")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Peptide gH625 enters into neuron and astrocyte cell lines and crosses the blood–brain barrier in rats
Authors Valiante S, Falanga A , Cigliano L, Iachetta G, Busiello RA, La Marca V, Galdiero M, Lombardi A, Galdiero S
Received 19 November 2014
Accepted for publication 29 December 2014
Published 10 March 2015 Volume 2015:10(1) Pages 1885—1898
DOI https://doi.org/10.2147/IJN.S77734
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Salvatore Valiante,1,* Annarita Falanga,2,3,* Luisa Cigliano,1 Giuseppina Iachetta,1 Rosa Anna Busiello,1 Valeria La Marca,1 Massimiliano Galdiero,4 Assunta Lombardi,1 Stefania Galdiero1,2
1Department of Biology, 2Department of Pharmacy, 3DFM Scarl, University of Naples Federico II, 4Department of Experimental Medicine, II University of Naples, Naples, Italy
*These authors contributed equally to this paper and are considered joint first authors
Abstract: Peptide gH625, derived from glycoprotein H of herpes simplex virus type 1, can enter cells efficiently and deliver a cargo. Nanoparticles armed with gH625 are able to cross an in vitro model of the blood–brain barrier (BBB). In the present study, in vitro experiments were performed to investigate whether gH625 can enter and accumulate in neuron and astrocyte cell lines. The ability of gH625 to cross the BBB in vivo was also evaluated. gH625 was administered in vivo to rats and its presence in the liver and in the brain was detected. Within 3.5 hours of intravenous administration, gH625 can be found beyond the BBB in proximity to cell neurites. gH625 has no toxic effects in vivo, since it does not affect the maximal oxidative capacity of the brain or the mitochondrial respiration rate. Our data suggest that gH625, with its ability to cross the BBB, represents a novel nanocarrier system for drug delivery to the central nervous system. These results open up new possibilities for direct delivery of drugs into patients in the field of theranostics and might address the treatment of several human diseases.
Keywords: drug delivery, neurons, astrocytes, blood–brain barrier, peptide
Introduction
Advances in molecular biology, nanotechnology, and pharmaceutical chemistry have led to the rapid evolving interdisciplinary field of nanomedicine, with the production of several new classes of imaging agents and highly potent drugs. One of the key topics in nanomedicine is targeted delivery of drugs to specific sites of disease in the body. Drugs are often characterized by physicochemical and/or biological features that make their use not ideal in humans; however, nanosized delivery systems (liposomes, micelles, and polymeric systems) can circumvent these problems by improving the therapeutic index.1,2 Apart from a few exceptions, nanocarriers have met with moderate success, mainly because of their behavior in vivo.3 Free diffusion of circulating molecules from the blood into the brain is made difficult by the presence of the blood–brain barrier (BBB), which is a complex physiological checkpoint in the central nervous system, preventing passage of both beneficial drugs and potentially toxic molecules.4,5 For this reason, many diseases affecting the brain still cannot be treated effectively. Therefore, development of novel systems that can significantly enhance the delivery of therapeutic agents across the BBB represents a fundamental challenge for the treatment of a number of brain diseases.6,7
Cationic cell-penetrating peptides are a group of short peptides able to cross the membrane bilayer using a mechanism mediated by endocytosis, and are well suited for development as drug delivery vehicles to penetrate the BBB. Their advantages as delivery enhancers are their relatively small molecular weight, ease of synthesis, relatively low cytotoxicity and immunogenicity, and degradation in vivo to naturally occurring compounds.8 The mechanism of their uptake is still debated, although endocytosis is believed to be the main pathway involved;9 thus, the cargo is trapped in endosomes, eventually ending in lysosomes, and its intracellular bioavailability is decreased. To be able to avoid the endocytic pathway, it is important to design new molecules operating under different mechanisms of uptake. The new challenge of recent research is the use of hydrophobic peptides; these have a key role in several processes involving temporary membrane destabilization and subsequent reorganization, such as crossing of biological membranes and promoting lipid membrane-reorganizing processes (fusion or pore formation).10,11 The intrinsic ability of virus-derived peptides to induce membrane perturbation may be useful in the design of delivery vehicles.12–14
The peptide gH625, previously identified as a membrane-perturbing domain in glycoprotein H (gH) of herpes simplex virus 1, has been demonstrated to be able to traverse the membrane bilayer.15 Further, gH625 has been used extensively for vector-mediated strategies that enable passage of a large variety of small molecules as well as proteins across cell membranes in vitro.16–20 Conjugation of gH625 to the surface of nanoparticles also enhances their transport across the BBB, as previously demonstrated in an in vitro model of the BBB.19
Whether or not multifunctional nanodevices, designed and tested in vitro, work properly in vivo in a mammalian host is still not fully understood. In vivo studies are necessary to address this issue, thus validating design strategies and facilitating optimization and further functionalization. Although numerous studies have shown that gH625 is an efficient carrier for bioactive cargoes in vitro,21 these results do not guarantee that gH625 can be developed into a useful pharmaceutical delivery platform.
The aim of the current study was to evaluate whether gH625 can enter into brain cells in vitro and whether it crosses the BBB when administered to rats in vivo. We investigated cell viability and mitochondrial functionality/maximal oxidative capacity on exposure of neuroblastoma and glioblastoma-astrocytoma cell lines to gH625. The passage of gH625 across the BBB in vivo was evaluated after its intravenous administration in rats. Our results may have importance for the treatment of brain diseases and tracking of nanosystems in vivo, bringing us a step closer to having available a nanocarrier for treatment as well as monitoring of brain-related disorders.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium and fetal bovine serum were purchased from BioWhittaker (Verseviers, Belgium). L-glutamine, penicillin, and streptomycin were from Gibco (Life Technologies, Monza, Italy). Cell culture flasks (25 cm2), 96-well cell culture plates, and sterile pipettes were obtained from Becton-Dickinson (Milan, Italy). MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] was from Sigma-Aldrich (St Louis, MO, USA). Cytochrome c, succinate rotenone, malonate, sodium ascorbate, and N,N,N’,N’-tetramethyl-p-phenylenediamine were also from Sigma-Aldrich.
Peptide synthesis
The gH625 peptide (H2N-HGLASTLTRWAHYNALIRAFGGG-CONH2; molecular weight 2,461 Da) was synthesized using the standard solid-phase-9-fluorenylmethoxycarbonyl (Fmoc) method as previously reported.13 The peptide was obtained with good yields (30%–40%). Labeling was performed on resin-bound peptides as previously reported by Rapaport and Shai.22 Specifically, 30–70 mg of resin-bound peptide (10–25 μmol) was treated with piperidine in N,N-dimethylformamide to remove the Fmoc-protecting group of the N-terminal amino acid. The resin-bound peptide was then reacted with NBD-Cl (4-chloro-7-nitrobenz-2-oxa-1,3-diazole) in N,N-dimethylformamide (3–4 equivalents). The resin-bound peptides were washed thoroughly after 24 hours with methylene chloride. The peptides were cleaved from the resin and purified as previously reported for non-labeled peptides. A major product was obtained with an NBD moiety attached to the peptide’s N-terminal amino group. The identity of this compound was confirmed by liquid chromatography-mass spectrometry. The peptide was dissolved in phosphate-buffered saline as vehicle.
Cell culture
The human neuroblastoma cell line (SH-SY5Y) was obtained from the American Type Culture Collection (Rockville, MD, USA). The human glioblastoma-astrocytoma cell line (U-87 MG) was kindly provided by Pasquale Barba (Institute of Genetics and Biophysics, Naples, Italy). Both cell lines (500,000 cells) were seeded in 50 mL tissue culture flasks (25 cm2 surface), and grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (complete medium) at 37°C under a humidified atmosphere of 5% CO2 in air. The medium was changed twice a week, and cells were subcultivated when confluent.
MTT assay for analysis of cell viability
The MTT assay was performed to evaluate the viability and metabolic state of the cells. MTT is taken up by cells and transformed into formazan by mitochondrial succinate dehydrogenase. Accumulation of formazan directly reflects the activity of the mitochondria, an indirect measurement of cell viability. In detail, in order to evaluate the effect of gH625 on the viability of neurons and astrocytes, SH-SY5Y (at a density of 8,000 cells/well) and U-87 MG (at a density of 5,000 cells/well) were seeded into 96 well-plates and incubated with increasing concentrations of peptide (0.5, 1, 5, 10, and 15 μM) for 20 hours. As negative controls, we used the phosphate-buffered saline. In particular, the same volumes (corresponding to the added peptide solutions) of the phosphate-buffered saline were added to the control wells; no significant differences were found. As a positive control, we added 400 or 800 μM of H2O2, which is known to induce cell death in these cell lines. At the end of each incubation, the medium was removed and the cells were incubated with MTT solution (0.5 mg/mL) for 3 hours at 37°C. The medium with MTT solution was then gently removed, and the formazan crystals were dissolved in isopropanol with, 0.1 M HCl. The optical density was detected with a microplate reader at 590 nm.
Fluorescence analysis in vitro
The SH-SY5Y and U-87 MG cell lines were then used to assess uptake of gH625. Cells were seeded at a density of 60,000/well on four-well chamber slides (x-well; Sarstedt AG & Co, Nümbrecht, Germany). Each well was treated with gH625 1 or 5 μM for 2 hours. The cells were then fixed in ice-cold methanol for 10 minutes and mounted with a coverslip in glycerol/phosphate-buffered saline 1:1. The cell nuclei were counterstained with DAPI (4′-6 diamidino-2-phenylindole) dilactate (Life Technologies). The slides were then analyzed using an epifluorescent microscope (Axioskop; Zeiss, Oberkochen, Germany), mounting the 40× objective with a numerical aperture of 1.3. Images were acquired using an AxioCam MR c5 (Zeiss) with differential interference contrast, a fluorescein isothiocyanate channel (λex 488 nm; λem 515 nm) and a DAPI channel (λex 350 nm; λem 461 nm).
Spectrofluorimetric analysis in vitro
To evaluate the percentage of internalized peptide, gH625 was administered at 1 or 5 μM for 2 hours to SH-SY5Y and U-87 MG cells previously seeded (5×104 cell/well) for 20 hours in a white-bottomed 96-multiwell plate without serum, suitable for spectrofluorimetric analysis. After incubation, the cells were rinsed twice with phosphate-buffered saline for 5 minutes and then lysed with 200 μL/well of lysis buffer in phosphate-buffered saline (Tris-HCl 10 mM, ethylenediaminetetraacetic acid 5 mM, NaCl 200 mM, 0.2% sodium dodecyl sulfate, proteinase K 100 μg/mL) in order to release the internalized gH625. The cell lysate was filtered through a 0.45 μm nylon membrane filter and transferred to new wells. The plates were analyzed with an Infinite 200 M spectrofluorimeter (Tecan, Männedorf, Switzerland) equipped with a fluorescence filter (λex 485 nm; λem 560 nm). The standard curve (R²=0.965) was obtained with different concentrations of gH625, ie, 0.625 μM, 1.25 μM, 2.5 μM, and 5 μM (data not shown) in order to obtain the accurate concentration of internalized peptide. Spectrofluorimetric assays were performed in biological and technical triplicates.
Animals
Male Wistar rats (250–300 g) living in a temperature-controlled room at 24°C were kept, one per cage, under an artificial lighting regime of 12 hours light and 12 hours darkness. A commercial mash was available ad libitum and the animals had free access to water. The study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments at the University of Napoli Federico II (Permit 2012/000180). All efforts were made to minimize suffering. The rats were divided in two groups, each consisting of three rats: one group received a single intravenous administration of vehicle and the other group received a single intravenous administration of gH625-NBD at a dose of 40 μg/100 g body weight or vehicle. The rats were anesthetized by intraperitoneal administration of Zoletil® 40 mg/100 g and then euthanized by decapitation 3.5 hours after administration of gH625-NBD. The liver and brain were rapidly dissected sagittally and divided in two symmetrical parts. One part was immediately processed for preparation of homogenate and the other was used for histological analysis.
Stability of gH625 in serum
For the plasma stability assay, 0.15 mg of gH625 was added to 200 μL of fresh rat serum (25% v/v in phosphate-buffered saline) in a microvial and the mixture was incubated at 37°C. Aliquots (25 μL) were taken at 0, 0.5, 1, 2, 2.5, 3, and 3.5 hours, and quenched by adding 25 μL of a 7 M urea solution at 95°C for 2 minutes. The proteins were precipitated with 50 μL of trichloroacetic acid (10% v/v) and centrifuged at 13,400 rpm for 15 minutes. The samples (8 μg) withdrawn were analyzed by high-performance liquid chromatography on a Kinetex 5 μ C18 100 Å 150×4.60 mm, using a linear gradient of 10% to 40% B (acetonitrile, 0.1% trifluoroacetic acid) in A (H2O, 0.1% trifluoroacetic acid) over 5 minutes, followed by a linear gradient of 40% to 80% B in A over 20 minutes (1.0 mL per minute).
Preparation of tissue homogenates
To evaluate the mitochondrial respiration rate and cytochrome oxidase activity, brain and liver fragments were gently homogenized an isolation medium consisting of 220 mM mannitol, 70 mM sucrose, 20 mM Tris-HCl, 1 mM ethylenediaminetetraacetic acid, 5 mM ethylene glycol tetraacetic acid and bovine serum albumin 1% (pH 7.4) with a Potter-Elvehjem homogenizer (dilution 1:5 weight/volume). The homogenates were left on ice until detection of cytochrome oxidase activity or mitochondrial respiration rate.
Fluorescence histological analysis ex vivo
As soon as the brains and livers were dissected, they were fixed in Bouin’s fluid (71% picric acid, 5% acetic acid, 24% formaldehyde) for 24 hours and embedded in paraffin wax (Carlo Erba, Milan, Italy). Sagittal 10 μm brain sections and 7 μm sections of liver were treated with 70% ethanol supplemented with 0.1% Sudan Black B for 20 minutes to minimize autofluorescence of tissues. The slides were then washed three times for 5 minutes with Tris-buffered saline containing 0.02% Tween 20 to remove the excess Sudan Black B.23,24 Sections were then stained with hemallum to evaluate tissue morphology. After dehydration, the sections were mounted with DPX mountant (Sigma-Aldrich). Images were acquired using the AxioCam MR c5 in bright field with a 488 nm ex/515 nm em filter. Images were then analyzed using ImageJ 1.48 software.11
Immunofluorescence analysis
Brain sections were dewaxed and hydrated. Antigen unmasking was performed with citrate buffer (pH 6.0) twice for 10 minutes in a microwave at 96°C (for anti-glial fibrillary acidic protein [GFAP] antibody, see below). Immunolocalizations were carried out with primary antibodies (negative technical controls were obtained by omitting primary antibodies) against specific endothelial protein found in areas with BBB (mouse monoclonal [SMI-71] anti-BBB), anti-GFAP (Abcam, Cambridge, MA, USA), or anti-tubulin III to determine if the gH625-NBD signal colocalized with brain endothelial cells, astrocytes, or neurons, respectively. To visualize antibodies, two secondary antibodies were used, ie, Dylight 594 (Abcam) to detect anti-BBB and AlexaFluor 594 (Life Technologies) to detect anti-GFAP and anti-tubulin III. Images were acquired using the AxioCam MR c5 with λex 488 nm/λem 515 nm and λex 594 nm/λem 618 nm filters. Five images for each experimental class were then analyzed with ImageJ 1.48 software or Zen 2012; the Deconvolutionlab plugin was used to deconvolve image channels using the Tikhonov-Miller algorithm;11 the Colocalization Colormap plugin was then used to evaluate the degree of correlation between pairs of pixels in the red and green channels, resulting in distribution of the values for the normalized mean deviation product (nMDP) and the index of correlation as the fraction of positively correlated pixels in the image.25 To evaluate the green fluorescence inside and outside the BBB vessels, the mean gray values of ten sections of controls (vehicle or gH625 without NBD; n=3) and treated animals (n=3) were measured within and outside the region of interest (ROI) of the BBB vessels. These values were then divided for the correspondent ROI area (μm2) and the ratio between the outside and inside values was indicated. The two-tailed unpaired t-test was used, and the values were considered statistically significant at P<0.01.
Detection of maximal oxidative capacity of cell lines and tissue homogenate
Cytochrome oxidase (COX) activity was used as a marker for the maximal oxidative capacity of the cells and tissues. In order to detect the effect of gH625 on COX activity, SH-SY5Y cells and U-87 MG cells were incubated with increasing concentrations of peptide (0.5 to 15 mM) for 20 hours, after which the cells were lysed and homogenized with a Polytron® for 5 seconds. To detect cytochrome oxidase activity in the liver and brain homogenates, the homogenates were diluted 1:2 (v/v) with Lubrol (100 mg/g tissue) in order to unmask the enzyme activity. The homogenates were then left standing in ice for 30 minutes.
Cytochrome oxidase activity was determined polarographically as described by Lanni et al.26 A Clark-type oxygen electrode (Rank Brothers, Botttisham, UK) with an incubation chamber maintained at 37°C was used for detection of oxygen consumption.
Cell lysates (corresponding to 45,000 cells) were incubated in 0.5 mL of reaction medium containing 30 μM cytochrome c, 4 μM rotenone, 0.5 mM dinitrophenol, 10 mm sodium malonate, and 75 mm HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer at pH 7.4. COX activity was measured as the difference between the rate of oxygen consumption observed after addition of substrate (4 mM sodium ascorbate with 0.3 mM N,N,N’,N’-tetramethyl-p-phenylenediamine) and cell lysate, and after addition of substrate alone, in order to take into account the auto-oxidation of ascorbate.
The homogenate (300 μg protein) was incubated in 1 mL of respiratory buffer (80 mM KCl, 50 mM HEPES, 1 mM ethylene glycol tetraacetic acid, 5 mM KH2PO4, 2 mM MgCl2, and 1% bovine serum albumin, pH 7.4). Oxidation of succinate, in the presence of rotenone, was used to detect mitochondrial respiration. To this end, the incubation medium was supplemented with 4 μM rotenone, and the homogenate was energized with 5 mM succinate in both the absence of adenosine diphosphate (ADP, to obtain an indication of basal respiration, in which there is no phosphorylation of ADP to adenosine triphosphate [ATP]) and in the presence of 200 μM ADP (to obtain indications on respiration in which phosphorylation of ADP is at the maximal rate [ADP-stimulated respiration]).
In order to be able to attribute the detected oxygen consumption to mitochondrial respiration or to cytochrome oxidase activity, 1 mM KCN (a known inhibitor of COX and mitochondrial respiration rate) was added to the assay medium. Complete inhibition of oxygen consumption by KCN was observed, indicating that oxygen consumption was due to the activity of cytochrome oxidase or to mitochondrial respiration rate.
Results
Peptide synthesis
The gH625 peptide was obtained with a good yield of the purified compound (30%–40%) and with purity higher than 98%. The peptide sequence was also obtained labeled at the N-terminus with the NBD group.
Effect of gH625 on viability and maximal oxidative capacity of SH-SY5Y or U-87 MG cells
The toxicity of gH625 in SH-SY5Y (neuroblastoma) cells and U-87 MG (astrocytoma) cells was tested using the MTT assay. Incubation of SH-SY5Y cells (Figure 1A) and U-87 MG cells (Figure 1B) to gH625 at a concentration of 0.5–15 μM for 20 hours had no significant effect on cell viability. These results suggest that the peptide is not toxic to brain cell populations at the concentrations herein utilized. As a positive control, we added H2O2 (400 or 800 μM) to the SH-SY5Y and U-87 MG cell lines; 400 μM H2O2 induced death of 30% and 22% of SH-SY5Y cells and U-87 MG cells, respectively. The higher (800 μM) concentration of H2O2 induced a death of 45% and 52% of SH-SY5Y and U-87 MG cells, respectively (data not shown).
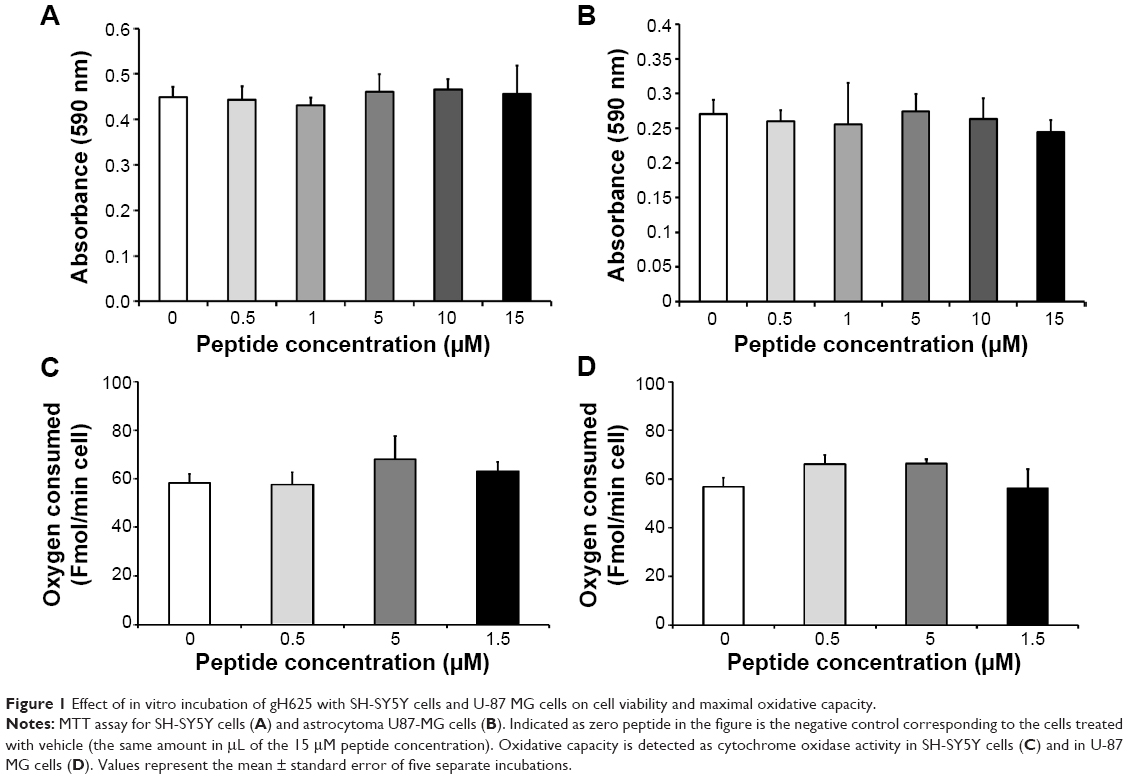 | Figure 1 Effect of in vitro incubation of gH625 with SH-SY5Y cells and U-87 MG cells on cell viability and maximal oxidative capacity. |
Exposure of SH-SY5Y cells to 0–15 μM gH625 for 20 hours did not significantly affect their maximal oxidative capacity, as detected as cytochrome oxidase activity (Figure 1C). gH625 was also ineffective when incubated with U-87 MG cells, regardless of the concentration used (Figure 1D).
Fluorescence analysis in vitro
To confirm internalization of gH625, we examined its cellular uptake by fluorescence microscopy. SH-SY5Y and U-87 MG cells showed significant uptake of gH625 after 2 hours of incubation. The peptide was efficiently internalized in both cell lines (Figure 2E and K). NBD alone was almost unable to penetrate into the cells, as previously reported.16
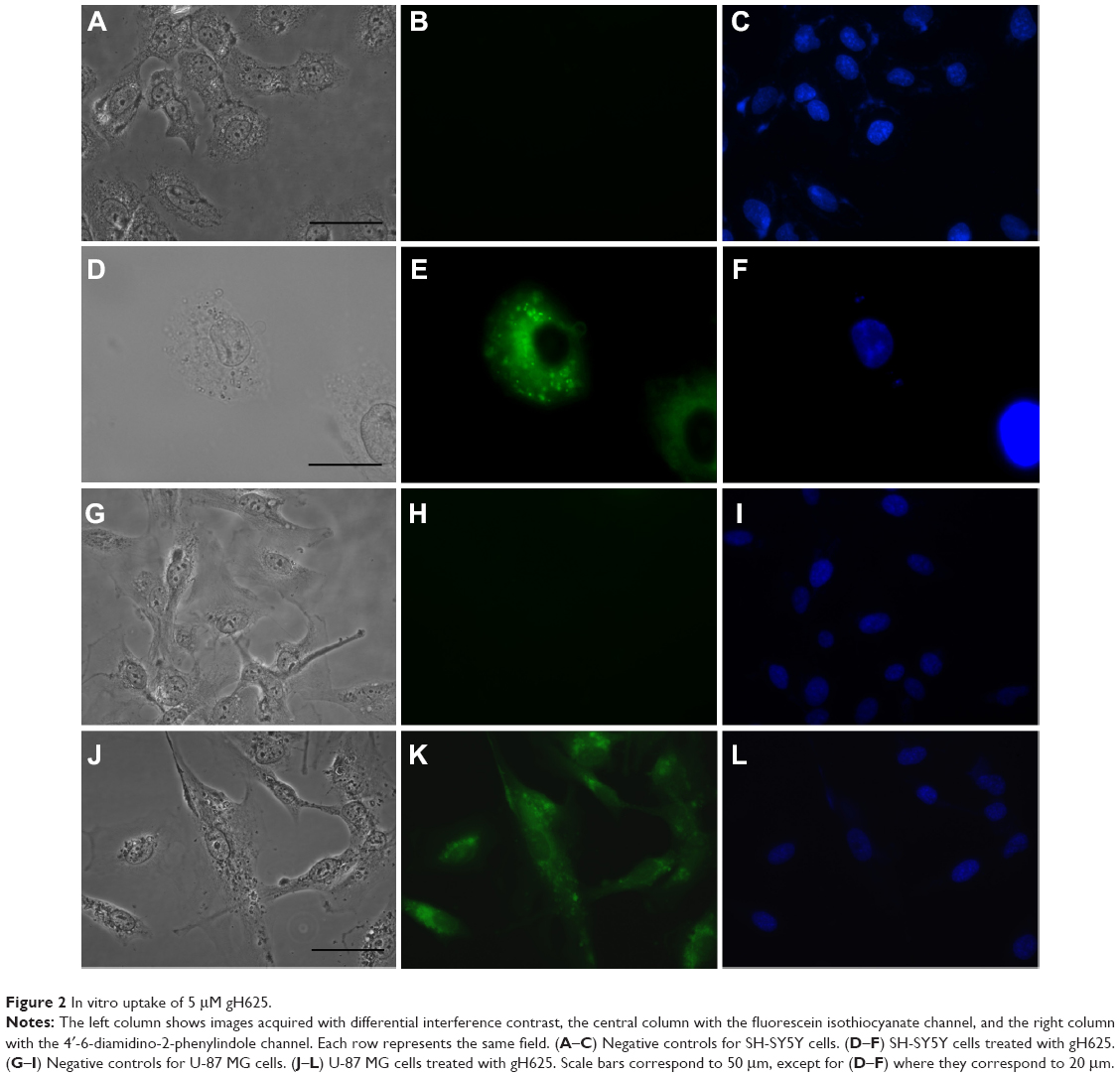 | Figure 2 In vitro uptake of 5 μM gH625. |
Spectrofluorimetric analysis in vitro
A spectrofluorimetric analysis was carried out to evaluate the uptake of gH625 in both cell lines in a quantitative manner. As shown in Figure 3, the data clearly confirm the results obtained by microscopy. Indeed, SH-SY5Y (Figure 3A) and U-87 MG (Figure 3B) cells efficiently accumulated gH625. At low concentration (1 μM), approximately 30% of gH625 was internalized, while uptake was nearly complete (80%–90%) at a concentration of 5 μM (Figure 3).
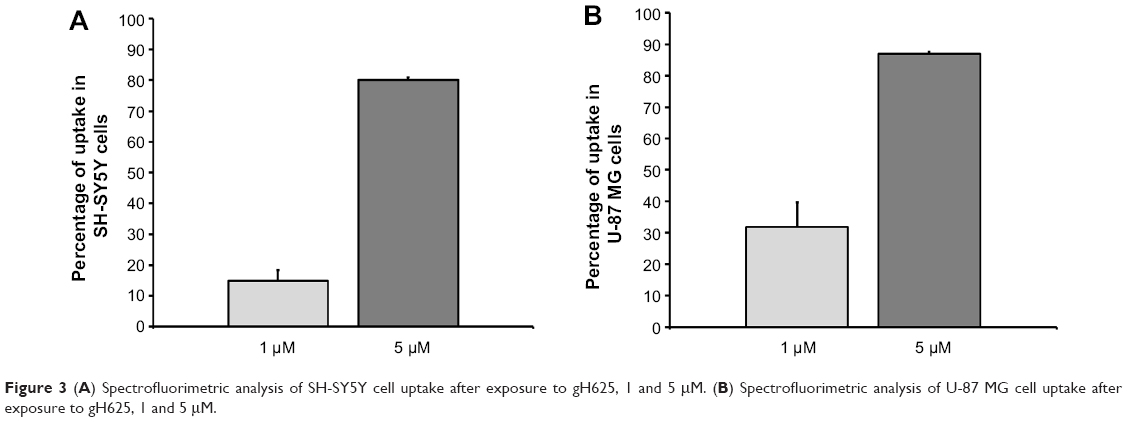 | Figure 3 (A) Spectrofluorimetric analysis of SH-SY5Y cell uptake after exposure to gH625, 1 and 5 μM. (B) Spectrofluorimetric analysis of U-87 MG cell uptake after exposure to gH625, 1 and 5 μM. |
Stability of gH625 in plasma
The main obstacle to the application of peptides for drug delivery is their instability due to their general susceptibility to proteolysis in blood. Herein, we investigated the stability of gH625 in rat plasma. gH625 was incubated at 37°C with fresh rat serum. Based on reverse-phase high-performance liquid chromatography analyses (Figure 4), it was evident that gH625 starts to be degraded after 1 hour of incubation but the intact peptide is still present after 3.5 hours of incubation (Figure 5).
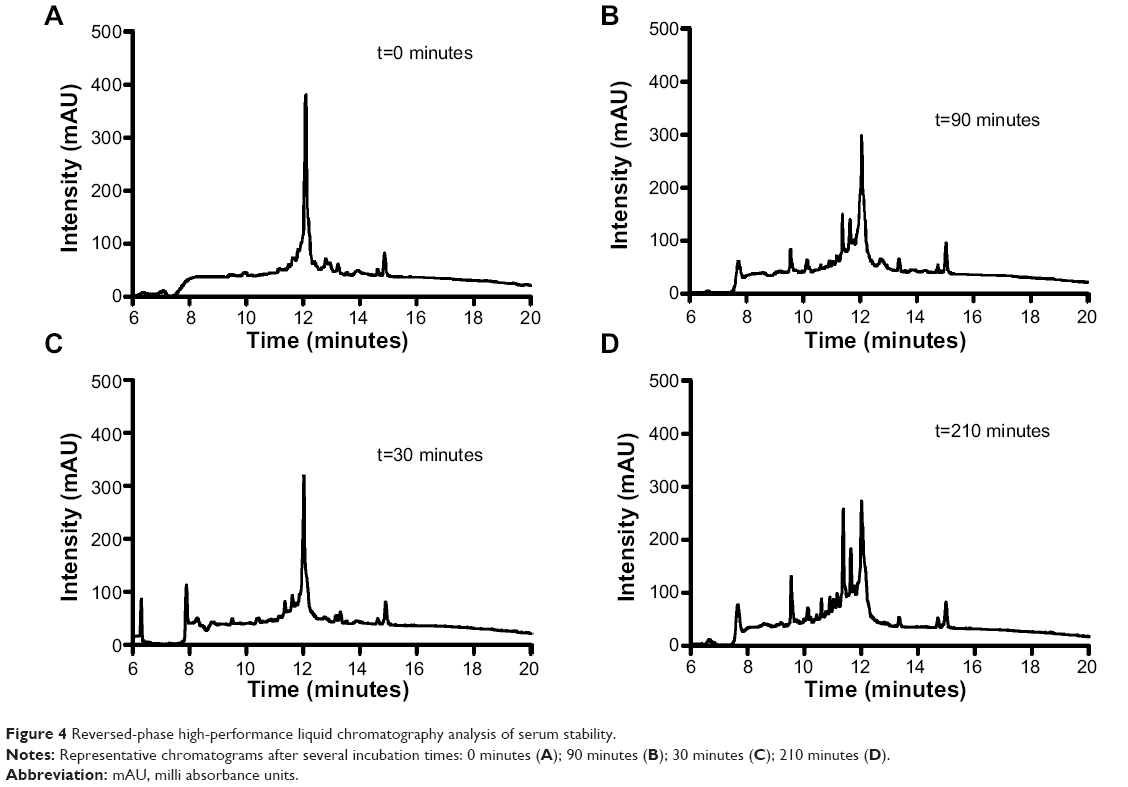 | Figure 4 Reversed-phase high-performance liquid chromatography analysis of serum stability. |
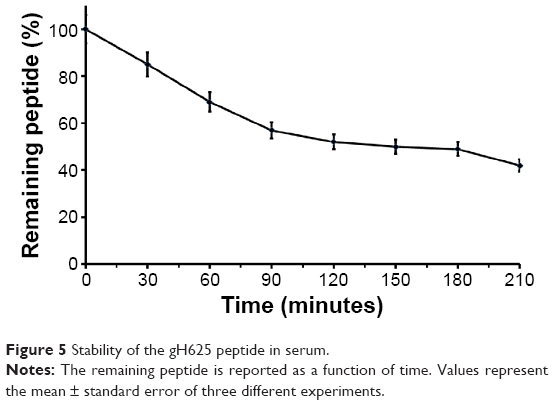 | Figure 5 Stability of the gH625 peptide in serum. |
Fluorescence histological analysis ex vivo
In order to perform the histological analysis, dissected brain and liver tissues were fixed and stained and images were acquired and analyzed. The fluorescence images clearly show that within 3.5 hours of its in vivo administration, gH625 was partly taken up by Kupffer cells in the liver, which are clearly distinguishable from hepatocytes because they possess the typical morphology of macrophages, and showed overfilled cytoplasm (Figure 6C and D). In addition, gH625 reaches the BBB and accumulates in the extensions of nerve cells (Figure 6G and H).
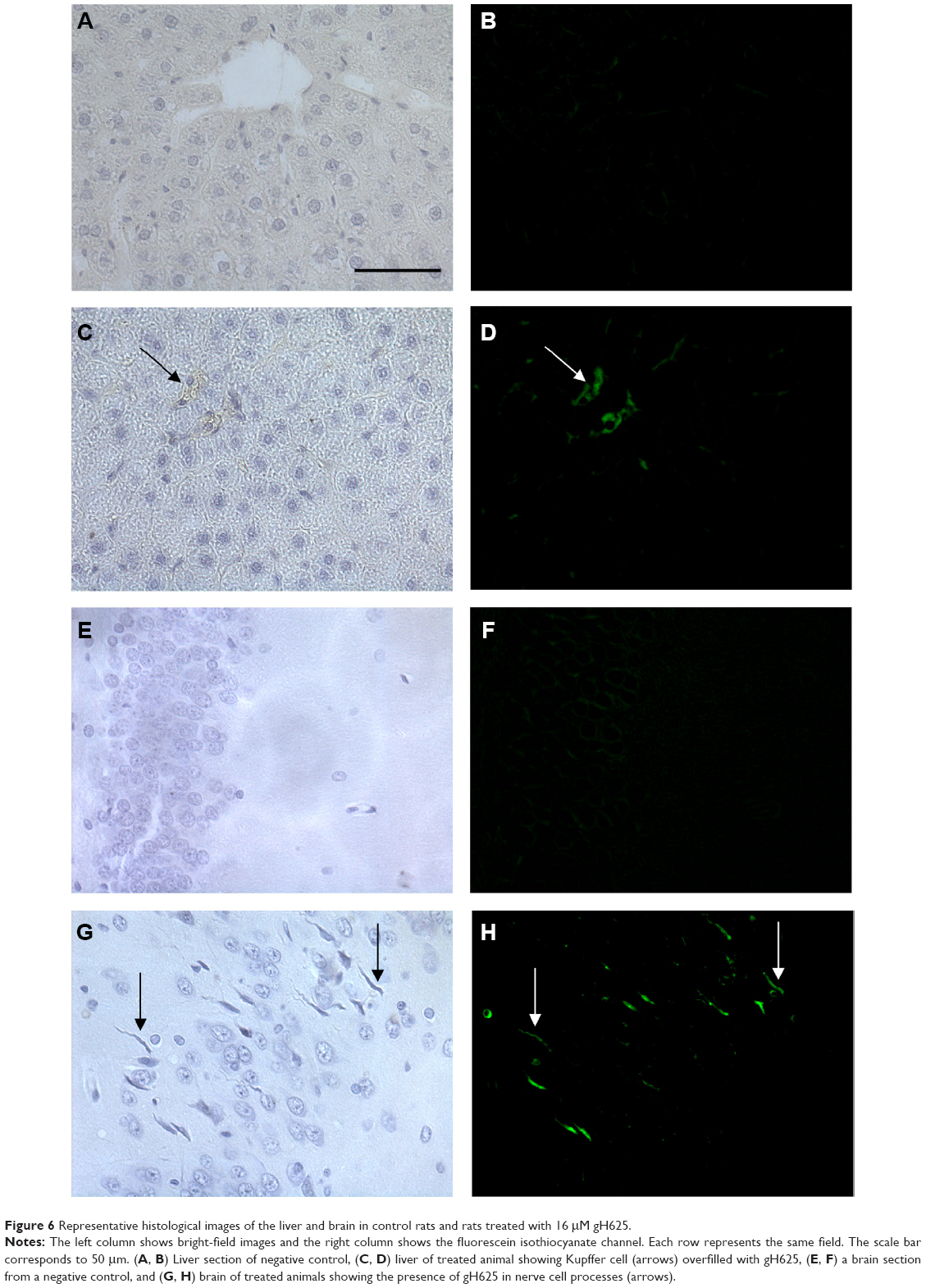 | Figure 6 Representative histological images of the liver and brain in control rats and rats treated with 16 μM gH625. |
Immunofluorescence analysis
By evaluation of fluorescence levels, we demonstrated that gH625 passes significantly through the BBB compared to controls (Figure 7). Immunofluorescence demonstrated that gH625 accumulates in the blood vessels of the brain in treated rats. No signal was found in the negative control for the immunofluorescence technique (Figure 8A–a4). Low spurious fluorescent signals arose from scattered red blood cells in the control animals (Figure 8B–b4). There was little or no colocalization between gH625 and astrocytes surrounding the BBB, as shown by immunofluorescence and nMDP color images (Figure 8C–c4). There was no colocalization between gH625 and the BBB in the control animals (Figure 8D–d4). More colocalization was found between gH625 and the endothelial BBB on immunofluorescence and nMDP color images (Figure 8E–e4). It is of interest that some cells in the cerebral cortex, which morphologically and by immunofluorescence can be ascribed to neurons and their processes beyond the BBB were labeled for gH625, indicating the passage of gH625 through the BBB (Figure 8F–f4, Figure 9). The nMDP distribution values (Figure 10A and C) and the index of correlation (Figure 10B and D) suggest that gH625 labeling is correlated mainly with BBB than to astrocytes.
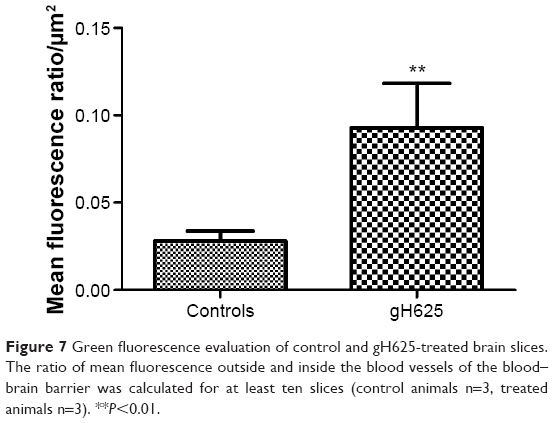 | Figure 7 Green fluorescence evaluation of control and gH625-treated brain slices. The ratio of mean fluorescence outside and inside the blood vessels of the blood–brain barrier was calculated for at least ten slices (control animals n=3, treated animals n=3). **P<0.01. |
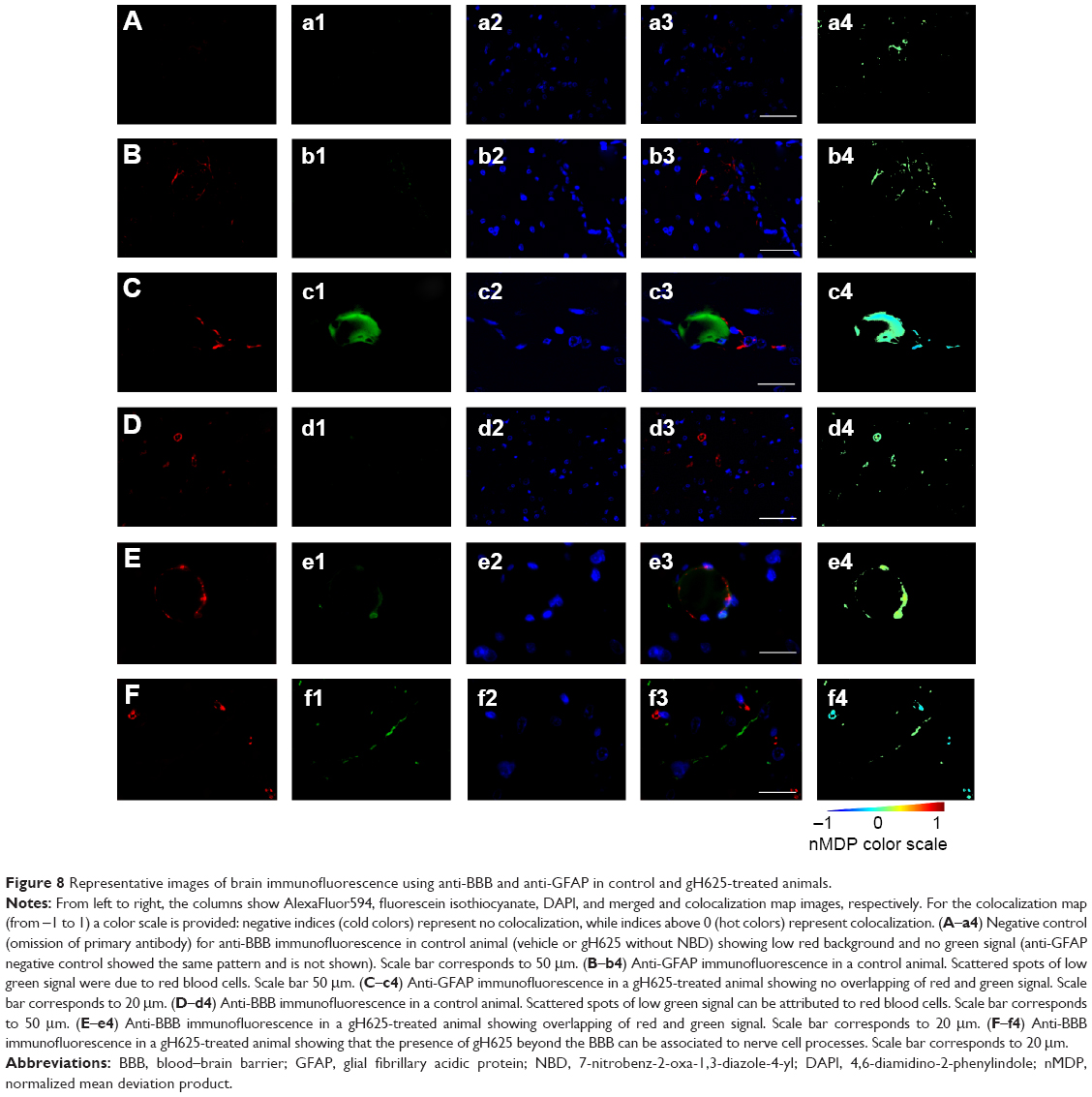 | Figure 8 Representative images of brain immunofluorescence using anti-BBB and anti-GFAP in control and gH625-treated animals. |
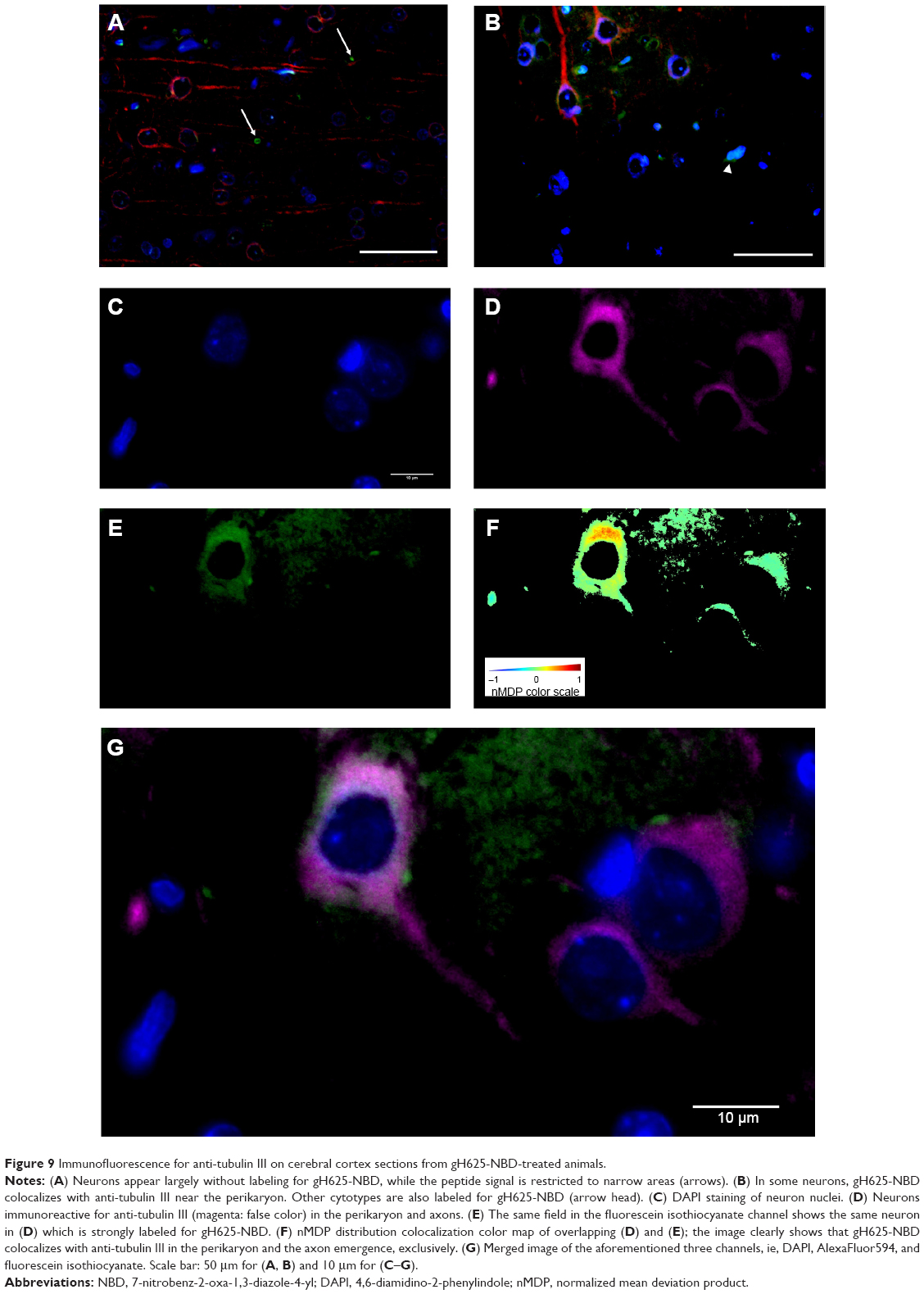 | Figure 9 Immunofluorescence for anti-tubulin III on cerebral cortex sections from gH625-NBD-treated animals. |
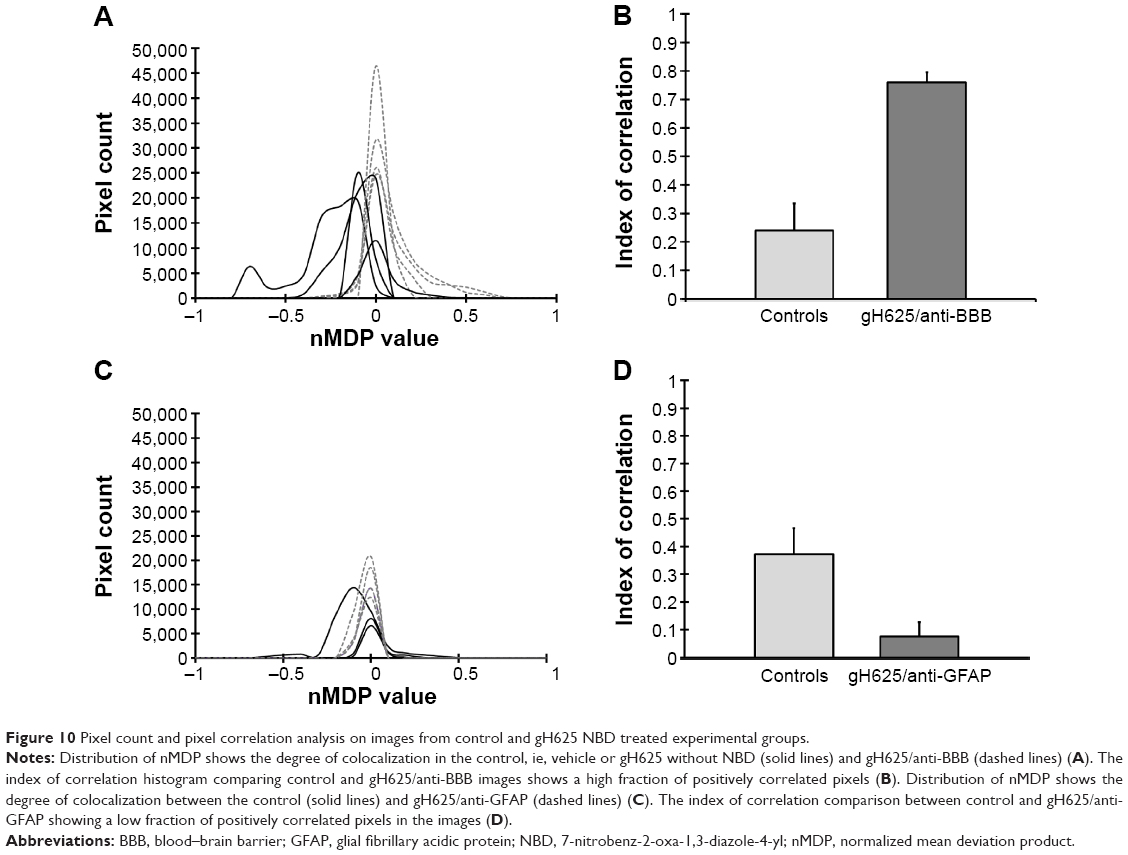 | Figure 10 Pixel count and pixel correlation analysis on images from control and gH625 NBD treated experimental groups. |
In vivo effect of gH625 on brain and liver mitochondrial respiration rates and maximal oxidative capacity
Following in vivo administration, gH625 did not induce significant changes in brain mitochondrial bioenergetic parameters; in fact, no changes in basal respiration (in which there is no synthesis of ATP), ADP-stimulated respiration (in which ATP synthesis proceeds at its maximal rate), and respiratory control ratio (index of oxidative phosphorylation efficiency) were detected (Figure 11). In accordance with these bioenergetic parameters, maximal oxidative capacity of the liver and brain were not affected by in vivo administration of gH625 (Figure 11). Overall, these data suggest that within 3.5 hours of administration in vivo, gH625 has no toxic effect at the mitochondrial level in either the liver or brain.
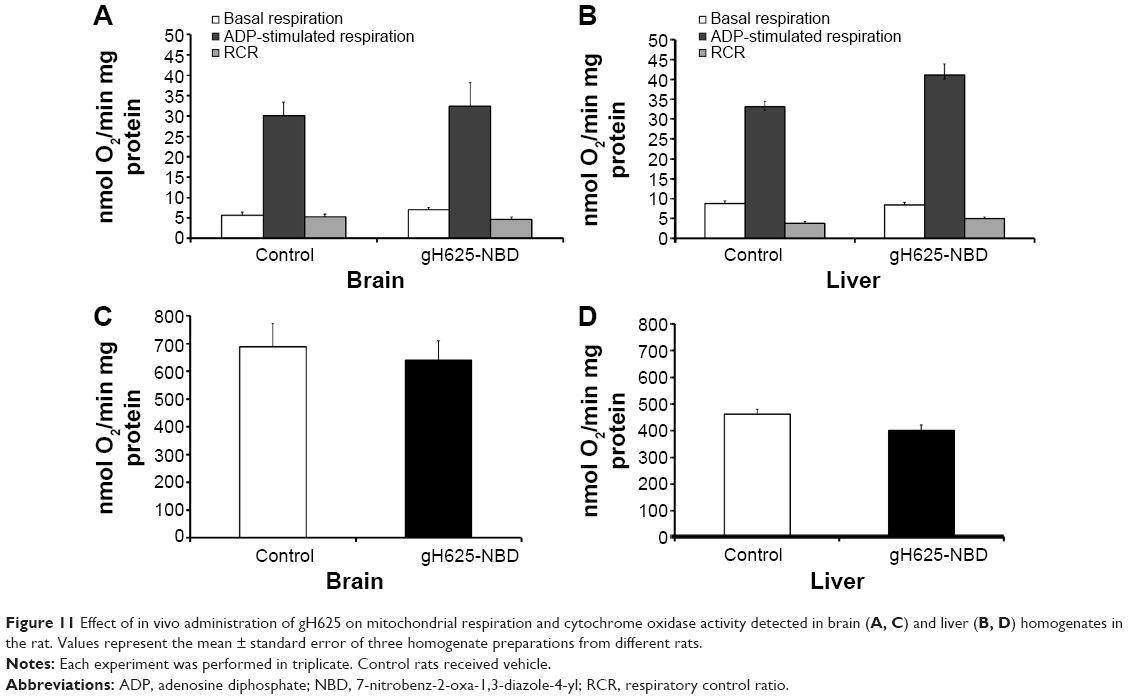 | Figure 11 Effect of in vivo administration of gH625 on mitochondrial respiration and cytochrome oxidase activity detected in brain (A, C) and liver (B, D) homogenates in the rat. Values represent the mean ± standard error of three homogenate preparations from different rats. |
Discussion
The BBB represents a major obstacle in the development of potential new therapies to treat disorders of the central nervous system due to the impermeability of the cell membranes in this compartment to several molecules.27,28 To date, numerous attempts have been made to identify and characterize an efficient drug delivery approach for the brain,29,30 and a number of strategies are now under investigation.27 These are mainly invasive procedures, including direct intraventricular administration of drugs by means of surgery and temporary disruption of the BBB via intracarotid infusion of hyperosmolar solutions; pharmacologically-based strategies that involve increasing the lipid solubility of the drug to facilitate its passage through the BBB; and physiologically-based strategies exploiting various carrier mechanisms.
Peptide-based carriers have attracted considerable attention in the field of targeted drug delivery due to their high binding affinity and specificity for targets. The presence of advanced technologies for peptide identification and modification further supports the extensive application of peptides in the field of theranostics research.31,32
We have previously demonstrated that gH625, a 19-residue peptide, is a membrane-perturbing domain13 that interacts with model membranes, contributing to their merging13 and is able to traverse the membrane bilayer and transport a cargo (quantum dots, liposomes, and dendrimers)16,18,20 into the cytoplasm and across an in vitro model of the BBB.19 Uptake studies suggested a nonactive translocation mechanism in crossing the lipid bilayer, which may vary depending on the cargo.15 Penetration by gH625 occurs in a rapid, concentration-dependent fashion that appears to be independent of receptors and transporters and instead is thought to target the lipid bilayer component of the cell membrane. Thus, in principle, all mammalian cell types should be susceptible to gH625 transduction, and indeed we have used this technology to transduce different cargoes into several cell types in vitro.21 However, as with any potential pharmacological approach, it is necessary to translate in vitro findings to animal models to determine whether gH625 is able to deliver molecules into cells of entire organisms and to keep track of the whole biological picture and the complex nano-biological interactions before any potential application.
Other peptides have also been proposed as drug delivery systems. It was demonstrated that transactivator of transcription (TAT) is able to enter tissues in vivo in mice,33 and an antennapedia-homeodomain-derived peptide (ANTP) was able to activate endogenous T-cells in mice.34 These peptides are highly positively charged, and absorptive-mediated transcytosis has been proposed as the mechanism for their transport across the BBB. A bradykinin analog was also able to increase the penetration of small molecules by transient opening of the BBB.35 Here, we report early results showing that a membranotropic peptide derived from herpes virus type 1 could be used as a potential delivery system for macromolecules in vivo. These results, coupled with previous in vitro data, support the view that gH625 enters the BBB without involving endocytic processes. Hence, the eventual cargo may be immediately and completely available.
The gH625 peptide has a significant ability to penetrate brain cells. This was confirmed by both in vitro exposure of neuron and astrocyte cell lines to gH625 and in vivo administration of the peptide in rats. In vitro studies showed that both SH-SY5Y and U-87 MG cells can actually take up more than 90% of administered gH625, indicating that internalization of gH625 into these cell lines is an efficient process. Uptake of the peptide was not associated with variation in cell viability or with alteration in maximal mitochondrial oxidative capacity, indicating that, at the dose tested, the peptide was devoid of cytotoxic effects. These data suggest that gH625 could potentially be used as a brain delivery system for macromolecules.
The results of in vivo administration of gH625 strongly support this possibility. Indeed, we demonstrated that, despite the blood filtration action of the liver, carried out through the gH625 uptake of Kupffer cells, gH625 substantially reaches the brain BBB vessels. Interestingly, gH625 could be found in the BBB endothelium, so this peptide can be efficiently accumulated in endothelial cells of the BBB. We also evaluated whether gH625 had actually crossed the BBB or was simply trapped within endothelial cells of the brain. In particular, although we did not detect any association with astrocytes, we were able to find the presence of gH625 in neurons. These data suggest that gH625 reaches the cortical perikarya and their processes, demonstrating that this peptide may be useful in the development of functionalized nanosystems for drug delivery to the brain.
We also evaluated the ability of gH625 to affect mitochondrial oxidative capacity in terms of the mitochondrial respiration rate and/or cytochrome oxidase activity (the latter being an index of the maximal oxidative capacity of a cell or tissue), in view of the fundamental role played by the mitochondria in the energy metabolism of the cell. The results of this study indicate that administration of gH625 to rats does not influence the mitochondrial respiration rate or maximal oxidative capacity in brain and liver homogenates, suggesting that this peptide does not influence processes critical to cell survival.
The presence of multiple metabolic barriers may restrict the application of such a peptide-based ligand for targeted drug delivery in vivo. Peptides alone or conjugated on the surface of nanocarriers are subject to proteolysis in the blood after systemic administration. In addition, the BBB is also a metabolic barrier due to the presence of various enzymes in brain capillary endothelial cells. In our study, gH625 started to be degraded after 1 hour of incubation, but the intact peptide was still present after 3.5 hours of incubation, and thus it has potential for extending brain targeting efficiency due to its resistance to proteolysis for 3.5 hours.
Due to its stability and targeting efficacy, we envisage that gH625 may be a promising candidate for designing a targeted delivery system. The exact molecular mechanism for the entry of gH625 remains to be established. However, the fact that it is a membranotropic peptide able to interact with the membrane bilayer and destabilize it locally and temporarily, suggests translocation across the membrane bilayer that does not seem to involve classical endocytosis mechanisms as previously demonstrated by inhibition of endocytosis in live cells.19
Our results show that gH625 may be used for mediating delivery to virtually any tissue and in particular across the BBB. gH625 could transport a wide variety of cargoes with intact bioactivity into almost any tissue or organ. Nonselective transduction is not a drawback because specific targeting units may be added to increase retention in specific tissues.
Conclusion
These results show for the first time that a membranotropic peptide can be incorporated in vitro by neuron or astrocyte cells and has the ability in vivo to accumulate in the brain and could be advantageously used as a new brain delivery system. Further work is underway with various therapeutic targets, including small molecules, macromolecules, and other pharmaceutical agents.
Acknowledgments
This work was supported by Progetto FARO (2012/0043756) and by the Ministero dell’Istruzione, dell’Università e della Ricerca (BAP114AMK). The authors thank Maurizio Amendola and Luca De Luca for their excellent technical assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Bertrand N, Leroux J-C. The journey of a drug-carrier in the body: an anatomo-physiological perspective. J Control Release. 2012;161(2):152–163. | ||
Dinda SC, Pattnaik G. Nanobiotechnology-based drug delivery in brain targeting. Curr Pharm Biotechnol. 2014;14(15):1264–1274. | ||
Moros M, Mitchell SG, Grazu V, de la Fuente JM. The fate of nanocarriers as nanomedicines in vivo: important considerations and biological barriers to overcome. Curr Med Chem. 2013;20(22):2759–2778. | ||
Bernacki J, Dobrowolska A, Nierwinska K, Malecki A. Physiology and pharmacological role of the blood-brain barrier. Pharmacol Rep. 2008;60(5):600–622. | ||
Alyautdin R, Khalin I, Nafeeza MI, Haron MH, Kuznetsov D. Nanoscale drug delivery systems and the blood–brain barrier. Int J Nanomedicine. 2014;9:795–811. | ||
Dominguez A, Suarez-Merino B, Goni-de-Cerio F. Nanoparticles and blood–brain barrier: the key to central nervous system diseases. J Nanosci Nanotechnol. 2014;14(1):766–779. | ||
Xu G, Mahajan S, Roy I, Yong KT. Theranostic quantum dots for crossing blood–brain barrier and providing therapy of HIV-associated encephalopathy. Front Pharmacol. 2013;4:140. | ||
Gabathuler R. Development of new peptide vectors for the transport of therapeutic across the blood–brain barrier. Ther Deliv. 2010;1(4):571–586. | ||
Jones AT. Gateways and tools for drug delivery: endocytic pathways and the cellular dynamics of cell penetrating peptides. Int J Pharm. 2008;354(1–2):34–38. | ||
Galdiero S, Vitiello M, Falanga A, Cantisani M, Incoronato N, Galdiero M. Intracellular delivery: exploiting viral membranotropic peptides. Curr Drug Metab. 2012;13(1):93–104. | ||
Vonesch C, Unser M. A fast thresholded landweber algorithm for wavelet-regularized multidimensional deconvolution. IEEE Trans Image Process. 2008;17(4):539–549. | ||
Galdiero S, Vitiello M, D’Isanto M, et al. The identification and characterization of fusogenic domains in herpes virus glycoprotein B molecules. Chembiochem. 2008;9(5):758–767. | ||
Galdiero S, Falanga A, Vitiello M, et al. The presence of a single N-terminal histidine residue enhances the fusogenic properties of a membranotropic peptide derived from herpes simplex virus type 1 glycoprotein H. J Biol Chem. 2010;285(22):17123–17136. | ||
Galdiero S, Falanga A, Vitiello M, et al. Analysis of a membrane interacting region of herpes simplex virus type 1 glycoprotein H. J Biol Chem. 2008;283(44):29993–30009. | ||
Galdiero S, Falanga A, Morelli G, Galdiero M. gH625: a milestone in understanding the many roles of membranotropic peptides. Biochim Biophys Acta. 2015;1848(1 Pt A):16–25. | ||
Falanga A, Vitiello MT, Cantisani M, et al. A peptide derived from herpes simplex virus type 1 glycoprotein H: membrane translocation and applications to the delivery of quantum dots. Nanomedicine. 2011;7(6):925–934. | ||
Smaldone G, Falanga A, Capasso D, et al. gH625 is a viral derived peptide for effective delivery of intrinsically disordered proteins. Int J Nanomedicine. 2013;8:2555–2565. | ||
Tarallo R, Accardo A, Falanga A, et al. Clickable functionalization of liposomes with the gH625 peptide from herpes simplex virus type I for intracellular drug delivery. Chemistry. 2011;17(45):12659–12668. | ||
Guarnieri D, Falanga A, Muscetti O, et al. Shuttle-mediated nanoparticle delivery to the blood–brain barrier. Small. 2013;9(6):853–862. | ||
Carberry TP, Tarallo R, Falanga A, et al. Dendrimer functionalization with a membrane-interacting domain of herpes simplex virus type 1: towards intracellular delivery. Chemistry. 2012;18(43):13678–13685. | ||
Falanga A, Tarallo R, Galdiero E, Cantisani M, Galdiero M, Galdiero S. Review of a viral peptide nanosystem for intracellular delivery. Int J Nanomedicine. 2013;7(1):071599. | ||
Rapaport D, Shai Y. Interaction of fluorescently labeled pardaxin and its analogues with lipid bilayers. J Biol Chem. 1991;266(35):23769–23775. | ||
Baschong W, Suetterlin R, Laeng RH. Control of autofluorescence of archival formaldehyde-fixed, paraffin-embedded tissue in confocal laser scanning microscopy (CLSM). J Histochem Cytochem. 2001;49(12):1565–1572. | ||
Viegas MS, Martins TC, Seco F, do Carmo A. An improved and cost-effective methodology for the reduction of autofluorescence in direct immunofluorescence studies on formalin-fixed paraffin-embedded tissues. Eur J Histochem. 2007;51(1):59–66. | ||
Jaskolski F, Mulle C, Manzoni OJ. An automated method to quantify and visualize colocalized fluorescent signals. J Neurosci Methods. 2005;146(1):42–49. | ||
Lanni A, Moreno M, Lombardi A, Goglia F. Calorigenic effect of diiodothyronines in the rat. J Physiol. 1996;494 Pt 3:831–837. | ||
Lu CT, Zhao YZ, Wong HL, Cai J, Peng L, Tian XQ. Current approaches to enhance CNS delivery of drugs across the brain barriers. Int J Nanomedicine. 2014;9:2241–2257. | ||
Patel MM, Goyal BR, Bhadada SV, Bhatt JS, Amin AF. Getting into the brain: approaches to enhance brain drug delivery. CNS Drugs. 2009;23(1):35–58. | ||
Demeule M, Régina A, Annabi B, Bertrand Y, Bojanowski M, Béliveau R. Brain endothelial cells as pharmacological targets in brain tumors. Mol Neurobiol. 2004;30(2):157–183. | ||
Pardridge WM. Blood–brain barrier delivery. Drug Discov Today. 2007;12(1–2):54–61. | ||
Doolittle ND, Miner ME, Hall WA, et al. Safety and efficacy of a multicenter study using intraarterial chemotherapy in conjunction with osmotic opening of the blood-brain barrier for the treatment of patients with malignant brain tumors. Cancer. 2000;88(3):637–647. | ||
Hall W, Doolittle N, Daman M, et al. Osmotic blood–brain barrier disruption chemotherapy for diffuse pontine gliomas. J Neurooncol. 2006;77(3):279–284. | ||
Fawell S, Seery J, Daikh Y, et al. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci U S A. 1994;91(2):664–668. | ||
Schutze-Redelmeier MP, Gournier H, Garcia-Pons F, et al. Introduction of exogenous antigens into the MHC class I processing and presentation pathway by Drosophila antennapedia homeodomain primes cytotoxic T cells in vivo. J Immunol. 1996;157(2):650–655. | ||
Borlongan CV, Emerich DF. Facilitation of drug entry into the CNS via transient permeation of blood–brain barrier: laboratory and preliminary clinical evidence from bradykinin receptor agonist, Cereport. Brain Res Bull. 2003;60(3):297–306. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.