Back to Journals » Infection and Drug Resistance » Volume 7
Peptidomimetics as a new generation of antimicrobial agents: current progress
Authors Méndez-Samperio P
Received 7 May 2014
Accepted for publication 17 June 2014
Published 30 August 2014 Volume 2014:7 Pages 229—237
DOI https://doi.org/10.2147/IDR.S49229
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Patricia Méndez-Samperio
Department of Immunology, National School of Biological Sciences, National Polytechnic Institute, Mexico City, Mexico
Abstract: Antibiotic resistance is an increasing public health concern around the world. Rapid increase in the emergence of multidrug-resistant bacteria has been the target of extensive research efforts to develop a novel class of antibiotics. Antimicrobial peptides (AMPs) are small cationic amphiphilic peptides, which play an important role in the defense against bacterial infections through disruption of their membranes. They have been regarded as a potential source of future antibiotics, owing to a remarkable set of advantageous properties such as broad-spectrum activity, and they do not readily induce drug-resistance. However, AMPs have some intrinsic drawbacks, such as susceptibility to enzymatic degradation, toxicity, and high production cost. Currently, a new class of AMPs termed "peptidomimetics" have been developed, which can mimic the bactericidal mechanism of AMPs, while being stable to enzymatic degradation and displaying potent activity against multidrug-resistant bacteria. This review will focus on current findings of antimicrobial peptidomimetics. The potential future directions in the development of more potent analogs of peptidomimetics as a new generation of antimicrobial agents are also presented.
Keywords: drug resistance, infection, antimicrobial peptides
Introduction
The growing resistance of pathogens is one of the biggest public problems worldwide.1 Multidrug-resistant bacterial strains can cause severe infections as they are no longer responsive to most conventional antibiotics.2,3 To combat these pathogens, efforts have been extended to develop a new generation of antibiotics. Antimicrobial peptides (AMPs), also termed “host defense peptides” for their immunomodulatory properties, are cationic amphiphilic peptides, which are the first line of defense to protect organisms from microbial infection.4–8
It has been demonstrated that naturally occurring or synthetic AMPs can be a new functional class of antibiotics.9,10 AMPs are antimicrobial agents based on their activity against the prokaryotic membrane. These agents adopt globally amphipathic conformations upon initial contact with bacterial membranes rich in anionic phospholipids. The conformations, which resemble detergent-induced micelle formation, result in total membrane disintegration in which their cationic and hydrophobic side groups segregate into distinct regions. This finding indicates that AMPs are potential antibiotic agents with a different antimicrobial mechanism, and that this activity mainly depends on their physical mechanism.11 The structural and sequence diversity of AMPs include amphipathic α-helices (eg, cathelicidins), β-sheets with 2–4 disulfide bridges (β defensins and protegrins), extended conformation (indolicidin), and beta-loop peptides (brevinin).12–15 Among the AMPs, human defensins and cathelicidins play an important role, linking innate and acquired immunities (Figure 1).8 Importantly, AMPs are therapeutic agents with a lower tendency to elicit antibiotic resistance than conventional antibiotics.
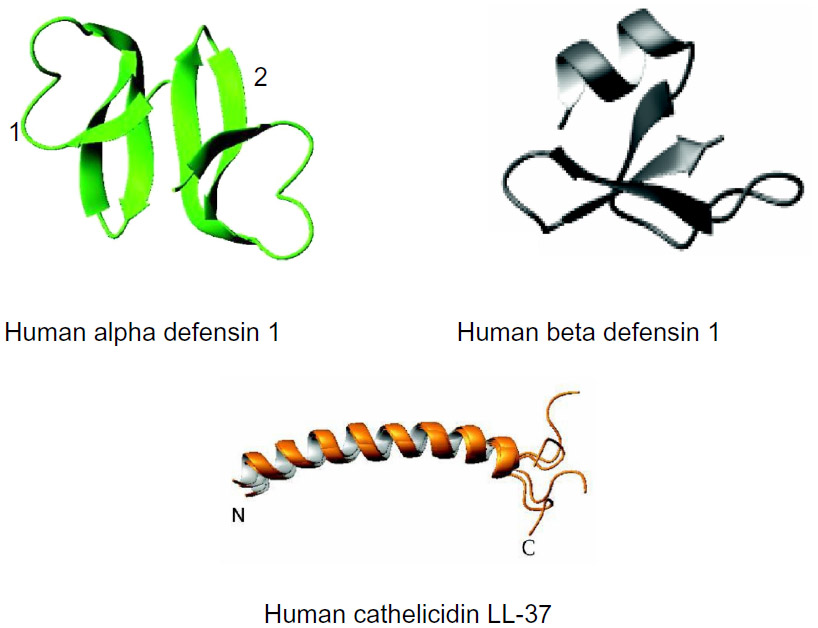 | Figure 1 Three-dimensional structures of human antimicrobial peptides. |
Currently, the main reasons for the limited practical application of AMPs include their very high susceptibility to proteolytic degradation by microbial enzymes, toxicity due to high amounts of drug needed for therapy, relatively short half-life, and their high production cost.16 The design and synthesis of peptide mimics (peptidomimetics) have been developed to mimic the structure, function, and mode of action of host-defense AMPs, which act on bacterial cell walls or membranes and can potentially circumvent those obstacles. Antimicrobial peptidomimetics display antibacterial activity against a broad-spectrum of bacteria, including drug-resistant strains, and are less susceptible to resistance development in bacteria. A number of antimicrobial peptidomimetics have been developed in the last decade, such as β-peptides,17–19 peptoids,20–25 arylamide oligomers,26,27 and β-turn mimetics.28,29 Recently, a new class of antimicrobial peptidomimetics termed “AApeptides” because they contain N-acylated-N-aminoethyl amino acid units derived from chiral peptide nucleic acid backbones have been developed. They are highly resistant to proteolytic degradation and their amphipathic structures can mimic the bactericidal mechanism of AMPs.30–32 Currently, different antimicrobial AApeptides have been developed, such as α-AApeptides and γ-AApeptides.33,34
This review aims to describe recent progress in the discovery of peptidomimetics as new generation antimicrobial agents and discusses future directions for antimicrobial peptidomimetics in the emergence of multidrug-resistant bacteria.
Molecular design and antibiotic activity of antimicrobial peptidomimetics
To improve the antimicrobial activity of peptidomimetics, the relationship between the structure and function of these peptides must be considered. Interestingly, antimicrobial peptidomimetics may be designed by joining amphiphilic peptide building blocks. In this regard, a potent and broad-spectrum antimicrobial activity can be fine-tuned by changing the ratio of cationic/hydrophobic groups via the introduction of hydrophobic building blocks, suggesting that the structure–activity relationships in antimicrobial peptidomimetics indicate the balance of forces required for bactericidal activity.35–38 To date, peptidomimetics have been designed by cyclization of linear peptides or coupling of stable unnatural amino acids. In addition, Hu et al,30 and Niu et al31,32 reported the development of a new class of peptidomimetics termed “AApeptides”, and depending on the position of the side chain (connected to either the α-C or γ-C in relation to the carbonyl group), two subclasses of these peptides (α-AApeptides and γ-AApeptides, respectively) have been designed (Figure 2). Previous studies have indicated that focused libraries of linear AApeptide (including both α-AApeptides and γ-AApeptides) sequences have been developed so that these sequences might mimic natural linear AMPs and adopt globally amphipathic conformations upon initial contact with bacterial membranes.39,40 Moreover, it has been reported that the antimicrobial activity of some AApeptides is still generally comparable, or even superior, to the AMP magainin as well as a previously reported linear α-AApeptide α1 against several bacterial strains.31 Interestingly, Padhee et al39 reported that a focused library of different linear α-AApeptide sequences such as α1 and α2 has been prepared to minimize the hemolytic activity, but with a potent and broad-spectrum antimicrobial activity to arrest the growth of both Gram-positive and Gram-negative bacterial pathogens. In addition, these authors showed that α1 and α2 are the most potent antimicrobial α-AApeptide peptidomimetics with broad-spectrum activity, especially toward clinically relevant strains including the multidrug-resistant strains vancomycin-resistant Enterococcus faecalis and methicillin-resistant Staphylococcus aureus (MRSA).39 Furthermore, a focused library of linear γ-AApeptide sequences containing γ-1, γ-2, γ-3, and γ-4 has been prepared.40 In this context, it has also been demonstrated that compared with γ-1, γ-2 and γ-3, γ-4 contains enhanced bactericidal activity against Gram-positive strains, indicating that some γ-AApeptides are very potent and supporting their potential development as antimicrobial agents to treat Gram-positive bacterial infections.40 However, they are quite toxic to blood cells as well as other mammalian cells. In fact, the antimicrobial activity of γ-AApeptides is likely to be enhanced if the overall hydrophobicity increases, which at the same time also leads to increased hemolytic activity and cytotoxicity. Interestingly, the hemolytic activity and cytotoxicity can be minimized by introducing more cationic residues.
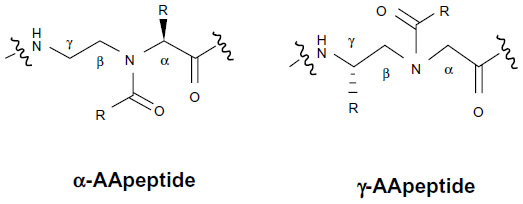 | Figure 2 Representative structures of α-AA and γ-AApeptides. |
On the other hand, focused libraries of lipo AApeptides (including both α-AApeptides and γ-AApeptides) sequences have been developed, and the lipid tails of these lipopeptides are important for biological activity to facilitate bacterial membrane interaction, giving them broad-spectrum activity against both Gram-positive and Gram-negative bacteria.30,31 A focused library of lipo antimicrobial α-AApeptides sequences has been prepared, including α3 and α4.32 Interestingly, the development of cyclic γ-AApeptides that mimic function of AMPs has been reported.41 These cyclic peptides have enhanced antimicrobial activity compared with their linear antimicrobial AApeptides, as their structures adopt a semirigid backbone conformation, resulting in a more stable amphipathic structure. Structure–activity relationships of cyclic antimicrobial γ-AApeptides incorporating a global distribution of cationic and hydrophobic residues are in development. In this context, group amphiphilic building blocks can be joined, and the resulting oligomers are cyclized.42
Since peptidomimetics interact nonspecifically with their target membranes, the addition of a positive charge by adding arginine, lysine, or histidine residues to the peptide sequence is required for initial electrostatic attraction with negatively charged bacterial membranes, whereas hydrophobic bulk guides insertion into the bacterial membrane. In addition, increasing hydrophobicity may increase antimicrobial activity. Recent developments in peptidomimetics that are formed through insertion into the amino acid backbone or heteroatom replacement indicate that several peptidomimetics form structural designs such as helices, sheets, turns, and loops via noncovalent interactions. To prepare AApeptides, the current literature indicates that different approaches have been developed.32,43 Originally, the synthesis of these peptides was achieved using the building block strategy (Figure 3).30–32 In this approach, 9-fluorenylmethyloxycarbonyl-protected peptide building blocks were prepared and then assembled on a solid support to provide the desired peptide sequences.44–47 Another approach termed “submonomeric” has been developed to prepare γ-AApeptides. This approach is a solid-phase synthesis of the peptides, which eliminates the need of building block preparation; thus, chemically diverse functional groups can be conveniently introduced into the desired peptide sequences.43
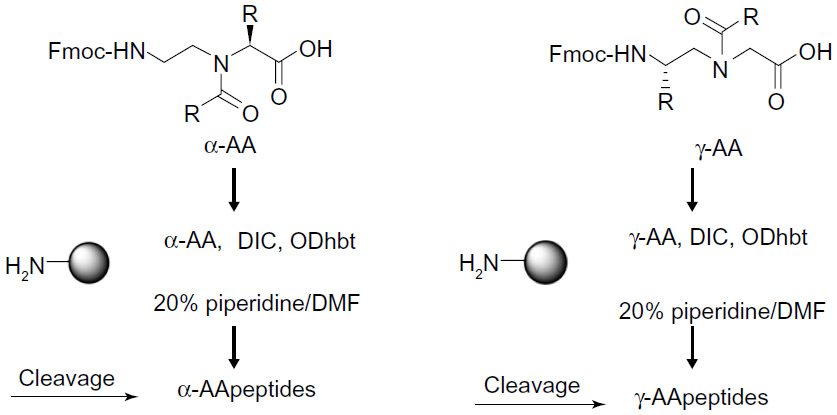 | Figure 3 General building block strategy for the synthesis of AApeptides. |
At present, it has also been reported that preorganized secondary structures including helical or sheet-like conformations within peptidomimetics are unnecessary in the antibacterial activities of these peptides.39,40,48–50 In contrast, the presence of backbones with certain flexibility can lead to a potent and broad-spectrum antimicrobial activity, indicating the importance of the conformational rigidity in the molecular design of antimicrobial peptidomimetics.51–53 In this context, peptidomimetics have more dihedral angles compared to canonical peptides, and this molecular design induces high flexibility.
Previous studies have indicated that in the molecular design of the amphipathic helical 21-mer peptide ([KAAKKAA]3; amino acid sequence: K A A K K A A K A A K K A A K A A K K A A), the peptide’s cytotoxic activity is highly dependent upon the spatial positions of tryptophan and cationic residues within the hydrophobic sector of an α-helix.54 More recently, the synthesis of enzymatically resistant versions of AMPs by partial substitution of L-residues with nonnatural D- or B-residues has been developed. In this regard, McGrath et al55 synthesized a lysine-leucine or klotho peptide known as (KLAKLAK)2, which had low toxicity toward mammalian cells, with high antimicrobial activity. Furthermore, these authors demonstrated that D(KLAKLAK)2, a variant of this molecule, is bactericidal against several Gram-negative species, including Escherichia coli, Klebsiella pneumoniae, and Acinetobacter baumannii. Interestingly, a strain of K. pneumoniae, which was resistant to conventional antibiotics, was susceptible to this peptide with the minimal inhibitory concentration of 75 μg/mL. In addition, this peptide has stronger fungicidal activity.55
In the remainder of this review, the discussion will highlight the discoveries that have led to our current understanding of the development of peptidomimetics in the context of their use as therapeutic agents.
Therapeutic potential of antimicrobial peptidomimetics
Peptidomimetics represent an important field in pharmacology as they circumvent the limitations of AMPs used in therapy. Therapeutic applications of antimicrobial peptidomimetics have also been considered in regard to their high resistance against enzymatic degradation.56–59 Regarding antimicrobial peptidomimetics which are currently in Phase II clinical trials, Choi et al60 have designed small arylamide foldamers that mimic AMPs. Importantly, these authors also demonstrated that hydrogen-bonded restraints in the structure of arylamide increase activity toward S. aureus and E. coli. On the other hand, the pharmaceutical company Lytix Biopharma AS (Tromsø, Norway) has recently commenced Phase I/IIa clinical trials with another antimicrobial peptidomimetic known as Lytixar TM (also known as LTX-109) for nasal decolonization of MRSA (http://www.lytixbiopharma.com). This peptidomimetic, containing a modified tryptophan derivate as lipophilic bulk, displayed a combination of high antibacterial activity against methicillin-resistant Staphylococci and staphylococcal biofilms.57 Another antimicrobial peptidomimetic, which is currently in Phase II clinical trials for the broad spectrum treatment of MRSA infections, is brilacidin (also known as PMX-30063).61 This peptidomimetic was synthesized by PolyMedix (Radnor, PA, USA) and was then acquired by Cellceutix (Beverly, MA, USA) in September 2013. It is important to consider that this peptide is a potent bactericidal with broad-spectrum activity, not only against Gram-positive bacteria such as S. aureus and Enterococcus faecium but also against Gram-negative species such as E. coli. Interestingly, in a Phase II study involving patients with acute skin infections, 215 patients were treated with three different doses of brilacidin, and brilacidin’s favorable profile was noted in 65%–87% of patients.61 Another antimicrobial peptidomimetic known as POL7080 has also been developed. POL7080 is active in the nanomolar range against Gram-negative Pseudomonas spp. Researchers at the University of Zurich and Polyphor Ltd (Allschwill, Switzerland) have developed this peptidomimetic by means of a novel approach to peptide preparation named protein epitope mimetic technology. This peptidomimetic specifically targets Pseudomonas aeruginosa through a mode of action that is different from the membrane-disrupting activity of the parent compound.29 Currently, POL7080 is being prepared by the company Polyphor, and in preclinical studies, this antimicrobial peptidomimetic was highly active on a broad panel of clinical isolates, including multidrug resistant Pseudomonas bacteria, with a potent bactericidal activity in a mouse septicemia infection model. P. aeruginosa is an opportunistic bacteria which causes serious infections in patients with reduced immune systems (eg, those having acquired immunodeficiency syndrome or cancer). At present, Phase I clinical studies29 of the peptidomimetic POL7080 have been completed in healthy individuals in Europe, demonstrating the clinical safety and tolerability of this peptide (http://www.polyphor.com/products/pol7080).
Regarding an important role of antimicrobial peptidomimetics in the prevention of virus infections and the treatment of cancer metastasis, antimicrobial peptidomimetics that inhibit virus replication as well as possess antitumor activity against different cancer cell lines have been designed. Importantly, the structure of these peptidomimetics has been explored by using a peptidomimetic library in order to obtain higher plasma and metabolic stabilities. Furthermore, the use of nanotechnology as delivery tool for both classes of peptides will be presented later in the review. As shown in previous reports, some γ-AApeptides are effective in arresting the growth of both Gram-positive and Gram-negative drug-resistant bacterial pathogens.50 In addition, it has been demonstrated that γ-AApeptides can mimic the human immunodeficiency virus (HIV) Tat peptide by binding to HIV-1 RNA with a high affinity, comparable to their peptide counterparts.44,46 Moreover, it is well known that the CXC chemokine receptor 4 (CXCR4) is a coreceptor of HIV-1 infection in human cells.62,63 In this context, development of peptidomimetic ligands for CXCR4 as therapeutic agents for HIV-1 infection and cancer has been reported.64–66 In fact, the peptidomimetic Arg(*)-Arg-Nal(2)-Cys(1x)-Tyr-Gln-Lys-(d-Pro)-Pro-Tyr-Arg-Cit-Cys(1x)-Arg-Gly-(d-Pro)(*) (POL3026) is a novel specific beta-hairpin mimetic CXCR4 antagonist, with potent anti-HIV activity.67 This peptide has a ten-fold increase in potency, with a good bioavailability profile following subcutaneous administration.67 Interestingly, ligand binding site mapping using a panel of CXCR4 mutants demonstrated that the new analog D(1-10)-vMIP-II-(9-68)-SDF-1 (RCP222) share the interactive amino acids on CXCR4 with HIV-1 glycoprotein 120.68,69 To date, peptidomimetic ligands have served as inhibitors of stromal cell-derived factor-1 since these peptidomimetics are involved in the interactions of HIV-1 envelope glycoprotein and stromal cell-derived factor-1 with membrane ligands of CD4+ human cells.70–74 Another peptide with anti-HIV activity is ALX40-4C. The structure of this peptide was designed from the basic HIV-1 transactivation domain for the inhibition of Tat–TAR interaction.75 The anti-HIV effects of this peptide are elicited through selective binding to CXCR4.76 In addition, Zhou et al77 reported that the human APJ, a G protein-coupled seven-transmembrane receptor, is essential for its coreceptor activity for HIV-1; thus, it is an alternative target of ALX40-4C to block HIV glycoprotein 120 from binding to the cellular membrane. On the other hand, regarding the development and improvement of nanoparticles for stabilization and delivery of antiviral peptides, a nanoformulation of the amphiphatic α-helical peptide p41 (a positively-charged analog of C5A peptide, derived from the hepatitis C virus protein) has been designed to treat HIV/hepatitis C virus coinfection, indicating the potential of this nanoformulation for stabilization and delivery of antiviral peptides, while maintaining their functional activity.78
Currently, cancer is a major concern in relation to human mortality, and all types of cancer are characterized by irregular cell growth. Antitumor drugs are subject to differences in target tissue and absorption, which can be particular to each patient. In addition, acquired drug resistance is considered the widespread cause for tumor recurrence.79 At present, some radiolabeled γ-AApeptides have been used as tracers for positron emission tomography, indicating a therapeutic application as anticancer agents.45 In addition, peptidomimetics have been the basis for a number of studies performed to discover new novel anticancer agents.79,80 In this regard, the in vivo inhibitory effects on the growth of tumor cell xenografts in nude mice by the cyclic pentapeptide FC092 ([D-Arg2]-FC131), a CXCR4 antagonist, have been reported.81,82 The intrinsic relationship between its structure and its high specificity to tumor cells is likely playing the key role in the cytotoxicity of peptidomimetics. These characteristics allow the peptidomimetics to bind to cancer cells and disrupt the negatively-charged tumor cell membrane, which is derived from a greater than normal expression of anionic molecules such as sialic acid-rich glycoproteins or phosphatidylserine.83 Importantly, these chemical differences aid the electrostatic interaction of the positively-charged peptide and the negatively-charged tumor cell membranes.80 Studies have reported of AMPs that are effective against bacteria and cancer cells but not against normal mammalian cells such as cecropins from insects and magainins from amphibians.84,85 On the other hand, signal transducer and activator of transcription (STAT) proteins are a family of cytoplasmic transcription factors. Phosphorylation induces their homo- or heterodimerization, and an important function of these dimers is to control gene expression. STAT3 is frequently activated in many human cancer cell lines and is involved in cancer development and progression. Importantly, dysregulation of STAT3 can lead to increase in its activity and contribute to tumorigenesis. Currently, peptidomimetics have been utilized to directly target STAT3 signaling. In this regard, it has been reported that an oxazole-based small-molecule STAT3 inhibitor, which modulates STAT3 stability, induces significant antitumor cellular effects.86 One primary goal of drug delivery for cancer therapy is to increase the amount of drug delivered to the tumor site and decrease its exposure to healthy tissues.87 Recent advances in microencapsulation technologies have been used to enhance drug protectivity, availability, and distribution by employing different biodegradable delivery platforms like liposomes, dendrimers, nanoemulsions, polymeric nanocarriers, and nanoparticles. These nanoformulations can be used to control drug/molecule release and enhance targeted delivery and effectiveness.88 In this regard, Wang and Zhang89 encapsulated a polypeptide isolated from the unicellular green algae Chlorella pyrenoidosa, which exhibited the highest inhibitory activity on human liver HepG2 cancer cells (49%), and they named the polypeptide Chlorella pyrenoidosa antitumor polypeptide. The main mechanism of action of this peptidomimetic is condensation/fragmentation of nuclear chromatin.89 The in vitro release of this peptide against gastric cancer cells provided a basis for the development of encapsulated antitumor peptides. The peptidomimetics KLAKLAKKLAKLAK and the isoAsp-Gly-Arg (or isoDGR) peptides serve as potent tools for developing new antitumor peptides. They can selectively kill CD13−/αvβ3+ breast cancer cells in both in vitro and in vivo experiments by inhibiting angiogenesis by binding to αvβ3+, which is increased on tumor cells.90 Currently, the antitumor role of the analgesic-antitumor peptide (AGAP) isolated from the scorpion Buthus martensii has been reported. This protein, consisting of a small ubiquitin-related modifier linked with a hexahistidine tag from E. coli, was used as an antitumor peptide, and the main mechanism of action of this peptidomimetic is through cell cycle arrest.91 The recombinant system AGAP showed considerable inhibition of lymphoma and glioma propagation.91 Interestingly, using SW480 human colon cancer cells, it was proposed that recombinant AGAP induces cell cycle arrest in the G0/G1 phase, attended by the decrease in the S phase without significant change in the G2/M phase.91 Together, these studies strongly suggest that the use of peptidomimetics is a potent tool for developing new antitumor peptides. The main limitations in the use of these peptides are their poor bioavailability due to insolubility related to their intrinsic physicochemical properties, potential toxicity to host cells, tissue distribution, and poor pharmacokinetic issues. Despite these disadvantages, antitumor peptidomimetics have potential due to their high potency and specificity against malignant cells.
It is important to consider that further studies are needed to investigate the cost of large scale production of peptidomimetics and the transition of these peptides from the laboratory to the clinic to confirm that they provide an effective new class of therapeutic agents. However, the combination of the therapeutic use of peptidomimetics and conventional therapy against cancer (eg, chemotherapy, radiotherapy, or surgical procedures) can help in overcoming drug resistance in cancer cells. Increased funding and innovative research approaches to prepare peptidomimetics are required for practical use of these peptides as therapeutic agents.
Future directions for peptidomimetics research as a new generation of antimicrobial agents
Substantial progress has been achieved in the past decade with respect to the development of antimicrobial peptidomimetics that mimic the bactericidal activity and mode of action of AMPs. Since several classes of peptidomimetics have great potential as a new generation of antimicrobial agents due to their low immunogenicity and enhanced stability compared with AMPs, in the near future, it will be important to resolve issues of hemolytic activity and cytotoxicity of some antimicrobial peptidomimetics. Elucidating these concerns will ensure their future application in vivo. In this regard, the use of more cationic charges is likely to decrease toxicity. It will also be possible to gain further insight into the development of molecular design in peptidomimetics and to explore its medical and pharmacological applications. In fact, fine-tuning the biological activity of peptidomimetics may be readily achieved with the introduction of a variety of additional hydrophobic building blocks. In addition, mechanistic studies are needed to evaluate the possibility for antimicrobial peptidomimetics to induce drug resistance in bacteria, and research will be focused on the development of antimicrobial peptidomimetics against Gram-negative bacteria, as they are generally more difficult to kill than Gram-positive bacteria. Finally, an important challenge over the next decade will be to develop new potential drug delivery systems for peptidomimetics. In this context, nanoformulation approaches have emerged as an important tool to improve the delivery and stability of antimicrobial peptidomimetics.
In conclusion, peptidomimetics possess significant properties that support their inclusion in the generation of new antimicrobial agents. These antimicrobial molecules show potent bactericidal activity against drug-resistant bacterial strains. Moreover, antimicrobial peptidomimetics have several advantages over AMPs, including enhanced stability, cell specificity, and better tolerability. Furthermore, the synthetic flexibility of these molecules allows fast structure modifications to create novel antimicrobial peptidomimetics, having particular pharmacological properties. Finally, structure–activity relationships clear the way to establish peptidomimetic libraries, which can lead to the development of novel antimicrobial agents. A better understanding of the structural properties of peptidomimetics will potentially facilitate the practical use of these peptides as important therapeutic agents.
Acknowledgment
PMS is a researcher of Comisión de Operación y Fomento de Actividades Académicas, Estimulos al Desempeño de Investigadores, and Sistema Nacional de Investigadores.
Disclosure
The author reports no conflicts of interest in this work.
References
Livermore DM. Current epidemiology and growing resistance of gram-negative pathogens. Korean J Intern Med. 2012;27(2):128–142. | |
Scott RW, DeGrado WF, Tew GN. De novo designed synthetic mimics of antimicrobial peptides. Curr Opin Biotechnol. 2008;19(6):620–627. | |
Tew GN, Scott RW, Klein ML, Degrado WF. De novo design of antimicrobial polymers, foldamers, and small molecules: from discovery to practical applications. Acc Chem Res. 2010;43(1):30–39. | |
Wu M, Maier E, Benz R, Hancock RE. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry. 1999;38(22):7235–7242. | |
Friedrich CL, Moyles D, Beveridge TJ, Hancock RE. Antibacterial action of structurally diverse cationic peptides on gram-positive bacteria. Antimicrob Agents Chemother. 2000;44(8):2086–2092. | |
Zhang L, Dhillon P, Yan H, Farmer S, Hancock RE. Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2000;44(12):3317–3321. | |
Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24(12):1551–1557. | |
Méndez-Samperio P. Role of antimicrobial peptides in host defense against mycobacterial infections. Peptides. 2008;29(10):1836–1841. | |
Hancock RE, Lehrer R. Cationic peptides: a new source of antibiotics. Trends Biotechnol. 1998;16(2):82–88. | |
Marr AK, Gooderham WJ, Hancock RE. Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr Opin Pharmacol. 2006;6(5):468–472. | |
Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3(3):238–250. | |
Shai Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta. 1999;1462(1–2):55–70. | |
Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers. 2002;66(4):236–248. | |
Huang Y, Huang J, Chen Y. Alpha-helical cationic antimicrobial peptides: relationships of structure and function. Protein Cell. 2010;1(2):143–152. | |
Lehrer RI, Lu W. α-Defensins in human innate immunity. Immunol Rev. 2012;245(1):84–112. | |
Peters BM, Shirtliff ME, Jabra-Rizk MA. Antimicrobial peptides: primeval molecules or future drugs? PLoS Pathog. 2010;6(10):e1001067. | |
Karlsson AJ, Pomerantz WC, Weisblum B, Gellman SH, Palecek SP. Antifungal activity from 14-helical beta-peptides. J Am Chem Soc. 2006;128(39):12630–12631. | |
Karlsson AJ, Pomerantz WC, Neilsen KJ, Gellman SH, Palecek SP. Effect of sequence and structural properties on 14-helical beta-peptide activity against Candida albicans planktonic cells and biofilms. ACS Chem Biol. 2009;4(7):567–579. | |
Karlsson AJ, Flessner RM, Gellman SH, Lynn DM, Palecek SP. Polyelectrolyte multilayers fabricated from antifungal β-peptides: design of surfaces that exhibit antifungal activity against Candida albicans. Biomacromolecules. 2010;11(9):2321–2328. | |
Chongsiriwatana NP, Patch JA, Czyzewski AM, et al. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc Natl Acad Sci U S A. 2008;105(8):2794–2799. | |
Olsen CA, Ziegler HL, Nielsen HM, Frimodt-Møller N, Jaroszewski JW, Franzyk H. Antimicrobial, hemolytic, and cytotoxic activities of beta-peptoid-peptide hybrid oligomers: improved properties compared to natural AMPs. Chembiochem. 2010;11(10):1356–1360. | |
Kapoor R, Wadman MW, Dohm MT, Czyzewski AM, Spormann AM, Barron AE. Antimicrobial peptoids are effective against Pseudomonas aeruginosa biofilms. Antimicrob Agents Chemother. 2011;55(6):3054–3057. | |
Chongsiriwatana NP, Miller TM, Wetzler M, et al. Short alkylated peptoid mimics of antimicrobial lipopeptides. Antimicrob Agents Chemother. 2011;55(1):417–420. | |
Kapoor R, Eimerman PR, Hardy JW, Cirillo JD, Contag CH, Barron AE. Efficacy of antimicrobial peptoids against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2011;55(6):3058–3062. | |
Huang ML, Shin SB, Benson MA, Torres VJ, Kirshenbaum K. A comparison of linear and cyclic peptoid oligomers as potent antimicrobial agents. ChemMedChem. 2012;7(1):114–122. | |
Hua J, Yamarthy R, Felsenstein S, Scott RW, Markowitz K, Diamond G. Activity of antimicrobial peptide mimetics in the oral cavity: I. Activity against biofilms of Candida albicans. Mol Oral Microbiol. 2010;25(6):418–425. | |
Hua J, Scott RW, Diamond G. Activity of antimicrobial peptide mimetics in the oral cavity: II. Activity against periopathogenic biofilms and anti-inflammatory activity. Mol Oral Microbiol. 2010;25(6):426–432. | |
Obrecht D, Robinson JA, Bernardini F, et al. Recent progress in the discovery of macrocyclic compounds as potential anti-infective therapeutics. Curr Med Chem. 2009;16(1):42–65. | |
Srinivas N, Jetter P, Ueberbacher BJ, et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science. 2010;327(5968):1010–1013. | |
Hu Y, Li X, Sebti SM, Chen J, Cai J. Design and synthesis of AApeptides: a new class of peptide mimics. Bioorg Med Chem Lett. 2011;21(5):1469–1471. | |
Niu Y, Wu H, Li Y, et al. AApeptides as a new class of antimicrobial agents. Org Biomol Chem. 2013;11(26):4283–4290. | |
Niu Y, Hu Y, Wu H, Cai J. Synthesis of AApeptides. Methods Mol Biol. 2013;1081:35–46. | |
Ishitsuka Y, Arnt L, Majewski J, et al. Amphiphilic poly(phenyleneethynylene)s can mimic antimicrobial peptide membrane disordering effect by membrane insertion. J Am Chem Soc. 2006;128(40):13123–13129. | |
Claudon P, Violette A, Lamour K, et al. Consequences of isostructural main-chain modifications for the design of antimicrobial foldamers: helical mimics of host-defense peptides based on a heterogeneous amide/urea backbone. Angew Chem Int Ed Engl. 2010;49(2):333–336. | |
Dragulescu-Andrasi A, Rapireddy S, Frezza BM, Gayathri C, Gil RR, Ly DH. A simple gamma-backbone modification preorganizes peptide nucleic acid into a helical structure. J Am Chem Soc. 2006;128(31):10258–10267. | |
Tedeschi T, Sforza S, Dossena A, Corradini R, Marchelli R. Lysine-based peptide nucleic acids (PNAs) with strong chiral constraint: control of helix handedness and DNA binding by chirality. Chirality. 2005;17 Suppl:S196–S204. | |
Tedeschi T, Chiari M, Galaverna G, et al. Detection of the R553X DNA single point mutation related to cystic fibrosis by a “chiral box” D-lysine-peptide nucleic acid probe by capillary electrophoresis. Electrophoresis. 2005;26(22):4310–4316. | |
Inokuchi E, Yamada A, Hozumi K, et al. Design and synthesis of amidine-type peptide bond isosteres: application of nitrile oxide derivatives as active ester equivalents in peptide and peptidomimetics synthesis. Org Biomol Chem. 2011;9(9):3421–3427. | |
Padhee S, Hu Y, Niu Y, et al. Non-hemolytic α-AApeptides as antimicrobial peptidomimetics. Chem Commun (Camb). 2011;47(34):9729–9731. | |
Niu Y, Padhee S, Wu H, et al. Identification of γ-AApeptides with potent and broad-spectrum antimicrobial activity. Chem Commun (Camb). 2011;47(44):12197–12199. | |
Walsh PS, Kusaka R, Buchanan EG, et al. Cyclic constraints on conformational flexibility in γ-peptides: conformation specific IR and UV spectroscopy. J Phys Chem A. 2013;117(47):12350–12362. | |
James WH, Buchanan EG, Müller CW, et al. Evolution of amide stacking in larger γ-peptides: triamide H-bonded cycles. J Phys Chem A. 2011;115(47):13783–13798. | |
Wu H, Amin MN, Niu Y, et al. Solid-phase synthesis of γ-AApeptides using a submonomeric approach. Org Lett. 2012;14(13):3446–3449. | |
Niu Y, Jones AJ, Wu H, Varani G, Cai J. γ-AApeptides bind to RNA by mimicking RNA-binding proteins. Org Biomol Chem. 2011;9(19):6604–6609. | |
Yang Y, Niu Y, Hong H, et al. Radiolabeled γ-AApeptides: a new class of tracers for positron emission tomography. Chem Commun (Camb). 2012;48(63):7850–7852. | |
Niu Y, Bai G, Wu H, et al. Cellular translocation of a γ-AApeptide mimetic of Tat peptide. Mol Pharm. 2012;9(5):1529–1534. | |
Bai G, Padhee S, Niu Y, et al. Cellular uptake of an α-AApeptide. Org Biomol Chem. 2012;10(6):1149–1153. | |
Schmitt MA, Weisblum B, Gellman SH. Interplay among folding, sequence, and lipophilicity in the antibacterial and hemolytic activities of alpha/beta-peptides. J Am Chem Soc. 2007;129(2):417–428. | |
Mowery BP, Lee SE, Kissounko DA, et al. Mimicry of antimicrobial host-defense peptides by random copolymers. J Am Chem Soc. 2007;129(50):15474–15476. | |
Niu Y, Padhee S, Wu H, et al. Lipo-γ-AApeptides as a new class of potent and broad-spectrum antimicrobial agents. J Med Chem. 2012;55(8):4003–4009. | |
Ivankin A, Livne L, Mor A, et al. Role of the conformational rigidity in the design of biomimetic antimicrobial compounds. Angew Chem Int Ed Engl. 2010;49(45):8462–8465. | |
Kuroda K, Caputo GA. Antimicrobial polymers as synthetic mimics of host-defense peptides. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2013;5(1):49–66. | |
Takahashi H, Palermo EF, Yasuhara K, Caputo GA, Kuroda K. Molecular design, structures, and activity of antimicrobial peptide-mimetic polymers. Macromol Biosci. 2013;13(10):1285–1299. | |
Rekdal Ø, Haug BE, Kalaaji M, et al. Relative spatial positions of tryptophan and cationic residues in helical membrane-active peptides determine their cytotoxicity. J Biol Chem. 2012;287(1):233–244. | |
McGrath DM, Barbu EM, Driessen WH, et al. Mechanism of action and initial evaluation of a membrane active all-D-enantiomer antimicrobial peptidomimetic. Proc Natl Acad Sci U S A. 2013;110(9):3477–3482. | |
Barbu EM, Shirazi F, McGrath DM, et al. An antimicrobial peptidomimetic induces Mucorales cell death through mitochondria-mediated apoptosis. PLoS One. 2013;8(10):e76981. | |
Haug BE, Stensen W, Kalaaji M, Rekdal ø, Svendsen JS. Synthetic antimicrobial peptidomimetics with therapeutic potential. J Med Chem. 2008;51(14):4306–4314. | |
Lu F, Chi SW, Kim DH, Han KH, Kuntz ID, Guy RK. Proteomimetic libraries: design, synthesis, and evaluation of p53-MDM2 interaction inhibitors. J Comb Chem. 2006;8(3):315–325. | |
Niu Y, Wang RE, Wu H, Cai J. Recent development of small antimicrobial peptidomimetics. Future Med Chem. 2012;4(14):1853–1862. | |
Choi S, Isaacs A, Clements D, et al. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc Natl Acad Sci U S A. 2009;106(17):6968–6973. | |
Polymedix, Inc. Brilacidin (PMX-30063) Antibiotic Fact Sheet. Randor, PA: PolyMedix, Inc.; 2013. Available from: http://files.shareholder.com/downloads/ABEA-4ITCYZ/1997024712x0x611205/FEA66C67-D02F-4FF2-B230-AA711F46ED7F/PMX-30063_Antibiotic_Fact_Sheet_Nov_2012_.pdf. Accessed February 10, 2013. | |
Blanco J, Jacotot E, Cabrera C, et al. The implication of the chemokine receptor CXCR4 in HIV-1 envelope protein-induced apoptosis is independent of the G protein-mediated signalling. AIDS. 1999;13(8):909–917. | |
Tanaka G, Nakase I, Fukuda Y, et al. CXCR4 stimulates macropinocytosis: implications for cellular uptake of arginine-rich cell-penetrating peptides and HIV. Chem Biol. 2012;19(11):1437–1446. | |
Tamamura H, Fujii N. The therapeutic potential of CXCR4 antagonists in the treatment of HIV infection, cancer metastasis and rheumatoid arthritis. Expert Opin Ther Targets. 2005;9(6):1267–1282. | |
Tsutsumi H, Tanaka T, Ohashi N, et al. Therapeutic potential of the chemokine receptor CXCR4 antagonists as multifunctional agents. Biopolymers. 2007;88(2):279–289. | |
Tamamura H, Tsutsumi H, Masuno H, Fujii N. Development of low molecular weight CXCR4 antagonists by exploratory structural tuning of cyclic tetra- and pentapeptide-scaffolds towards the treatment of HIV infection, cancer metastasis and rheumatoid arthritis. Curr Med Chem. 2007;14(1):93–102. | |
Moncunill G, Armand-Ugón M, Clotet-Codina I, et al. Anti-HIV activity and resistance profile of the CXC chemokine receptor 4 antagonist POL3026. Mol Pharmacol. 2008;73(4):1264–1273. | |
Choi WT, Tian S, Dong CZ, et al. Unique ligand binding sites on CXCR4 probed by a chemical biology approach: implications for the design of selective human immunodeficiency virus type 1 inhibitors. J Virol. 2005;79(24):15398–15404. | |
LaLonde JM, Kwon YD, Jones DM, et al. Structure-based design, synthesis, and characterization of dual hotspot small-molecule HIV-1 entry inhibitors. J Med Chem. 2012;55(9):4382–4396. | |
Mbemba E, Benjouad A, Saffar L, Gattegno L. Glycans and proteoglycans are involved in the interactions of human immunodeficiency virus type 1 envelope glycoprotein and of SDF-1alpha with membrane ligands of CD4(+) CXCR4(+) cells. Virology. 1999;265(2):354–364. | |
Tanaka T, Nomura W, Narumi T, et al. Structure-activity relationship study on artificial CXCR4 ligands possessing the cyclic pentapeptide scaffold: the exploration of amino acid residues of pentapeptides by substitutions of several aromatic amino acids. Org Biomol Chem. 2009;7(18):3805–3809. | |
Demmer O, Dijkgraaf I, Schumacher U, et al. Design, synthesis, and functionalization of dimeric peptides targeting chemokine receptor CXCR4. J Med Chem. 2011;54(21):7648–7662. | |
Gravel S, Malouf C, Boulais PE, et al. The peptidomimetic CXCR4 antagonist TC14012 recruits beta-arrestin to CXCR7: roles of receptor domains. J Biol Chem. 2010;285(49):37939–37943. | |
Veldkamp CT, Ziarek JJ, Peterson FC, Chen Y, Volkman BF. Targeting SDF-1/CXCL12 with a ligand that prevents activation of CXCR4 through structure-based drug design. J Am Chem Soc. 2010;132(21):7242–7243. | |
O’Brien WA, Sumner-Smith M, Mao SH, Sadeghi S, Zhao JQ, Chen IS. Anti-human immunodeficiency virus type 1 activity of an oligocationic compound mediated via gp120 V3 interactions. J Virol. 1996;70(5):2825–2831. | |
Doranz BJ, Grovit-Ferbas K, Sharron MP, et al. A small-molecule inhibitor directed against the chemokine receptor CXCR4 prevents its use as an HIV-1 coreceptor. J Exp Med. 1997;186(8):1395–1400. | |
Zhou N, Zhang X, Fan X, et al. The N-terminal domain of APJ, a CNS-based coreceptor for HIV-1, is essential for its receptor function and coreceptor activity. Virology. 2003;317(1):84–94. | |
Zhang J, Mulvenon A, Makarov E, et al. Antiviral peptide nanocomplexes as a potential therapeutic modality for HIV/HCV co-infection. Biomaterials. 2013;34(15):3846–3857. | |
Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5(3):219–234. | |
Hoskin DW, Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim Biophys Acta. 2008;1778(2):357–375. | |
Kim JM, Lee YH, Ku CR, Lee EJ. The cyclic pentapeptide d-Arg3FC131, a CXCR4 antagonist, induces apoptosis of somatotrope tumor and inhibits tumor growth in nude mice. Endocrinology. 2011;152(2):536–544. | |
Shin DY, Kim KJ, Ku CR, et al. Different CXCR4 expression according to various histologic subtype of papillary thyroid carcinoma. Endocr Pathol. 2013;24(4):169–176. | |
Jenssen H, Hamill P, Hancock RE. Peptide antimicrobial agents. Clin Microbiol Rev. 2006;19(3):491–511. | |
Papo N, Shai Y. Host defense peptides as new weapons in cancer treatment. Cell Mol Life Sci. 2005;62(7–8):784–790. | |
Schweizer F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur J Pharmacol. 2009;625(1–3):190–194. | |
Siddiquee KA, Gunning PT, Glenn M, et al. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem Biol. 2007;2(12):787–798. | |
McDaniel JR, Callahan DJ, Chilkoti A. Drug delivery to solid tumors by elastin-like polypeptides. Adv Drug Deliv Rev. 2010;62(15):1456–1467. | |
Onwulata CI. Encapsulation of new active ingredients. Annu Rev Food Sci Technol. 2012;3:183–202. | |
Wang X, Zhang X. Separation, antitumor activities, and encapsulation of polypeptide from Chlorella pyrenoidosa. Biotechnol Prog. 2013;29(3):681–687. | |
Hou L, Zhao X, Wang P, Ning Q, Meng M, Liu C. Antitumor activity of antimicrobial peptides containing CisoDGRC in CD13 negative breast cancer cells. PLoS One. 2013;8(1):e53491. | |
Gu Y, Liu SL, Ju WZ, Li CY, Cao P. Analgesic-antitumor peptide induces apoptosis and inhibits the proliferation of SW480 human colon cancer cells. Oncol Lett. 2013;5(2):483–488. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.