosen based on their orientation and chemical features in relation to the 3D structure. The PRED was also calculated in order to obtain more insights into the crucial protein residues involved in ligand binding. This will potentially aid in the design of potent inhibitors that bind to these HCAAR. The results presented in this study present a map to the innovation and design of potent drug candidates against different biological targets. Results have shown that choosing highly contributing FBE candidates from a library of compounds generated from a PRED target-bound pharmacophore map is a more reliable and accurate approach.
Acknowledgments
Ethics committee approval was not required by our institute for this study because it did not involve human nor animal participants. The authors would like to acknowledge the School of Health Science, University of KwaZulu-Natal for financial support. Computational resources granted by the Center for High Performance Computing (http://www.chpc.ac.za) are highly appreciated.
Disclosure
The authors report no conflicts of interest in this work.
References
Friedrich BM, Dziuba N, Li G, Endsley MA, Murray JL, Ferguson MR. Host factors mediating HIV-1 replication. Virus Res. 2011;161(2):101–114. | ||
Jonckheere H, De Clercq E, Anné J. Fidelity analysis of HIV-1 reverse transcriptase mutants with an altered amino-acid sequence at residues Leu74, Glu89, Tyr115, Tyr183 and Met184. Eur J Biochem. 2000;267(9):2658–2665. | ||
amfAR. [webpage on the Internet]. Statistics: Worldwide: The Foundation for AIDS Research: HIV/AIDS Research. Available from http://www.amfar.org/worldwide-aids-stats/. Accessed March 8, 2016. | ||
Moonsamy S, Dash RC, Soliman ME. Integrated computational tools for identification of CCR5 antagonists as potential HIV-1 entry inhibitors: homology modeling, virtual screening, molecular dynamics simulations and 3D QSAR analysis. Molecules. 2014;19(4):5243–5265. | ||
Singh K, Marchand B, Kirby KA, Michailidis E, Sarafianos SG. Structural aspects of drug resistance and inhibition of HIV-1 reverse transcriptase. Viruses. 2010;2(2):606–638. | ||
Abad MJ, Bedoya LM, Bermejo P. Yi-Wei Tang (editor). HIV-screening strategies for the discovery of novel HIV-inhibitors. Recent Translational Research in HIV/AIDS. 2007;22:457–468. | ||
Jacobo-Molina A, Arnold E. HIV reverse transcriptase structure-function relationships. Biochemistry. 1991;30(26):6351–6356. | ||
Chong P, Sebahar P, Youngman M, et al. Rational design of potent non-nucleoside inhibitors of HIV-1 reverse transcriptase. J Med Chem. 2012;55(23):10601–10609. | ||
Usach I, Melis V, Peris JE. Non-nucleoside reverse transcriptase inhibitors : a review on pharmacokinetics, pharmacodynamics, safety and tolerability. J Int AIDS Soc. 2013;16:1–14. | ||
Barreca ML, Rao A, De Luca L, et al. Discovery of novel benzimidazolones as potent non-nucleoside reverse transcriptase inhibitors active against wild-type and mutant HIV-1 strains. Bioorg Med Chem Lett. 2007;17(7):1956–1960. | ||
Distinto S, Esposito F, Kirchmair J, et al. Identification of HIV-1 reverse transcriptase dual inhibitors by a combined shape-, 2D-fingerprint- and pharmacophore-based virtual screening approach. Eur J Med Chem. 2012;50:216–229. | ||
Keller PA, Birch C, Leach SP, Tyssen D, Griffith R. Novel pharmacophore-based methods reveal gossypol as a reverse transcriptase inhibitor. J Mol Graph Model. 2003;21(5):365–373. | ||
Penta A, Ganguly S, Murugesan S. Design and synthesis of tetrahydrophthalimide derivatives as inhibitors of HIV-1 reverse transcriptase. Org Med Chem Lett. 2013;3(1):8. | ||
Ren J, Milton J, Weaver KL, Short SA, Stuart DI, Stammers DK. Structural basis for the resilience of efavirenz (DMP-266) to drug resistance mutations in HIV-1 reverse transcriptase. Structure. 2000;8(10):1089–1094. | ||
Zavodszky MI, Rohatgi A, Van Voorst JR, Yan H, Kuhn LA. Scoring ligand similarity in structure-based virtual screening. J Mol Recognit. 2009;22(4):280–292. | ||
Soliman ME. A hybrid structure/pharmacophore-based virtual screening approach to design potential leads: A computer-aided design of South African HIV-1 subtype C protease inhibitors. Drug Dev Res. 2013;74(5):283–295. | ||
Gu W. Impact of virtual screening on HIV reverse transcriptase inhibitor discovery. Jacobs Journal of AIDS/HIV. 2014;14–15. | ||
Vyas V, Jain A, Jain A, Gupta A. Virtual screening: A fast tool for drug design. Sci Pharm. 2008;76:333–360. | ||
Skelton AA, Maharaj YR, Soliman ME. Target-bound generated pharmacophore model to improve the pharmacophore-based virtual screening: Identification of G-protein human CCR2 receptors inhibitors as anti-inflammatory drugs. Cellular and Molecular Bioengineering. 2014;7(1):45–57. | ||
Lavecchia A, Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Curr Med Chem. 2013;20(23):2839–2860. | ||
Young DC. Computational Drug Design: A Guide for Computational and Medicinal Chemists. Wiley-Interscience; 2009. | ||
Yang SY. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today. 2010;15(11–12):444–450. | ||
Mallipeddi PL, Kumar G, White SW, Webb TR. Recent advances in computer-aided drug design as applied to anti-influenza drug discovery. Curr top Med Chem. 2014;14(16):1875–1889. | ||
Willett P. Enhancing the effectiveness of ligand-based virtual screening using data fusion. QSAR Comb Sci. 2006;25(12):1143–1152. | ||
Drwal MN, Griffith R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov Today Technol. 2013;10(3):e395–e401. | ||
Khanna M, Wang F, Jo I, et al. Targeting multiple conformations leads to small molecule inhibitors of the uPAR·uPA protein-protein interaction that block cancer cell invasion. ACS Chem Biol. 2011;6(11):1232–1243. | ||
Amaro RE, Baron R, McCammon JA. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J Comput Aided Mol Des. 2008;22(9):693–705. | ||
Okimoto N, Futatsugi N, Fuji H, et al. High-performance drug discovery: computational screening by combining docking and molecular dynamics simulations. PLoS Comput Biol. 2009;5(10):e1000528. | ||
Wang L, Gu Q, Zheng X, et al. Discovery of new selective human aldose reductase inhibitors through virtual screening multiple binding pocket conformations. J Chem Inf Model. 2013;53(9):2409–2422. | ||
Rastelli G. Emerging topics in structure-based virtual screening. Pharm Res. 2013;30(5):1458–1463. | ||
Islam MA, Pillay TS. Exploration of the structural requirements of HIV-protease inhibitors using pharmacophore, virtual screening and molecular docking approaches for lead identification. J Mol Graph Model. 2015;56:20–30. | ||
Shoichet BK, McGovern SL, Wei B, Irwin JJ. Lead discovery using molecular docking. Curr Opin Chem Biol. 2002;6(4):439–446. | ||
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform. 2012;4(1):1–17. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. | ||
Case DA, Berryman JT, Betz RM, et al. AMBER 2015, University of California, San Francisco. Available from ambermd.org/doc12/Amber15.pdf. | ||
Götz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized Born. J Chem Theory Comput. 2012;8(5):1542–1555. | ||
Duan Y, Wu C, Chowdhury S, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24(16):1999–2012. | ||
Antony Dhivyan JE, Anoop MN. Virtual screening and lead optimization to identify novel inhibitors for HDAC-8. 2012;9:1–58. Available from http://arxiv.org/ftp/arxiv/papers/1209/1209.2793.pdf. | ||
Harvey MJ, De Fabritiis G. An implementation of the Smooth Particle Mesh Ewald Method on GPU Hardware. J Chem Theory Comput. 2009;5(9):2371–2377. | ||
Wu X, Brooks BR. Self-guided Langevin dynamics simulation method. Chem Phys Lett. 2003;381:512–518. | ||
Kräutler V, Van Gunsteren WF, Hünenberger PH. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J Comput Chem. 2001;22(5):501–508. | ||
Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
mgltools.scripps.edu [homepage on the Internet]. MGLTools Website – Welcome – MGLTools. Available from: http://mgltools.scripps.edu/. Accessed February 14, 2016. | ||
Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19(14):1639–1662. | ||
Hou T, Wang J, Li Y, Wang W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J Comput Chem. 2011;32(5):866–877. | ||
Lyne PD, Lamb ML, Saeh JC. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J Med Chem. 2006;49(16):4805–4808. | ||
Koes DR, Camacho CJ. ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res. 2012;40(Web Server issue):409–414. | ||
Irwin JJ, Shoichet BK. ZINC – a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–182. | ||
Kawasaki Y, Freire E. Finding a better path to drug selectivity. Drug Discov Today. 2011;16:985–990. | ||
Kroeger Smith MB, Rouzer CA, Taneyhill LA, et al. Molecular modeling studies of HIV-1 reverse transcriptase nonnucleoside inhibitors: total energy of complexation as a predictor of drug placement and activity. Protein Sci. 1995;4(10):2203–2222. | ||
Rodgers DW, Gamblin SJ, Harris BA, et al. The structure of unliganded reverse transcriptase from the human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1995;92(4):1222–1226. | ||
Das K, Martinez SE, Bandwar RP, Arnold E. Structures of HIV-1 RT-RNA/DNA ternary complexes with dATP and nevirapine reveal conformational flexibility of RNA/DNA: insights into requirements for RNase H cleavage. Nucleic Acids Res. 2014;42(12):8125–8137. | ||
Madrid M, Jacobo-Molina A, Ding J, Arnold E. Major subdomain rearrangement in HIV-1 reverse transcriptase simulated by molecular dynamics. Proteins. 1999;35(3):332–337. | ||
Temiz NA, Bahar I. Inhibitor binding alters the directions of domain motions in HIV-1 reverse transcriptase. Proteins. 2002;49(1):61–70. |
Supplementry materials
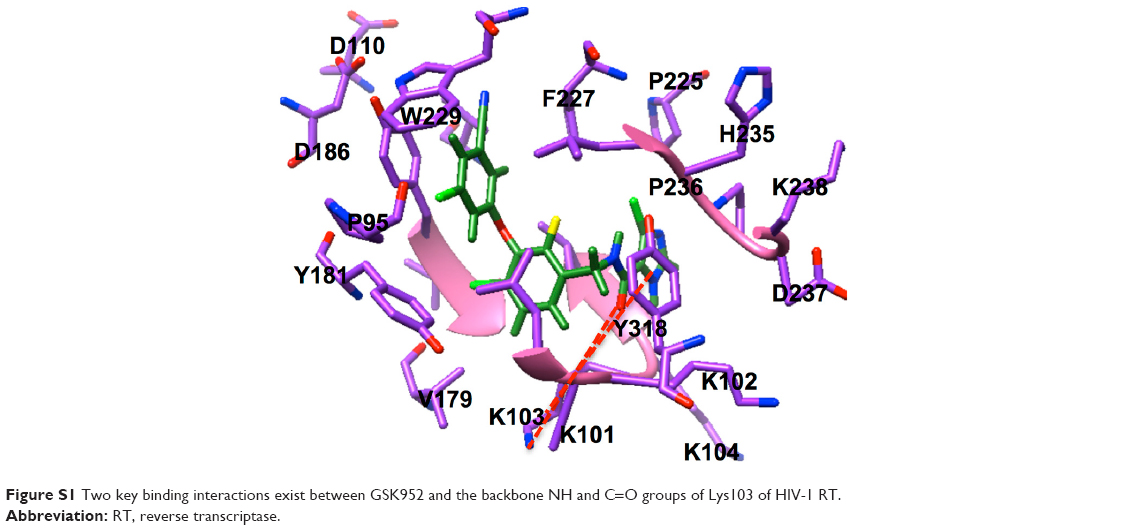 | Figure S1 Two key binding interactions exist between GSK952 and the backbone NH and C=O groups of Lys103 of HIV-1 RT. |
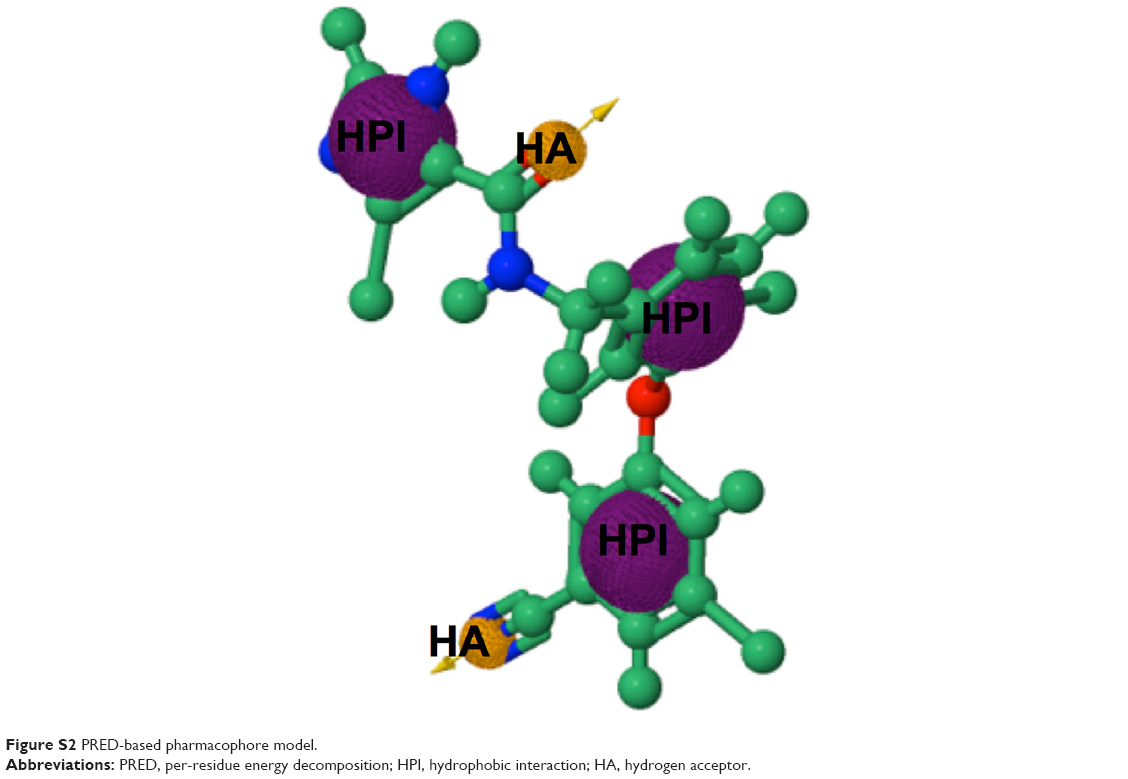 | Figure S2 PRED-based pharmacophore model. |
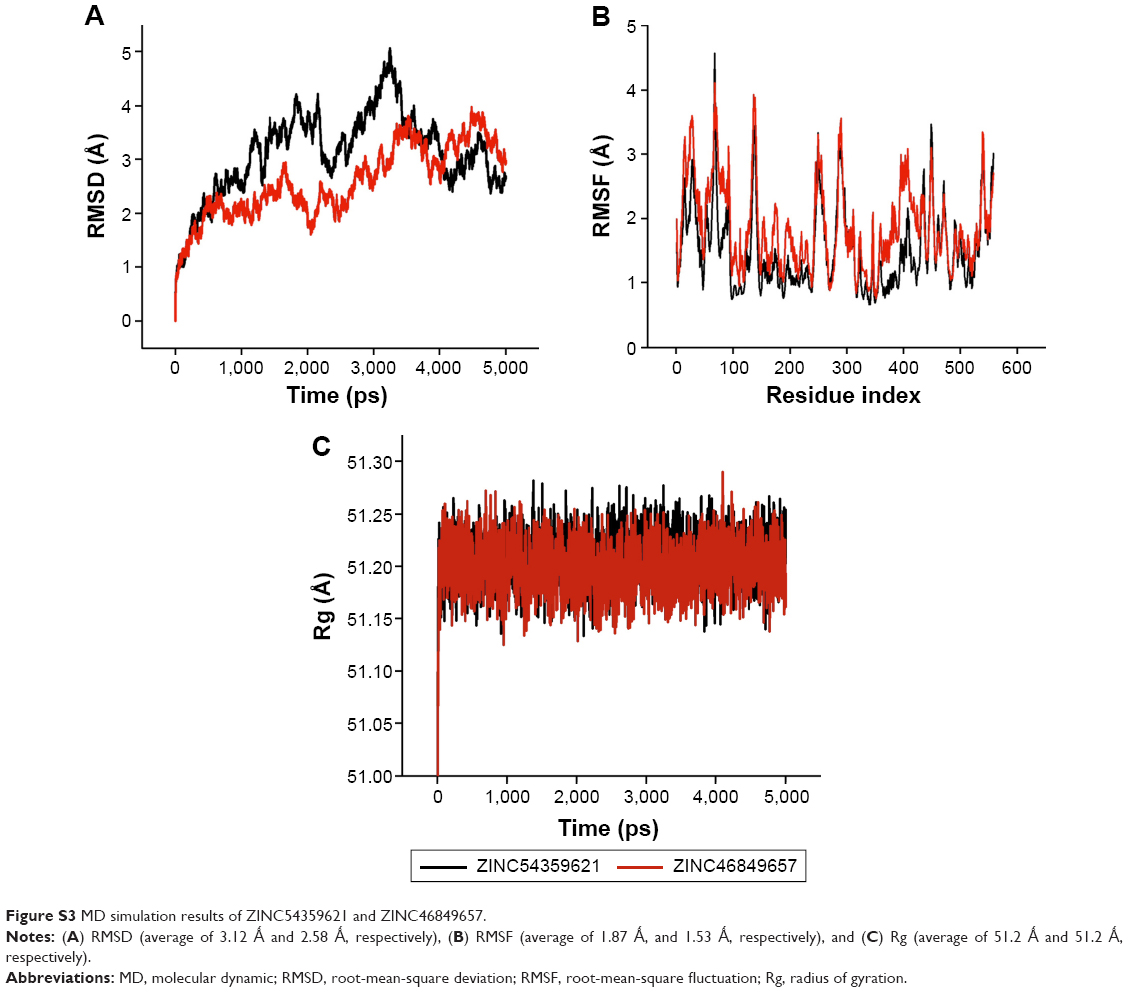 | Figure S3 MD simulation results of ZINC54359621 and ZINC46849657. |
 | Table S1 PRED of highly interacting residues |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.