Back to Journals » International Journal of Nanomedicine » Volume 14
Pemetrexed-loaded nanoparticles targeted to malignant pleural mesothelioma cells: an in vitro study
Authors Cova E, Pandolfi L, Colombo M ![]() , Frangipane V
, Frangipane V ![]() , Inghilleri S, Morosini M, Mrakic-Sposta S
, Inghilleri S, Morosini M, Mrakic-Sposta S ![]() , Moretti S, Monti M
, Moretti S, Monti M ![]() , Pignochino Y
, Pignochino Y ![]() , Benvenuti S, Prosperi D
, Benvenuti S, Prosperi D ![]() , Stella G, Morbini P, Meloni F
, Stella G, Morbini P, Meloni F
Received 7 September 2018
Accepted for publication 13 November 2018
Published 23 January 2019 Volume 2019:14 Pages 773—785
DOI https://doi.org/10.2147/IJN.S186344
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Emanuela Cova,1 Laura Pandolfi,1 Miriam Colombo,2 Vanessa Frangipane,1 Simona Inghilleri,1 Monica Morosini,1 Simona Mrakic-Sposta,3 Sarah Moretti,3 Manuela Monti,4 Ymera Pignochino,5 Silvia Benvenuti,5 Davide Prosperi,2,6 Giulia Stella,1 Patrizia Morbini,7 Federica Meloni8
1Clinic of Lung Diseases, IRCCS Foundation Policlinico San Matteo, Pavia, Italy; 2Deparment of Biotechnology and Bioscience, University of Milano - Bicocca, Milan, Italy; 3National Council of Research, Institute of Bioimaging and Molecular Physiology, Segrate, Milan, Italy; 4Laboratory of Biotechnology, Research Center of Rigenerative Medicine, IRCCS Foundation Policlinico San Matteo, Pavia, Italy; 5Experimental Clinical Molecular Oncology, IRCCS Candiolo Cancer Institute-FPO, Candiolo, Turin, Italy; 6Laboratory of Nanomedicine, Clinical Institute of Maugeri, S.p.A., Pavia, Italy; 7Department of Molecular Medicine, Pathology Unit, IRCCS Foundation Policlinico San Matteo, Pavia, Italy; 8Department of Internal Medicine, Pneumology Unit, University of Pavia, Pavia, Italy
Purpose: Malignant pleural mesothelioma (MPM) is an aggressive tumor characterized by poor prognosis. Its incidence is steadily increasing due to widespread asbestos exposure. There is still no effective therapy for MPM. Pemetrexed (Pe) is one of the few chemotherapeutic agents approved for advanced-stage disease, although the objective response to the drug is limited. The use of gold nanoparticles (GNPs) as a drug delivery system promises several advantages, including specific targeting of malignant cells, with increased intracellular drug accumulation and reduced systemic toxicity, and, in the case of MPM, direct treatment administration into the pleural space. This study aims at exploring CD146 as a potential MPM cell-specific target for engineered Pe-loaded GNPs and to assess their effectiveness in inhibiting MPM cell line growth.
Methods: MPM cell lines and primary cultures obtained by pleural effusions from MPM patients were assayed for CD146 expression by flow cytometry. Internalization by MPM cell lines of fluorescent dye-marked GNPs decorated with a monoclonal anti CD146 coated GNPs (GNP-HC) was proven by confocal microscopy. The effects of anti CD146 coated GNPs loaded with Pe (GNP-HCPe) on MPM cell lines were evaluated by cell cycle (flow cytometry), viability (MTT test), clonogenic capacity (soft agar assay), ROS production (electric paramagnetic resonance), motility (wound healing assay), and apoptosis (flow cytometry).
Results: GNP-HC were selectively uptaken by MPM cells within 1 hour. MPM cell lines were blocked in the S cell cycle phase in the presence of GNP-HCPe. Both cell viability and motility were significantly affected by nanoparticle treatment compared to Pe. Apoptotic rate and ROS production were significantly higher in the presence of nanoparticles. Clonogenic capacity was completely inhibited following nanoparticle internalization.
Conclusion: GNP-HCPe treatment displays in vitro antineoplastic action and is more effective than Pe alone in inhibiting MPM cell line malignant phenotype. The innovative use of specifically targeted GNPs opens the perspective of local intrapleural administration to avoid normal cell toxicity and enhance chemotherapy efficacy.
Keywords: gold nanoparticles, nanodrug delivery, mesothelioma, pemetrexed, intrapleural delivery
Introduction
Malignant pleural mesothelioma (MPM) is an aggressive tumor characterized by poor prognosis.1 The incidence of MPM is expected to raise until the year 2020 in Europe, especially in those areas where environmental and occupational exposure to asbestos fibers was more frequent.2 There is still no effective therapeutic regimen for MPM, and, as a consequence, patient median survival is approximately 1 year.3 Although a relevant number of genomic alterations are known to drive epithelial carcinogenesis, very few data have so far been reported about mesothelial cell transformation. For this reason, at present no actionable targets can be exploited to effectively treat MPM, and conventional chemotherapy represents the only feasible therapeutic approach. The association of pemetrexed (Pe) and platinum is the current treatment for advanced disease.4 Although this schedule has fostered a major improvement in MPM treatment, prognosis is still poor.5 Besides, conventional chemotherapy entails remarkable toxic side effects disproportionate with clinical benefits. Poor therapeutic results could reasonably be ascribed to drug inability to reach the site of disease and adequate intracellular drug concentrations at the therapeutic dose, while systemic toxicity does not allow to further increase the dosage. To address this unmet medical need, we planned to investigate novel delivery strategies based on the enormous potentiality offered by nanotechnology, with the aim to specifically target mesothelial tumor cells, increase intracellular drug uptake, and deliver the drug directly in the pleural space.
Among the nanomaterials used as drug delivery vehicles, gold nanoparticles (GNPs) have great potential, thanks to their unique size-dependent electronic and optical properties, biocompatibility, and to the possibility of customization. GNPs are produced by wet chemistry, and surface functionalization with amphiphilic polymer coating is needed to provide them with high biocompatibility and low cytotoxicity, resulting in prolonged half-life in vivo. In addition, various ligands can be attached to the GNP surface with the purpose to target specific cells.
We have previously developed gold nanocarriers for the treatment of chronic lung allograft dysfunction (CLAD) and proved their in vitro efficacy in inhibiting mesenchymal cells, which are responsible for CLAD onset,6 and in vivo feasibility7 of safe administration by inhalatory route. Nanocarriers were decorated with antibodies specific for an antigen selectively expressed by mesenchymal cells and were loaded with mTOR inhibitor everolimus. On the basis of these previous results, we extended this innovative approach to design a new therapeutic option for MPM treatment.
For this purpose, we identified CD146 as a protein specifically expressed by MPM cells, suitable for nanoparticle functionalization, and selected Pe as a drug to be loaded in engineered GNPs. As already performed for CLAD experiments, we assayed the in vitro efficacy of our nanovectors to inhibit MPM cell lines to prove the feasibility of the intrapleural delivery route and use them as a novel therapeutic approach for MPM treatment.
Materials and methods
Nanoparticle preparation
Biocompatible GNPs functionalized with the half chain of anti-CD146 monoclonal antibody (moAb) and loaded with Pe (anti CD146 coated GNPs loaded with Pe [GNP-HCPe]) were synthesized by adapting the protocol described by Cova et al.7 GNPs (around 6 nm in diameter; Figure S1) were synthesized in organic solvent by following the protocol described by Brust et al8 and transferred into water solution with an amphiphilic polymer, obtained by condensation of poly-(isobutylene-alt-maleic anhydride) and dodecylamine (PMA).9 The resulting water-soluble nanoparticles (500 μL of a 4 μM solution) were shaken for 2 hours with 2,2-(ethylenedioxy)bis(ethylamine) (EDBE, 80 μL 0.05 M in dH2O) in the presence of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, 20 μL 1 M in dH2O). Next, the nanoparticle dispersion was concentrated and washed two additional times with water.
Five hundred microliters of N-succinimidyl-3-[2-pyridyldithio]-propionate (SPDP 10 mg/mL in dimethyl sulfoxide [DMSO]) were added and reacted for 4 hours. Next, the half-chains of anti-CD146 moAb (0.4 mg; Affymetrix EBIO; Thermo Fisher Scientific, Waltham, MA, USA) and MeO-PEG500-SH (PEG) were added and incubated at room temperature for 1 hour, resulting in the antibody-functionalized nanoparticles (anti CD146 coated gold nanoparticles [GNP-HC]). Pe (427.4 g/mol – Alimta®; Eli Lilly & Co., Indianapolis, IN, USA) dissolved in DMSO (0.65 mg) was added to 1 mg of the washed and recollected GNP-HC solution and incubated for 2 hours. Finally, the nanoparticles were concentrated at 3,500 rpm for 10 minutes and washed with PBS solution obtaining Pe-loaded nanoparticles (GNP-HCPe) devoid of DMSO.
The quantification of Pe was based on absorption spectrophotometry at λ=225 nm, and the loading efficiency (LE) was calculated as:
|
|
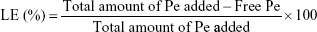
where, total amount of Pe added is the start quantity of drug added for the loading reaction. Free Pe is the quantity of the unreacted Pe collected in the washing solution after nanoparticle centrifugation.
Dye-labeled nanoparticles were obtained using 0.5 M PMA labeled with Alexa Fluor 488, obtained by the reaction of 5 mL of 0.5 M PMA in CHCl3 with 1.0 M Alexa Fluor 488 (0.5 mL) in ethanol overnight at room temperature.
Hydrodynamic diameter of nanoparticles was measured by dynamic light scattering ZetaSizer Nano Instrument from Malvern Instruments (Malvern, UK) operating at 4 mW of a He Ne 633 nm laser, using a scattering angle of 90°. The zeta potential analysis of nanoparticles in 10 mM NaCl was performed at 25°C using the same instrument.
Dynamic light scattering data of synthesized nanoparticles are summarized in Table S1.
Cell line culture, characterization, and treatments
The human MPM cell lines MSTO-211H (biphasic) and NCI-H2452 (epithelioid) were gently provided by Ymera Pignochino and Silvia Benvenuti, IRCCS Candiolo Cancer Institute, who bought both cell lines from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were seeded in high-glucose RPMI-1640 with 10% fetal calf serum (FCS), 1% L-glutamine, and 100 units/mL penicillin (P) and 100 μg/mL streptomycin (S) solution in a humidified atmosphere of 5% CO2 at 37°C. For in vitro experiments, we treated cells under these experimental conditions: 1) GNP-HCPe (25 μg/mL of nanoparticles and 100 nM of Pe), nanoparticle concentration was selected according to the previously published experimental evidence (Cova et al7); 2) Pe (final concentration 100 nM in PBS), Pe concentration was chosen from literature data;10,11 and 3) medium only (control [CTR]).
Primary cell lines were isolated from pleural exudates of MPM patients after obtaining informed consent. This procedure was approved by ethical committee of IRCCS Policlinico San Matteo (protocol number 20140002533, June 10, 2014), and this study was carried out by following the ethical principles of WMA Declaration of Helsinki. Pleural exudate was centrifuged, washed twice in PBS, and finally 3×106 cells/mL were seeded in the same culture medium of two commercial cell lines for 48 hours. Adherent cells were then washed and cultured until confluence in the same medium for 2 weeks to remove the inflammatory cells. Cells were then treated by trypsin-EDTA for 5 minutes at 37°C. After obtaining pure population, cells were collected, characterized as mesothelial with routine immunocytochemistry, and evaluated for CD146 expression.
Primary cell characterization and CD146 expression evaluation
To confirm the mesothelial nature of primary cell lines, aliquots of trypsin-EDTA-treated cells were fixed in 10%-buffered formalin, centrifuged on glass slides, and immunoreacted with antibodies against calretinin (clone DAK-Calret 1; Dako North America, Carpinteria, CA, USA) and podoplanin (clone D2-40; Dako North America) followed by revelation with avidin–biotin–peroxidase and diaminobenzidine precipitation. Surface expression of CD146 was studied by flow cytometry analysis using a FACSCanto II (BD, Franklin Lakes, NJ, USA). Primary cells and cell lines were trypsinized, washed in PBS, and incubated with antibody anti-CD146 PerCp-Cy5-5 (BD) for 30 minutes. The appropriate mouse immunoglobulin isotype was used as control. Analyses were performed by using BD FACS Diva Software (BD). CD146 expression in archival MPM biopsies was investigated by immunohistochemistry with routine protocols.
Effect of GNP-HCPe on MSTO-211H and NCI-H2452 cells
For technical and reproducibility reasons, experiments with nanoparticles were performed on MSTO-211H and NCI-H2452 cell lines.
Cell uptake
GNP-HC uptake was evaluated by confocal microscopy. Briefly, cells were seeded on collagen precoated glass slides and used at subconfluence. Entrance of Alexa Fluor 488-labeled GNP-HC (25 μg/mL) was assayed at 2 hours at 37°C in medium. At the end of the incubation periods, slides were fixed with 4% paraformaldehyde and incubated for 10 minutes with 0.1 M glycine in PBS solution. Samples were treated with a blocking solution, and slides were stained with DAPI. Microscopic analysis was performed using confocal laser microscope (FLUOVIEW, FUIOI; Olympus Corporation, Tokyo, Japan). Uptake specificity was proved in the same experimental conditions by using A549 adenocarcinoma cell line, lacking CD146 surface marker, and dye-labeled nanoparticles that were not functionalized with moAb anti-D146 (GNP-PEG).
Cell viability assay
MTT test was used to assay cell viability. Briefly, 1×103 cells were seeded in 96-well plates. After 2 hours of incubation with 25 μg/mL GNP-HCPe and 100 nM Pe, the medium was changed. At 24, 48, 72, and 96 hours from treatment cells were incubated with RPMI-1640 w/o red phenol + 10% MTT (Sigma-Aldrich Co., St Louis, MO, USA) for 4 hours, followed by the addition of DMSO after removal of all medium. Absorbance (550 nm) was measured using a microplate reader (BIO-RAD Model 680). Results were expressed as percentage of variation vs untreated cells.
Apoptotic assay
Apoptosis was detected by means of Annexin V/7-AAD staining (BD Pharmingen, BD Bioscience, San Jose, CA, USA) and analyzed by flow cytometry. MSTO-211H and NCI-H2452 cells were seeded in six-well plates (1.0×105 cells per well) and after 24 hours were incubated with 25 μg/mL GNP-HCPe and 100 nM Pe for 24 and 48 hours. Next, the cells were washed twice with cold PBS, carefully trypsinized, and resuspended in Annexin binding buffer in the presence of phycoerythrin labeled-Annexin V and 7-AAD. Sample acquisition was performed using FACSCanto II (BD) and analyzed with BD FACS Diva Software (BD).
Cell cycle analysis
Cells were seeded in 24-well plates and incubated for 2 hours with 25 μg/mL GNP-HCPe and 100 nM Pe, washed, and cultured for 24 and 48 hours. Then, cells were harvested, washed twice with ice-cold PBS, and fixed in cold 70% ethanol. Afterward, cells were washed with PBS and incubated in the dark with a staining solution containing 0.002% 4-nonylphenyl-polyethylene glycol (NP-40; Sigma-Aldrich Co.), 20 μg/mL of RNase A (Sigma-Aldrich Co.), and propidium iodide (PI, 50 μg/mL; Sigma-Aldrich Co.) in PBS at room temperature. The fluorescence emitted by the PI–DNA complex was quantified by flow cytometry (BD Biosciences, San Jose, CA, USA), and the percentage of cells in each phase of cell cycle was calculated using FlowJo 9.3.0 software.
ROS detection
ROS levels were detected at t0 (baseline) and after 6, 24, and 48 hours of incubation with 25 μg/mL GNP-HCPe and 100 nM Pe by electron paramagnetic resonance (EPR) spectroscopy (Bruker Optik GmbH, Ettlingen, Germany) operating at the common X-Band microwave frequency (~9.8 GHz). For ROS assessment, fluids extra-cells were incubated with 1 mM 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH; Noxygen, Elzach, Germany) probe prepared in Krebs–Hepes buffer containing 25 μM deferroxamine methanesulfonate salt (DF; Noxygen) chelating agent and 5 μM sodium diethyldithiocarbamate trihydrate (DETC; Noxygen) at pH 7.4.12
All the spectra were recorded at 37°C, adopting the same acquisition parameters, and analyzed using a standard EPR software supplied by Bruker Optik GmbH (Win EPR 2.11 version).13 Three repetitions were performed for each experiment.
Colony-forming capacity
To evaluate the colony-forming capacity of MSTO-211H and NCI-H2452 cell lines, 1×103 cells were suspended in DMEM containing 10% FBS and 0.5% low-melting point agarose (Sigma-Aldrich Co.) and seeded on a bottom layer containing 1% agarose in 24-well plates.
Briefly, 24-well plates were coated with culture medium containing 1% agarose and allowed to solidify. We detached MSTO-211H and NCI-H2452 cells and treated with GNP-HCPe or Pe for 2 hours in suspension. Afterward, a mixture of 0.5% agar/culture medium and 2×103 pre-treated cells was plated above the soft agar coat. After solidification, DMEM with 10% FBS was added, and plates were incubated at 37°C, 5% CO2. Medium was changed twice a week, and after 21 days, the cells were marked with 0.5 mg/mL MTT. Each experiment was repeated in triplicate. The total number of colonies was analyzed by digital image capture and analysis software (Cell^F Olympus; http://matrixoptics.com/html/software.aspx?PID=169&ID=66&Cat=False&Repost=True).
Wound healing assay
Cells were grown in 35 mm Petri dishes (Greiner, Frickenhausen, Germany) to 100% confluence and treated with 25 μg/mL GNP-HCPe and 100 nM Pe for 2 hours. A scratch line (wounds) was made with sterile 200 μL pipette tips in each Petri dish. Dislodged cells and debris were gently removed by washing with medium. Images (at least five per well) of the same spots were captured every 30 minutes after scratch for 5 hours. The distance between wound borders was measured using Cell F software (Olympus Corporation) in blind by two different operators and expressed in micrometer advancement using a reference ruler. For each plate, ten fields were selected and cell migration measured.
Statistical analyses
Statistical differences between untreated cells, cells treated with GNP-HCPe, and Pe were analyzed by one-way ANOVA followed by Newman–Keuls’ test. All analyses were carried out with Graph Prism 5.0 statistical program. A P-value of <0.05 was considered statistically significant.
Results
Study of CD146 expression
In preliminary experiments, we found that CD146 was selectively expressed by MPM cells and not by reactive mesothelium. We confirmed this observation in MPM biopsies, primary cells, and two commonly used mesothelial cell lines. Primary cells were isolated from patients affected by MPM by following the standard protocol,14 and their mesothelial origin was confirmed by immunocytochemistry with the expression of mesothelial markers (calretinin and D240). Flow cytometry demonstrated that CD146 was expressed at 96.97%±2.0% by primary MPM cells and 99.9%±1.8% and 94.4%±1.3% by MSTO-211H and NCI-H2452, respectively. Immunohistochemical staining of MPM biopsy samples showed that neoplastic cells constantly expressed CD146 on the cell membrane (Figure 1A), whereas reactive mesothelial cells were negative (Figure 1B), supporting the choice of CD146 as target for selective nanoparticle-based therapy, as suggested by other authors.15,16
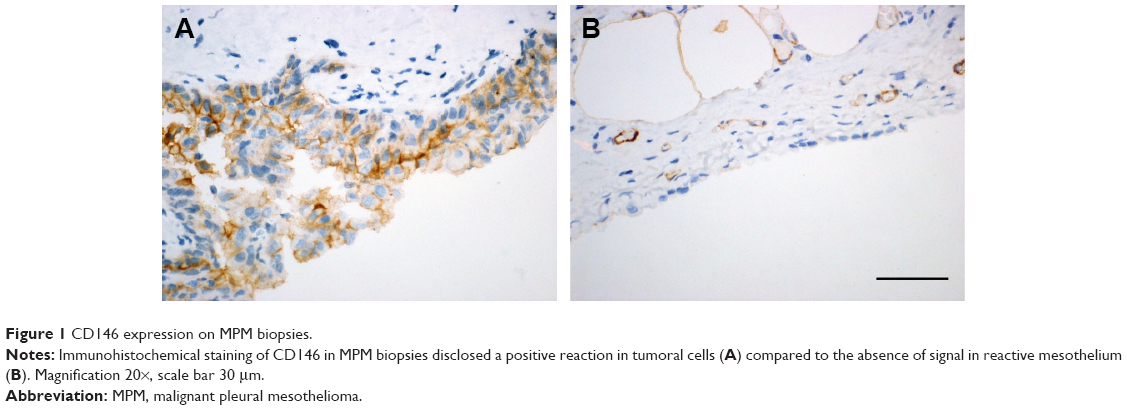 | Figure 1 CD146 expression on MPM biopsies. |
Nanoparticle synthesis and characterization
GNPs functionalized with the half chain of anti-CD146 moAb and loaded with Pe (GNP-HCPe) were synthesized as reported in the “Materials and methods” section. GNP-HCPe showed a hydrodynamic diameter of 53.4±2.2 nm, measured by dynamic light scattering (Table S1). GNPs were efficiently loaded with Pe. Thanks to UV quantification of the unreacted Pe collected in the washing solution, we assessed that the amount of Pe adsorbed to GNP-HCPe was 4 nmol for 1 mg of nanoparticle. Pe LE corresponded to 12%.
Cell uptake
Confocal microscopy documented the different uptake of GNP-HC by MSTO-211H (CD146-positive) and A549 cells (CD146-negative). GNP-HC were internalized only by MPM cells within 2 hours (Figure 2A). Inert dye-labeled GNP-PEG were not internalized by MPM cells (Figure 2B). A549 cells, which do not express CD146 surface marker, did not show any GNP-HC fluorescent signal (Figure 2C) as well as GNP-PEG (Figure 2D). These observations support the hypothesis that the entrance of nanovehicles is mediated by CD146 protein and that the functionalization of GNP with moAb is necessary to allow the efficient and fast internalization of GNP by cells.
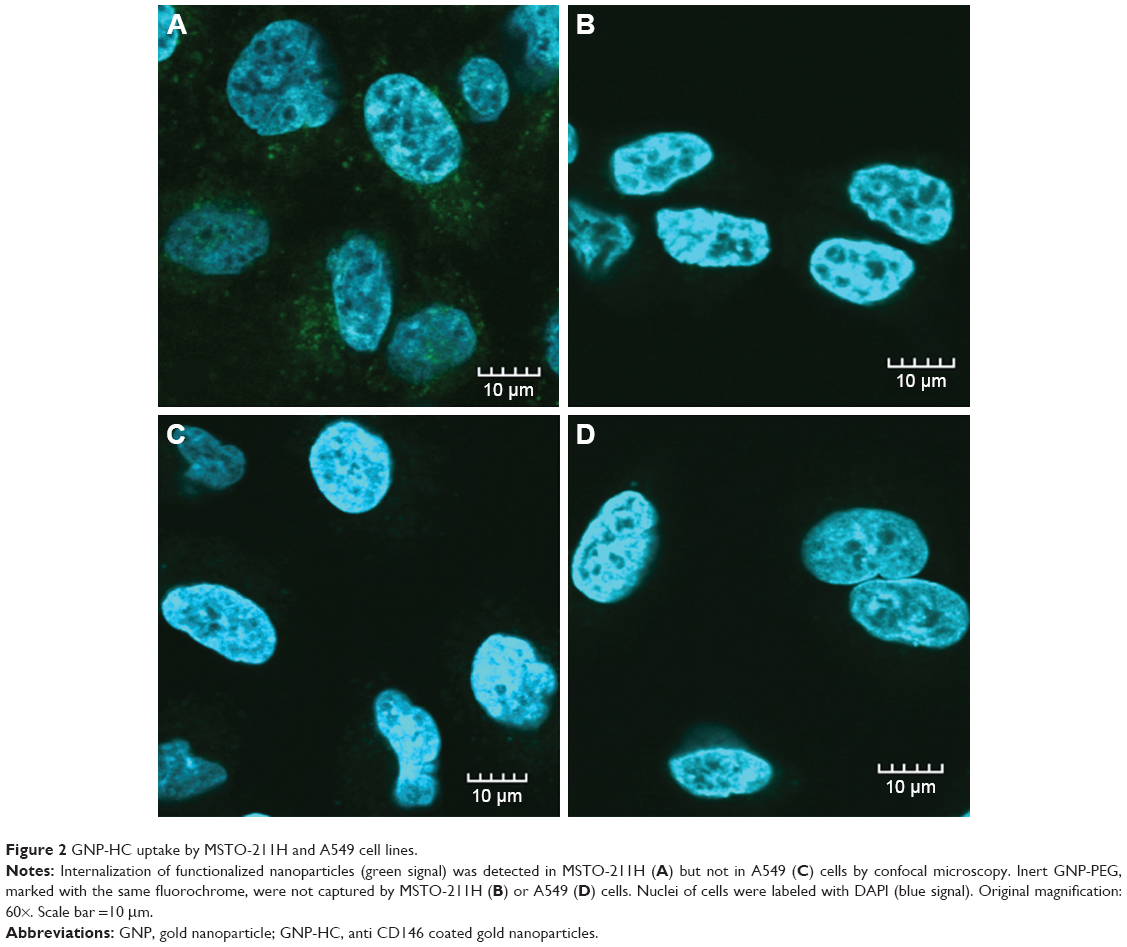 | Figure 2 GNP-HC uptake by MSTO-211H and A549 cell lines. |
Cell viability
GNP-HCPe-treated MSTO-211H (Figure 3A) and NCI-H2452 (Figure 3B) cells showed significantly reduced viability at 24 hours after treatment (65.6%±21.4% and 64.3%±6.4%, respectively) as compared with untreated cells (100%) (Figure 3A and B). In addition, the effect was long lasting since after 96 hours GNP-HCPe reduced MSTO-211H and NCI-H2452 cell viability down to 65.7%±16.3% and 59.4%±15.5%, respectively. For both cell lines, the peak of inhibition was recorded at 72 hours. The treatment with drug alone in the same experimental conditions only showed a modestly significant inhibition of cell viability at 48 hours for MSTO-211H (about 20%) and at 72 hours for NCI-H2452 (about 10%) (Figure 3A and B). It is important to note that we treated cells with the same concentration of Pe either alone or loaded inside nanoparticles (100 nM).
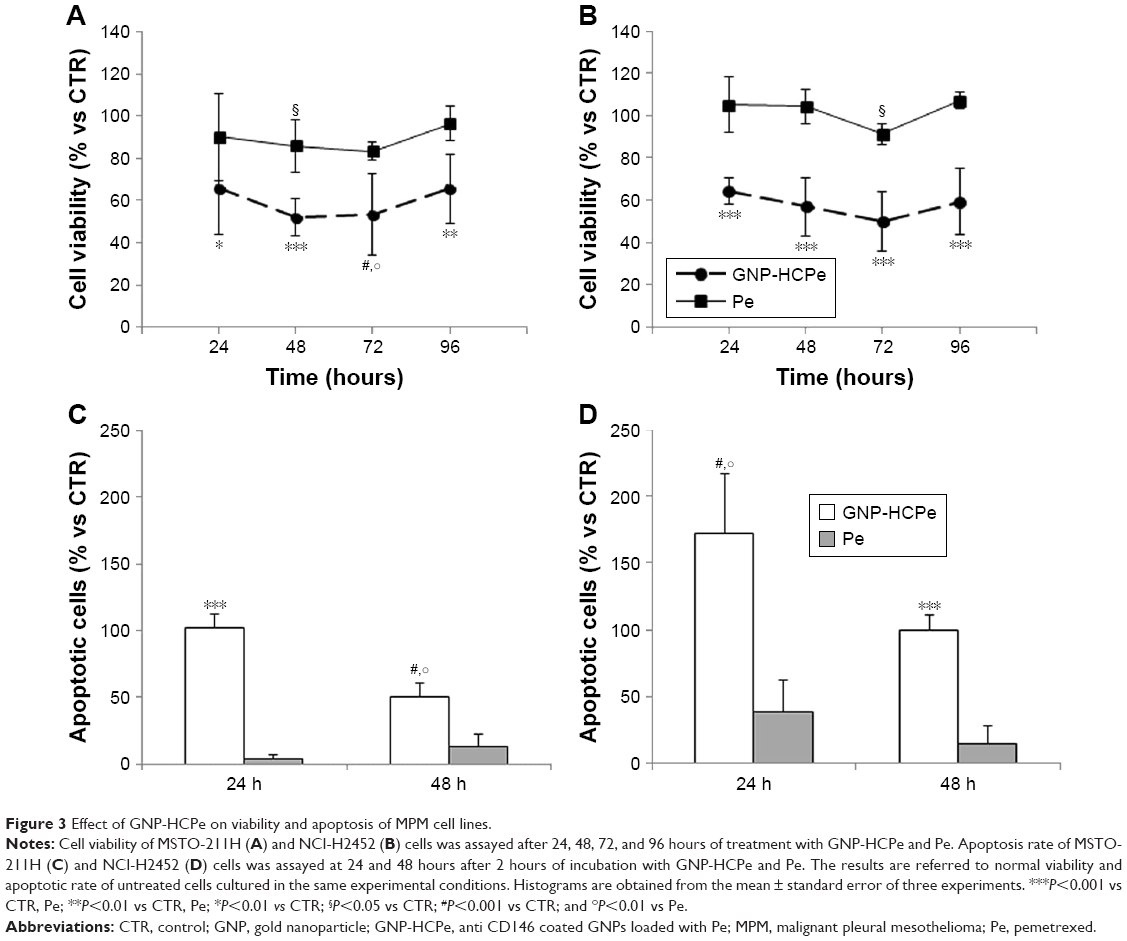 | Figure 3 Effect of GNP-HCPe on viability and apoptosis of MPM cell lines. |
Apoptotic rate
In order to understand the mechanism underlying the decrease in cell viability observed after GNP-HCPe treatment, we analyzed apoptotic rate by flow cytometry. GNP-HCPe treatment significantly increased apoptotic cell rate as compared to Pe in both cell lines (Figure 3C and D). The effect was more relevant for NCI-H2452 cells, both after 24 and 48 hours. These cells also showed higher susceptibility to drug treatment especially at 24 hours in contrast to MSTO-211H cells. These data confirm that internalization of GNP-HCPe inside MPM cells decreases cell viability through the induction of apoptosis.
Cell cycle
It is known that Pe has a cytostatic activity against malignant cells inhibiting DNA synthesis, causing the accumulation of cells in the S phase.17,18 In order to evaluate if our nanovehicle maintained the same activity, MSTO-211H and NCI-H2452 were incubated with GNP-HCPe and Pe for 24 and 48 hours. Cell cycle analysis showed a deregulation of normal cell cycle phase distribution in both cell lines after GNP-HCPe and drug incubation (Figure 4). In particular, in MSTO-211H cell line, we observed that GNP-HCPe caused an accumulation of the cells in the S phase after 24 hours of treatment, compared to Pe alone, followed by G2/M phase accumulation after 48 hours (Figure 4A and C). In NCI-H2452, both GNP-HCPe and Pe showed the same behavior causing an accumulation of the cells in the S phase at 24 hours, but GNP-HCPe showed a long-lasting effect up to 48 hours of treatment (Figure 4B and D). These data confirmed that the nanoformulation of Pe enhanced the inhibition of cell cycle progression activity of the drug, and this effect was more relevant in MSTO-211H cells.
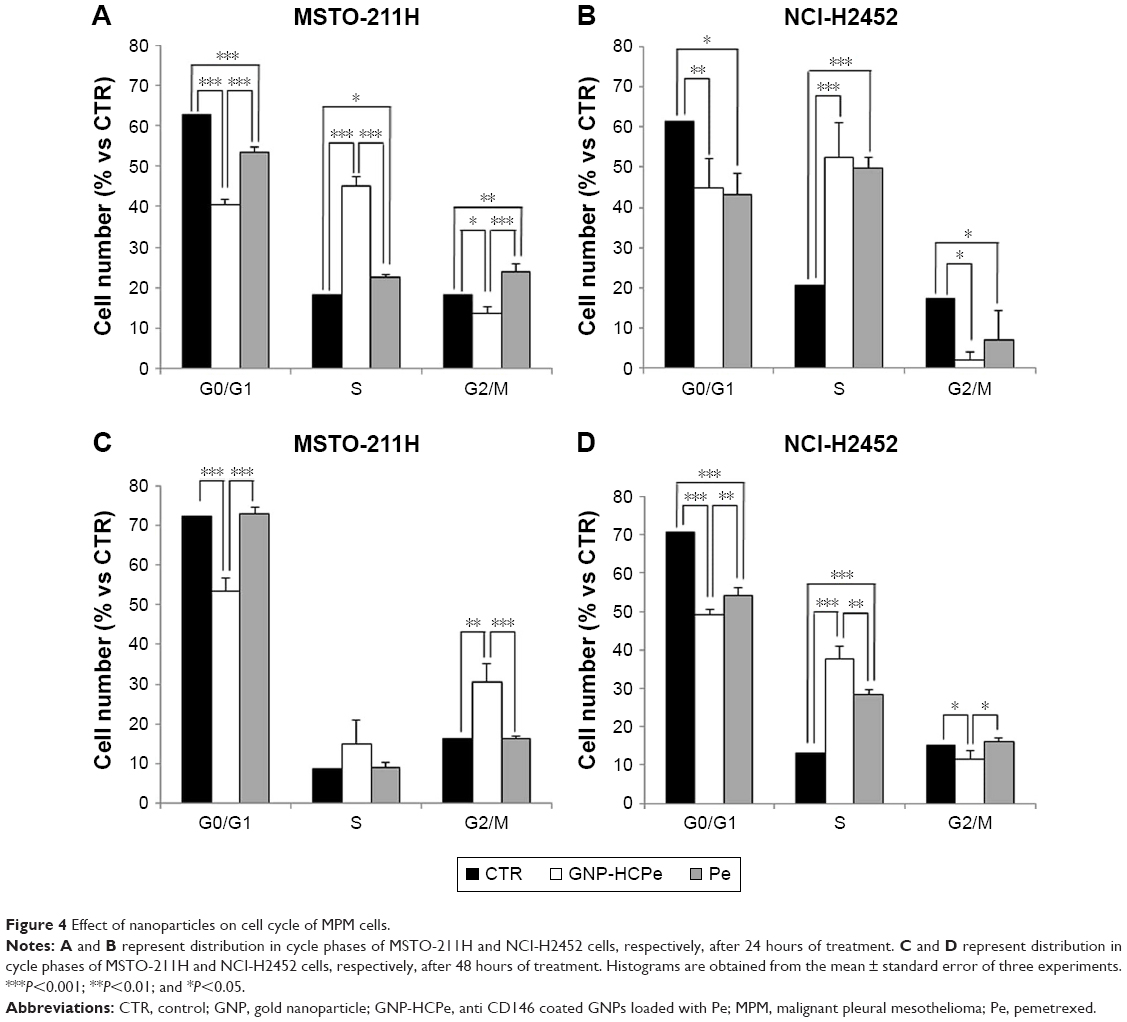 | Figure 4 Effect of nanoparticles on cell cycle of MPM cells. |
ROS production
GNP-HCPe and Pe significantly increased ROS production in culture media (Figure 5). Drug-loaded nanoparticles were more effective and, as already observed for cell viability and apoptosis, their effect was more persistent than with drug alone. After 48 hours of incubation, the amount of ROS in the extracellular compartment was still elevated, slightly higher with GNP-HCPe than with Pe alone, in MSTO-211H cells (Figure 5A), and considerably higher in NCI-H2452 cells (Figure 5B).
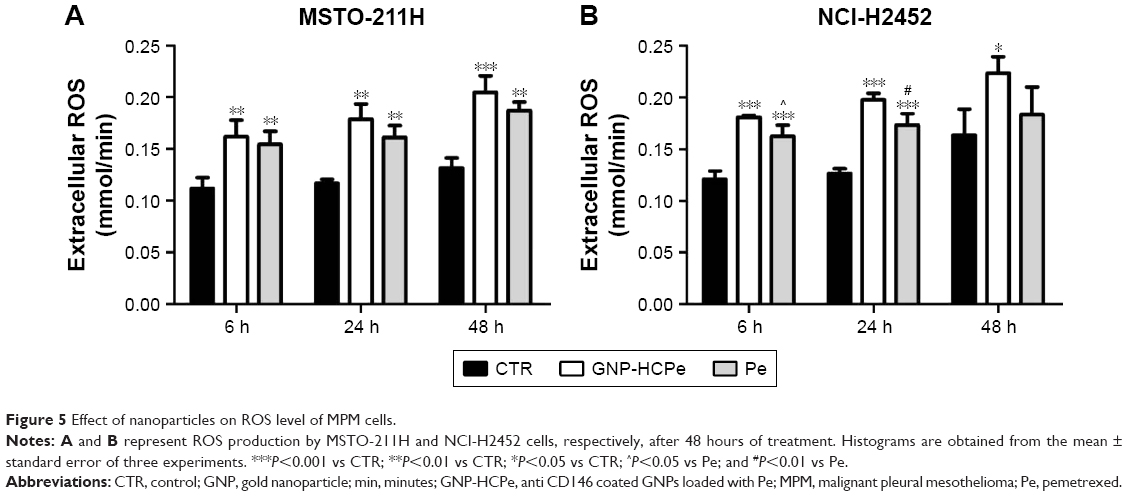 | Figure 5 Effect of nanoparticles on ROS level of MPM cells. |
Anchorage-independent growth and cell motility
The effect of nanoparticles in interfering with the clonogenic potential of cells, which is highly related to tumorigenicity,19 was evaluated by investigating cell growth on a soft support. The experiments showed that GNP-HCPe completely inhibited anchorage-independent growth after 15 days of incubation (Figure S2). Instead, treatment with Pe alone did not reduce cell clonogenic activity (139±25 in MSTO-211H and 619±72 in NCI-H2452) as compared with untreated sample (142±20 in MSTO-211H and 874±42 in NCI-H2452) (Figure S2).
We also evaluated the effect on motility of MSTO-211H and NCI-H2452 cells, assessed by continuous recording of wound healing after scratching the cell cultures up to 5 hours. In the presence of both GNP-HCPe and Pe, migration of cells was significantly affected, with respect to untreated cells (Figure S3). These results might not seem to be in line with other experiments in which we demonstrated that nanoformulation of Pe increases the therapeutic effect of the drug. However, it is important to note that migration assay was performed within 5 hours of incubation, and so we can speculate that in a short time period free and encapsulated Pes have similar effect. At late, free Pe is metabolized by cells more efficiently than GNPs that continue to release drug into the cells. This hypothesis is supported by literature data obtained with other nanoformulated drugs.20
Discussion
The diagnosis of MPM entails an end-stage disease, since median survival after its identification is 9–12 months.21 Currently recommended therapeutic strategies for advanced-stage diseases are based on standard chemotherapy.22,23 Pe is an antifolate agent that inhibits multiple folate-dependent enzymes.24 Although Pe in combination with platinum remains the cornerstone of therapy,25 the increase in survival rates of treated patients is modest.26 This poor efficacy is mostly due to the inability of Pe – as well as of all standard chemoagents – to efficiently reach and concentrate in the tumor/pleural space without causing excessive systemic toxicity.27 In order to enhance the pharmacological activity and pharmacodynamics of Pe, different strategies are adopted by researchers such as conjugation of drug with enhancer peptides28,29 or nanoformulation.30 The use of nanoparticles as innovative drug delivery vehicles that specifically targeted against a range of disease-responsible cells represents a huge opportunity to selectively challenge tumor cells, with the advantage of increasing intracellular drug concentration limiting systemic toxicity. Thus, based on previous experience, we engineered Pe-loaded GNPs that targeted against MPM cells. This strategy entails several advantages. First, GNPs are chemically inert and after functionalization reach an overall size of 47 nm, which is considered safe in terms of biodistribution. Second, GNP can be efficiently functionalized to target cells with an antibody directed against a cell surface marker reducing the possibility to damage the near positioned normal cells.6 Finally, GNP can be safely locally administered without eliciting any unwanted toxic/inflammatory reaction.7 To develop our strategy for MPM, we selected a surface MPM cell marker that is suitable for nanoparticle targeting. Among published markers,31–33 we identified CD146, a transmembrane glycoprotein belonging to the immunoglobulin superfamily, whose overexpression has been documented in several tumor cells (prostate, ovary, melanoma, and triple negative breast cancers) and linked to a poor prognosis. CD146 has been reported to not be expressed by normal or reactive mesothelium and is currently expressed in normal conditions by vascular endothelial cells and smooth muscle cells, with a recently identified role in the promotion of angiogenesis.15,34,35 In this study, we confirmed that this surface marker is highly expressed by primary cells isolated from pleural liquid of patients affected by MPM and by two commercially available MPM cell lines. In addition, we also observed that MPM biopsies express CD146 while normal/reactive mesothelium is negative for this marker. Therefore, we designed and engineered GNPs functionalized with half chain-goat anti human CD146 moAb and loaded them with Pe (GNP-HCPe). As discussed earlier, the main critical issues related to inefficient performance of conventional chemotherapeutic agents are related to the lack of treatment specificity and insufficient intracellular drug accumulation at safe treatment dose. Both these issues can be overcome successfully by targeted nanovehicles, which will allow local treatment of MPM cells by providing high intracellular drug accumulation while sparing normal and inflammatory cells. This approach is more promising in comparison to the conjugation of Pe with enhancer peptides, even if authors demonstrated that oral administration of the Pe–peptide complex markedly reduced microvessel density, proliferation, and increased apoptosis in the tumor tissues.28
We then aimed to confirm that GNP-HCPe are able to specifically target MPM cells even after a short treatment period (2 hours) and to compare the biological activity of GNP-HCPe in vitro with that of Pe alone. We demonstrated that GNP-HCPe were efficiently internalized by MPM cell lines, whereas nanoparticles lacking anti-CD146 moAb were not. Moreover, once GNP-HCPe were internalized, the Pe released inside the cell was significantly more effective and its action was more sustained over time as compared with the free drug.
This study provides experimental evidence that our targeted nanovehicle significantly impairs the malignant phenotype of transformed mesothelial cells. Results are even more relevant when compared to the biological responses (cell viability, cell cycle progression, and clonogenicity) obtained by exposing MPM cells to Pe alone. Only for cell motility, GNP-HCPe were as effective as the drug alone, probably because the given concentration of free Pe (100 nM) was sufficient to inhibit cell migration in a short time period (5 hours of treatment). Concerning ROS production, our results demonstrating a significant increase in ROS generation with GNP-HCPe, are in agreement with previous observations, indicating that Pe induces apoptosis in MPM and lung cancer cells through the activation of ROS and inhibition of sirtuin 1.35
It is worth to note that only drug-loaded nanoparticles were able to completely inhibit the capacity to form colonies on a soft substrate. This issue is remarkable since anchorage-independent growth is the hallmark of malignancy and is highly correlated with tumorigenicity in animals.36
This study clearly represents a preliminary proof-of-concept to be further developed with more extensive studies and in vivo experiments on animal models of disease. Interestingly, while targeted nanoparticles were always more effective than Pe alone, different response rates were observed in the two selected cell lines, suggesting individual variability. It will be necessary to explore the causes of this variability in order to identify potential predictive factors.
On the basis of these preliminary results, we believe that nanoparticles represent a promising approach for the development of a highly effective local treatment for MPM, because they allow us to directly address malignant cells and deliver high level of drugs over a sustained period of time, while sparing normal cells. Approaches of intrapleural drug delivery have been described in the literature,37–39 including Pe loaded nanoparticles40 but, to our knowledge, we have shown for the first time that specific targeting of MPM cells by nanoparticles is feasible and effective. Furthermore, CD146 targeting offers the possibility of new approaches that could be translated to the treatment of other cancers or metastasis affecting the pleural or peritoneal space.41,42
Acknowledgments
This work was supported by Crowdfunding Universitiamo of the University of Pavia and the Association of Asbestos Victims, Associazione Vittime Amianto Nazionale Italiana. An abstract of the paper “Pemetrexed-loaded nanoparticles targeted to malignant pleural mesothelioma cells: an in-vitro study” was presented at the ERS International Congress as an oral presentation; Amsterdam, 26–30 September, 2015. The talk’s abstract has been published.43
Disclosure
The authors report no conflicts of interest in this work.
References
Carbone M, Kratzke RA, Testa JR. The pathogenesis of mesothelioma. Semin Oncol. 2002;29(1):2–17. | ||
Røe OD, Stella GM. Malignant pleural mesothelioma: history, controversy and future of a manmade epidemic. Eur Respir Rev. 2015;24(135):115–131. | ||
Ramalingam SS, Belani CP. Recent advances in the treatment of malignant pleural mesothelioma. J Thorac Oncol. 2008;3(9):1056–1064. | ||
Vogelzang NJ, Rusthoven JJ, Symanowski J, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21(14):2636–2644. | ||
Remon J, Reguart N, Corral J, Lianes P. Malignant pleural mesothelioma: new hope in the horizon with novel therapeutic strategies. Cancer Treat Rev. 2015;41(1):27–34. | ||
Cova E, Colombo M, Inghilleri S, et al. Antibody-engineered nanoparticles selectively inhibit mesenchymal cells isolated from patients with chronic lung allograft dysfunction. Nanomedicine. 2015;10(1):9–23. | ||
Cova E, Inghilleri S, Pandolfi L, et al. Bioengineered gold nanoparticles targeted to mesenchymal cells from patients with bronchiolitis obliterans syndrome does not rise the inflammatory response and can be safely inhaled by rodents. Nanotoxicology. 2017;11(4):534–545. | ||
Brust M, Walker M, Bethell D, Schiffrin DJ, Whyman R. Synthesis of thiol-derivatised gold nanoparticles in a two-phase Liquid–Liquid system. J Chem Soc Chem Commun. 1994;7(7):801–802. | ||
Pellegrino T, Manna L, Kudera S, et al. Hydrophobic Nanocrystals Coated with an Amphiphilic Polymer Shell: A General Route to Water Soluble Nanocrystals. Nano Letters. 2004;4(4):703–707. | ||
Scott BR, Alexandra CR, Richard GM. Pemetrexed indirectly activates the metabolic kinase AMPK in human carcinomas. Cancer Res. 2010;70(24):10299–10309. | ||
Morfouace M, Shelat A, Jacus M, et al. Pemetrexed and gemcitabine as combination therapy for the treatment of Group3 medulloblastoma. Cancer Cell. 2014;25(4):516–529. | ||
Faoro V, Fink B, Taudorf S, et al. Acute in vitro hypoxia and high-altitude (4,559 m) exposure decreases leukocyte oxygen consumption. Am J Physiol Regul Integr Comp Physiol. 2011;300(1):R32–R39. | ||
Mrakic-Sposta S, Gussoni M, Montorsi M, Porcelli S, Vezzoli A. Assessment of a standardized ROS production profile in humans by electron paramagnetic resonance. Oxid Med Cell Longev. 2012;2012(3):1–10. | ||
Philippeaux MM, Pache JC, Dahoun S, et al. Establishment of permanent cell lines purified from human mesothelioma: morphological aspects, new marker expression and karyotypic analysis. Histochem Cell Biol. 2004;122(3):249–260. | ||
Sato A, Torii I, Okamura Y, et al. Immunocytochemistry of CD146 is useful to discriminate between malignant pleural mesothelioma and reactive mesothelium. Mod Pathol. 2010;23(11):1458–1466. | ||
Minato H, Kurose N, Fukushima M, et al. Comparative immunohistochemical analysis of IMP3, GLUT1, EMA, CD146, and desmin for distinguishing malignant mesothelioma from reactive mesothelial cells. Am J Clin Pathol. 2014;141(1):85–93. | ||
Dubey S, Schiller JH. Three emerging new drugs for NSCLC: pemetrexed, bortezomib, and cetuximab. Oncologist. 2005;10(4):282–291. | ||
Miklán Z, Orbán E, Bánóczi Z, Hudecz F. New pemetrexed-peptide conjugates: synthesis, characterization and in vitro cytostatic effect on non-small cell lung carcinoma (NCI-H358) and human leukemia (HL-60) cells. J Pept Sci. 2011;17(12):805–811. | ||
Mori S, Chang JT, Andrechek ER, et al. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene. 2009;28(31):2796–2805. | ||
Pandolfi L, Bellini M, Vanna R, et al. H-Ferritin enriches the curcumin uptake and improves the therapeutic efficacy in triple negative breast cancer cells. Biomacromolecules. 2017;18(10):3318–3330. | ||
Guazzelli A, Bakker E, Tian K, Demonacos C, Krstic-Demonacos M, Mutti L. Promising investigational drug candidates in phase I and phase II clinical trials for mesothelioma. Expert Opin Investig Drugs. 2017;26(8):933–944. | ||
Baas P, Fennell D, Kerr KM, et al. Malignant pleural mesothelioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(Suppl 5):v31–v39. | ||
Ettinger DS, Wood DE, Akerley W, et al. NCCN guidelines insights: malignant pleural mesothelioma, version 3.2016. J Natl Compr Canc Netw. 2016;14(7):825–836. | ||
Rollins KD, Lindley C. Pemetrexed: a multitargeted antifolate. Clin Ther. 2005;27(9):1343–1382. | ||
Vogelzang NJ, Rusthoven JJ, Symanowski J, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21(14):2636–2644. | ||
Nowak AK. Chemotherapy for malignant pleural mesothelioma: a review of current management and a look to the future. Ann Cardiothorac Surg. 2012;1(4):508–515. | ||
Bonelli MA, Fumarola C, La Monica S, Alfieri R. New therapeutic strategies for malignant pleural mesothelioma. Biochem Pharmacol. 2017;123:8–18. | ||
Foyez M, Ok-Cheol J, Farzana A, et al. Oral pemetrexed facilitates low-dose metronomic therapy and enhances antitumor efficacy in lung cancer. J Control Release. 2018;284:160–170. | ||
Yang W, Yang L, Xia Y, et al. Lung cancer specific and reduction-responsive chimaeric polymersomes for highly efficient loading of pemetrexed and targeted suppression of lung tumor in vivo. Acta Biomater. 2018;70:177–185. | ||
Stolarczyk EU, Stolarczyk K, Łaszcz M, Kubiszewski M, Leś A, Michalak O. Pemetrexed conjugated with gold nanoparticles – Synthesis, characterization and a study of noncovalent interactions. Eur J Pharm Sci. 2017;109:13–20. | ||
Kushitani K, Amatya VJ, Mawas AS, Miyata Y, Okada M, Takeshima Y. Use of anti-noxa antibody for differential diagnosis between epithelioid mesothelioma and reactive mesothelial hyperplasia. Pathobiology. 2016;83(1):33–40. | ||
Kinoshita Y, Takasu K, Yuri T, et al. Two cases of malignant peritoneal mesothelioma without asbestos exposure: cytologic and immunohistochemical features. Ann Diagn Pathol. 2013;17(1):99–103. | ||
Yuichi K, Kosho T, Takashi Y, et al. Cytologic-pathologic correlation two cases of malignant peritoneal mesothelioma without asbestos exposure: cytologic and immunohistochemical features. Ann Diagn Pathol. 2013;17(1):99–103. | ||
St Croix B. CD146: the unveiling of a pro-angiogenic netrin receptor. Cell Res. 2015;25(5):533–534. | ||
Hwang KE, Kim YS, Hwang YR, et al. Pemetrexed induces apoptosis in malignant mesothelioma and lung cancer cells through activation of reactive oxygen species and inhibition of sirtuin 1. Oncol Rep. 2015;33(5):2411–2419. | ||
Relan V, Morrison L, Parsonson K, et al. Phenotypes and karyotypes of human malignant mesothelioma cell lines. PLoS One. 2013;8(3):e58132. | ||
Sakaguchi H, Ishida H, Nitanda H, Yamazaki N, Kaneko K, Kobayashi K. Pharmacokinetic evaluation of intrapleural perfusion with hyperthermic chemotherapy using cisplatin in patients with malignant pleural effusion. Lung Cancer. 2017;104:70–74. | ||
Tada Y, Hiroshima K, Shimada H, et al. An intrapleural administration of zoledronic acid for inoperable malignant mesothelioma patients: a phase I clinical study protocol. Springerplus. 2016;5(1):195. | ||
Sterman DH, Alley E, Stevenson JP, et al. Pilot and feasibility trial evaluating immuno-gene therapy of malignant mesothelioma using intrapleural delivery of adenovirus-IFNα combined with chemotherapy. Clin Cancer Res. 2016;22(15):3791–3800. | ||
Ando H, Kobayashi S, Abu Lila AS, et al. Advanced therapeutic approach for the treatment of malignant pleural mesothelioma via the intrapleural administration of liposomal pemetrexed. J Control Release. 2015;220(Pt A):29–36. | ||
Orlandi E, Mordenti P, Zangrandi A, Cavanna L. Intrapleural trastuzumab therapy for malignant pleural effusion from her 2 overexpression in metastatic gastric cancer. Chemotherapy. 2014;60(5–6):321–324. | ||
Ohta Y, Shimizu Y, Matsumoto I, Watanabe G. Management of malignant pleural effusion by multimodality treatment including the use of paclitaxel administered by 24-hour intrathoracic infusion for patients with carcinomatous pleuritis. J Exp Clin Cancer Res. 2006;25(1):15–19. | ||
Stella G, Cova E, Inghilleri S, et al. Pemetrexed-loaded nanoparticles targeted to malignant pleural mesothelioma cells: an in vitro study [abstract]. Eur Respir J. 2015:46(Suppl 59). |
Supplementary materials
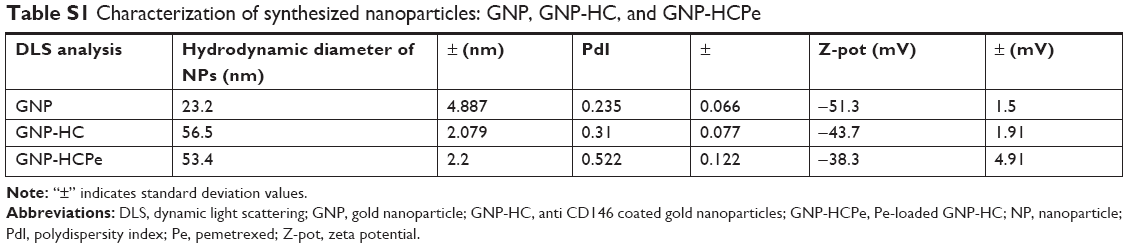 | Table S1 Characterization of synthesized nanoparticles: GNP, GNP-HC, and GNP-HCPe |
 | Figure S1 TEM image of unconjugated GNP. |
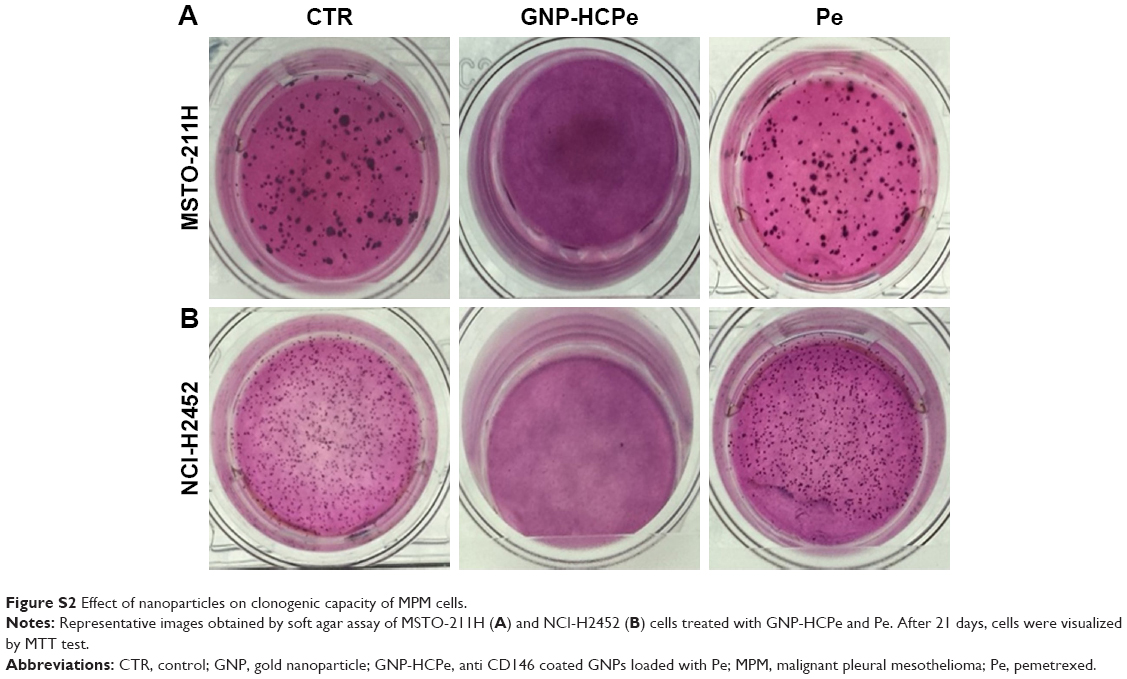 | Figure S2 Effect of nanoparticles on clonogenic capacity of MPM cells. |
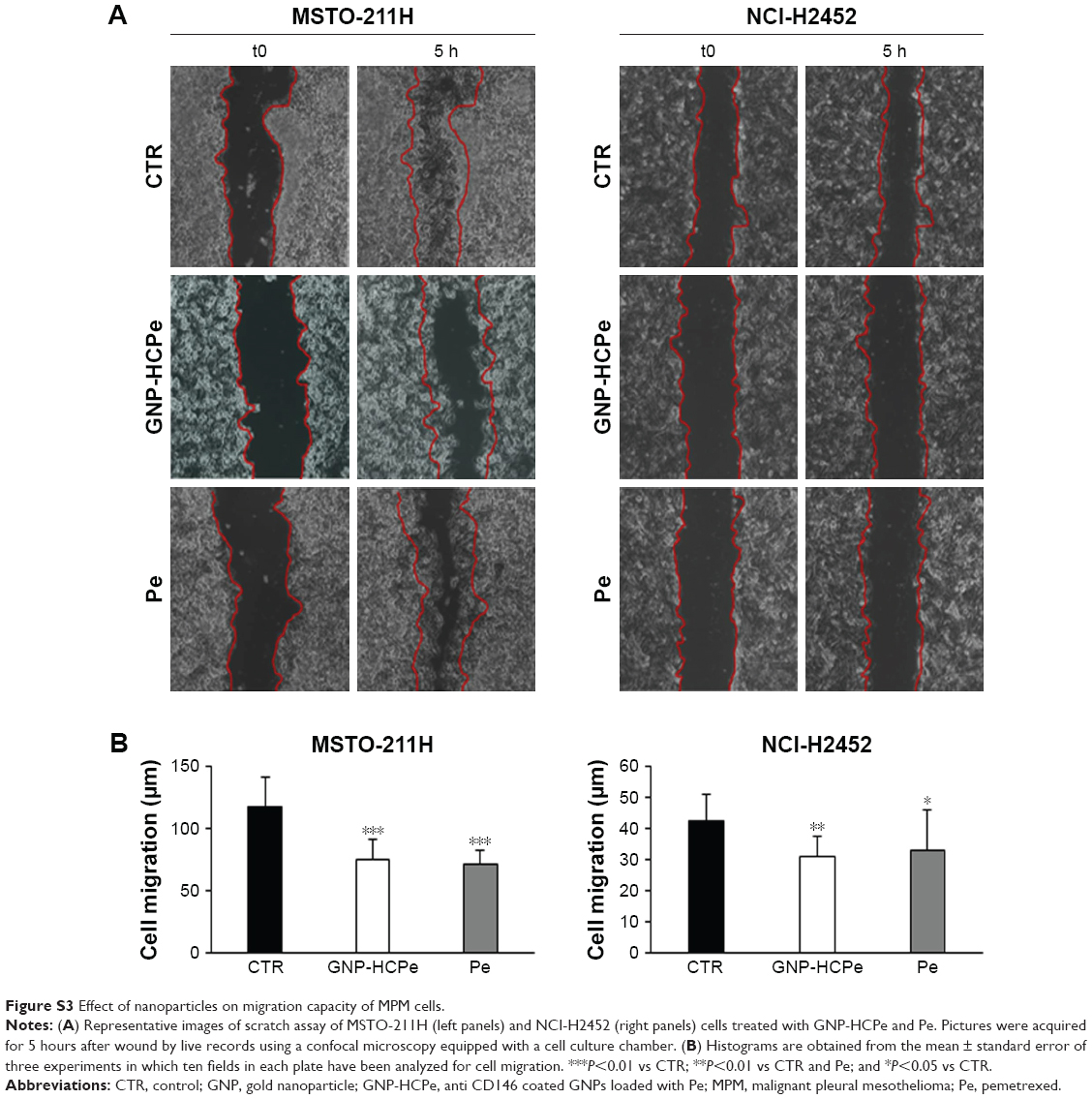 | Figure S3 Effect of nanoparticles on migration capacity of MPM cells. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.