")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
PEGylated apoptotic protein-loaded PLGA microspheres for cancer therapy
Authors Byeon HJ, Kim I, Choi JS, Lee ES, Shin BS, Youn YS
Received 14 October 2014
Accepted for publication 28 November 2014
Published 19 January 2015 Volume 2015:10(1) Pages 739—748
DOI https://doi.org/10.2147/IJN.S75821
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Carlos Rinaldi
Hyeong Jun Byeon,1 Insoo Kim,1 Ji Su Choi,1 Eun Seong Lee,2 Beom Soo Shin,3 Yu Seok Youn1
1Department of Pharmaceutical Sciences, School of Pharmacy, Sungkyunkwan University, Suwon, Republic of Korea; 2Division of Biotechnology, The Catholic University of Korea, Bucheon-si, Republic of Korea; 3Department of Pharmacy, College of Pharmacy, Catholic University of Daegu, Gyeongsan-si, Republic of Korea
Abstract: The aim of the current study was to investigate the antitumor potential of poly(D,L-lactic-co-glycolic acid) microspheres (PLGA MSs) containing polyethylene glycol (PEG)-conjugated (PEGylated) tumor necrosis factor–related apoptosis-inducing ligand (PEG-TRAIL). PEG-TRAIL PLGA MSs were prepared by using a water-in-oil-in-water double-emulsion method, and the apoptotic activities of supernatants released from the PLGA MSs at days 1, 3, and 7 were examined. The antitumor effect caused by PEG-TRAIL PLGA MSs was evaluated in pancreatic Mia Paca-2 cell-xenografted mice. PEG-TRAIL PLGA MS was found to be spherical and 14.4±1.06 µm in size, and its encapsulation efficiency was significantly greater than that of TRAIL MS (85.7%±4.1% vs 43.3%±10.9%, respectively). The PLGA MS gradually released PEG-TRAIL for 14 days, and the released PEG-TRAIL was shown to have clear apoptotic activity in Mia Paca-2 cells, whereas TRAIL released after 1 day had a negligible activity. Finally, PEG-TRAIL PLGA MS displayed remarkably greater antitumor efficacy than blank or TRAIL PLGA MS in Mia Paca-2 cell-xenografted mice in terms of tumor volume and weight, apparently due to increased stability and well-retained apoptotic activity of PEG-TRAIL in PLGA MS. We believe that this PLGA MS system, combined with PEG-TRAIL, should be considered a promising candidate for treating pancreatic cancer.
Keywords: Poly(D,L-lactic-co-glycolic acid), controlled release, PEGylation, TRAIL, pancreatic cancer
Introduction
Poly(D,L-co-glycolic acid) microspheres (PLGA MSs) have been extensively utilized for the controlled release of therapeutic peptides and proteins.1–3 Due to their biodegradability and biocompatibility, PLGA polymer products were approved for drug formulations and medical devices by the US Food and Drug Administration. Moreover, the MSs have excellent versatility in terms of availability of different molecular weights/compositions and variable degradability.1 To date, many therapeutic peptide-loaded PLGA formulations, such as Lupron Depot®, Trelstar Depot®, Zoladex® Depot, and Sandostatin LAR® Depot, have been made commercially available. Most PLGA formulations are injectable and therapeutically effective over a period of 1–3 months, which greatly decreases injection frequency.4
However, all such PLGA formulation products involve only peptides or synthetic chemical drugs, not protein drugs, because the optimal pharmaceutical application of proteins in PLGA depot systems has not been fully realized in practice.1,5 Although the first protein-loaded PLGA MS (recombinant human growth hormone, Nutropin Depot™) had been approved in 1998, this was withdrawn from the market in 2004 due to several technical problems. In general, protein-loaded PLGA MSs have been plagued with typical problems in terms of rapid initial burst release, physicochemical instability, and bioactivity loss.1,5,6 For these reasons, many reports have shown that PLGA MSs containing unmodified proteins exhibited undesirable release profiles and unsatisfactory therapy results in vivo5,7–9.
Tumor necrosis factor (TNF)–related apoptosis-inducing ligand (TRAIL) is one of the members of the TNF gene superfamily that induce caspase-dependent apoptosis through engagement of death receptors DR4 and DR5.10,11 TRAIL selectively kills tumors cells because such death receptors are expressed mostly on tumor cells, but barely expressed in normal cells.12,13 Therefore, this apoptotic protein has been regarded as a safe and attractive antitumor agent and has been studied extensively against a wide variety of cancer cells, including pancreatic tumor cells.13–15 Furthermore, several reports have demonstrated remarkable antitumor effects of TRAIL.16,17
PEGylation, the covalent modification with polyethylene glycol (PEG), is considered an effective way of improving many shortcomings of unmodified therapeutic proteins.18,19 Proteins are vulnerable to proteolysis by various enzymes, are cleared rapidly through the kidney due to hydrophilicity, and are easily taken up by organs such as the liver.20 On the contrary, it is well known that PEGylated proteins greatly resist proteolysis and renal filtration as compared with non-PEGylated counterparts, and thus display superior pharmacokinetic and pharmacodynamic properties in vivo.21 In our previous reports, we have documented that PEGylated TRAIL (PEG-TRAIL) is much more therapeutically effective than native TRAIL in various cancer models.18,22,23
Here, we have sought to develop a PLGA MS depot containing PEG-TRAIL to attain long-term antitumor effects in a pancreatic cancer model. In our previous studies,18,22,23 PEG-TRAIL displayed superior physicochemical stability, improved pharmacokinetics, and remarkable antitumor effects over unmodified TRAIL. In this study, many advantages of PEGylation technology for a TRAIL protein were combined with the sustained-release system of PLGA MS to acquire a synergistic therapeutic effect. To examine the effects of such a combination, the apoptotic activities of PEG-TRAIL-loaded PLGA MS were evaluated in Mia Paca-2 cells and its antitumor efficacy was examined in Mia Paca-2 cell-induced tumor-bearing mice. Additionally, the in vitro release profiles of PEG-TRAIL from PLGA MSs were examined and compared with that of unmodified TRAIL.
Materials and methods
Materials
TRAIL with a trimer-inducing domain and six histidines at its N-terminus (hTRAIL [114–281]) was produced as previously described.24,25 Briefly, monomethoxypolyethylene glycol aldehyde (mPEG-ALD; molecular weight [Mw]: 5,000 Da) was purchased from the NOF Corporation (Tokyo, Japan). PLGA (Mw: 10,000 Da; lactic acid:glycolic acid ratio =50:50) was purchased from Wako Pure Chemical (Tokyo, Japan). Polyvinyl alcohol (Mw: 30,000–70,000 Da) was purchased from Sigma (St Louis, MO, USA). In situ cell death detection kits were supplied by Roche Diagnostics (Mannheim, Germany). All other reagents, unless otherwise specified, were purchased from Sigma-Aldrich.
Experimental animals
BALB/c nu/nu mice (males, 5 weeks old) were obtained from the Hanlim Experimental Animal Laboratory (Seoul, South Korea). Animals were cared for according to the guidelines issued by the National Institutes of Health (NIH) for the care and use of laboratory animals (NIH publication 80-23, revised in 1996). Animals were housed in groups of four to six under a 12-hour light/dark cycle (lights on 6 am), allowed food and water ad libitum, and acclimatized for 1 week. This study was approved by the Ethical Committee on Animal Experimentation of Sungkyunkwan University.
Preparation of PEG-TRAIL
N-terminal mono-PEGylated TRAIL (PEG-TRAIL) was prepared as previously described.18,22,23 Briefly, 31.3 mg of mPEG-ALD was dissolved in 62.5 mL of 50 mM acetate buffer containing 100 mM NaCl and 60 mM sodium cyanoborohydride (pH 5.0), and this solution was mixed with 125 mL of TRAIL in 20 mM acetate buffer containing 100 mM NaCl (pH 5.0, 200 μg/mL). The PEGylation reaction was allowed to continue overnight at 4°C with gentle stirring. The solution was then concentrated using a Centricon-10 concentrator (Mw cutoff [CO]: 30 kDa; Millipore Amicon, Beverly, MA, USA). PEG-TRAIL was purified by size-exclusion chromatography on a HiPrep 16/60 Sephacryl S-200 HR column (16 mm ×60 cm, 25–75 μm; GE Healthcare, Piscataway, NJ, USA). Elution was carried out at a flow rate of 1.0 mL/minute using a 20 mM acetate buffer containing 100 mM NaCl (pH 5.0).
Preparation and characterization of PEG-TRAIL-loaded PLGA MSs
TRAIL and PEG-TRAIL PLGA MSs were prepared using the double emulsion solvent extraction method with slight modification.23 Briefly, 200 μL of protein solution (5 mg/mL) was emulsified in 2 mL of dichloromethane containing 150 mg PLGA by homogenization at 500 revolutions per minute (rpm) using a WiseTis homogenizer HG-15D (DAIHAN Scientific, Seoul, South Korea) for 30 seconds. The resulting water-in-oil emulsion was gradually injected using a glass syringe with a 22-gauge needle into 50 mL of an ice-cold 0.2% (w/v) of polyvinyl alcohol solution and then emulsified using a Silverson Laboratory Mixer (model L4RT) with a 5/8-inch head (Silverson Machines, Inc., East Longmeadow, MA, USA) for 2 minutes at 2,000 rpm to produce a water-in-oil-in-water (w/o/w) emulsion. The resultant w/o/w emulsion was then stirred for 6 hours to allow the dichloromethane to evaporate. The hardened MSs were centrifuged, washed three times, and lyophilized. Blank MSs were prepared in the same way without TRAIL or PEG-TRAIL. MS morphologies were observed using field-emission scanning electron microscopy (FE-SEM; Leo Supra 55, Genesis 2000) at an accelerating voltage of 15,000 V. Average particle sizes were determined using a laser diffraction particle size analyzer (Mastersizer; Malvern Instruments, Malvern, UK). Fluorescein-tagged PEG-TRAIL-loaded PLGA MSs were observed using confocal laser scanning microscopy ([CLSM] Meta LSM 510; Carl Zeiss, Jena, Germany) to investigate the inner distribution of PEG-TRAIL inside PLGA MSs.
Protein encapsulation efficiencies
The exact amount of protein loaded in MSs was determined using a modification of the two-step extraction method.23 TRAIL/PEG-TRAIL PLGA MSs (5 mg) were dissolved in 250 μL of a 0.2 M NaOH solution and then incubated at 37°C for 4 hours. After centrifugation, the amount of encapsulated protein was determined using the Micro-BCA Protein Assay Kit (Pierce Chemical Co, Rockford, IL, USA). Samples were assayed in triplicate.
In vitro release test
TRAIL or PEG-TRAIL PLGA MSs (5 mg) were suspended in 1 mL of 10 mM phosphate-buffered saline (PBS; pH 7.4) containing 0.02% (v/v) Tween-20 in Eppendorf tubes, which were gently rotated at 37°C. At predetermined times, the PLGA MSs were collected by centrifugation, and the amounts of TRAIL and PEG-TRAIL inside the PLGA MSs were determined using the Micro-BCA Protein Assay Kit (Pierce Chemical Co) in order to calculate the released percentage of TRAIL or PEG-TRAIL. In addition, PEG-TRAIL (5 mg) was reacted with 2 molar equivalents of N-hydroxysuccinimide-fluorescein (Pierce Chemical Co) in 10 mM of phosphate buffer (5 mL, pH 7.4), and the reaction was allowed to continue at room temperature for 1 hour. The mixture was dialyzed for at least 24 hours using a dialysis kit (MwCO: 3,500 Da; Gene Bio-Application Ltd, Kfar Hanagid, Israel), then concentrated at 5.0 mg/mL using a Centricon-30 concentrator (MwCO: 30 kDa; Millipore, Beverly, MA, USA). A portion of fluorescein-tagged PEG-TRAIL was loaded into PLGA MSs, and the release test was performed by using the same method described above. The morphology of fluorescein-tagged PEG-TRAIL PLGA MSs during drug release was investigated by SEM and CLSM.
In vitro TUNEL assay in pancreatic Mia Paca-2 cells
Apoptotic pancreatic Mia Paca-2 cells produced by TRAIL or PEG-TRAIL release (1, 3, and 7 days) from PLGA MSs were observed by terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine 5′ -triphosphate (dUTP) nick end labeling (TUNEL) using an in situ cell death detection kit (Fluorescein; Roche Diagnostics). Briefly, 2×105 cells were seeded on to glass slides by cytospinning (5 minutes, 200× g; Cytospin 3; Shandon, Frankfurt, Germany), air-dried, and fixed with paraformaldehyde (4% in PBS at pH 7.4) at room temperature for 1 hour. Slides were then rinsed with PBS, and cells were permeabilized by adding a solution of 0.1% Triton X-100 and 0.1% Na-citrate at room temperature for 2 minutes, rinsed twice with PBS, dried, and reacted with 50 μL of TUNEL reagent (containing TdT) at 37°C for 1 hour in the dark. Samples were then rinsed three times with PBS, dried, and treated with a 4′,6-diamidino-2-phenylindole (DAPI) solution containing nucleotides. Apoptotic cells were observed by CLSM using an excitation wavelength of 450–490 nm and a detection wavelength of 520 nm.
Cytotoxicity assessments of the protein drugs released from PLGA MSs
Cytotoxicity of the protein drugs released from PLGA MSs was investigated in a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, Mia Paca-2 cells were seeded at 1×104 cells per well in 96-well plates and preincubated for 24 hours in Dulbecco’s Modifed Eagle’s Medium (DMEM; Corning, NY, USA) containing 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin/streptomycin (Gibco). The medium was withdrawn from the well, and serum-free DMEM containing predetermined amounts of released TRAIL or PEG-TRAIL at days 1, 3, and 7 was added (final concentration: 500 ng/mL). After 24 hours, the viability and half-maximal inhibitory concentrations (IC50) were determined.
In vivo antitumor activity of PEG-TRAIL PLGA MSs in Mia Paca-2-bearing mice
To investigate the antitumor activities of PLGA MSs containing TRAIL or PEG-TRAIL, we used a BALB/c nu/nu mouse xenograft model. Mia Paca-2 cells (107 cells per mouse, 200 μL injection) were inoculated subcutaneously into the right dorsal flanks of male BALB/c nu/nu mice. When tumors reached volumes of ~300 mm3, mice were randomized into three groups (0 day). Mice in the first group received an intratumoral injection of blank PLGA MSs at 0 day as a control. The other two groups of xenografted mice were treated intratumorally with PLGA MSs containing either TRAIL or PEG-TRAIL (0.1 mg per mouse). Mice were treated with the respective PLGA MSs every 7 days for 2 weeks, that is, on days 0, 7, and 14. Tumor sizes were measured every 3 days using a digital caliper, and tumor volumes (in mm3) were calculated using the following formula: tumor volume = length × width2 ×0.5, as previously described.22 Further, mice were sacrificed at day 27 to measure tumor weights.
Histologies of mouse tumors
Histology of tumor tissues was evaluated using a modification of a previously described procedure.23,24 Excised tumor specimens were treated with formalin, paraffin embedded, and sectioned (at ~5 μm). Tumor sections were stained with hematoxylin and eosin, subjected to TUNEL assay (in situ cell death detection kit; Roche Diagnostics), and observed under a light microscope and a confocal laser scanning microscope. Histopathology and levels of apoptosis were compared across the treatment groups.
Statistical analysis
Data are presented as the means ± standard deviations. Statistical significances were determined using the Student’s t-test, and P-values <0.05 were considered statistically significant.
Results
Preparation and characterization of PLGA MS
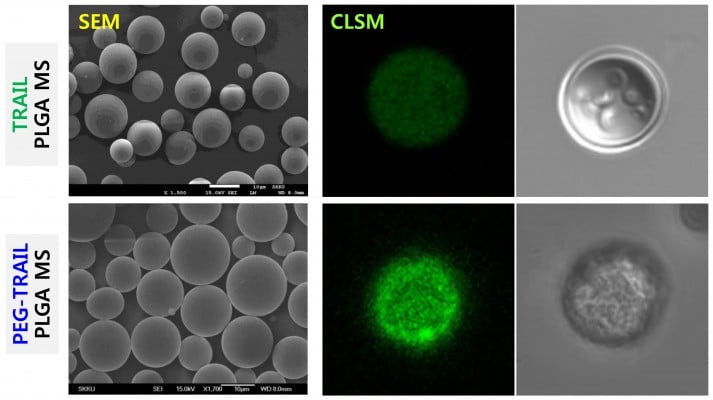
TRAIL or PEG-TRAIL PLGA MSs were prepared by a double emulsification/solvent evaporation technique, and their external surface morphologies were observed by SEM and CLSM. Both TRAIL and PEG-TRAIL PLGA MSs appeared to have a spherical shape with smooth surface morphology (Figure 1). The average diameter of lyophilized PEG-TRAIL PLGA MSs was found to be slightly greater than that of TRAIL PLGA MSs (14.4±1.8 μm vs 12.1±0.9 μm), probably due to the increased hydrodynamic volume of PEG-TRAIL. All external surface characteristics of PEG-TRAIL PLGA MSs had negligible differences from those of TRAIL PLGA MSs. However, the encapsulation efficiency of TRAIL PLGA MSs (43.3%±10.9%) was much lower than that of PEG-TRAIL PLGA MSs (85.7%±4.1%), which was demonstrated by a stronger green signal of fluorescein-tagged PEG-TRAIL PLGA MSs vs TRAIL PLGA MSs.
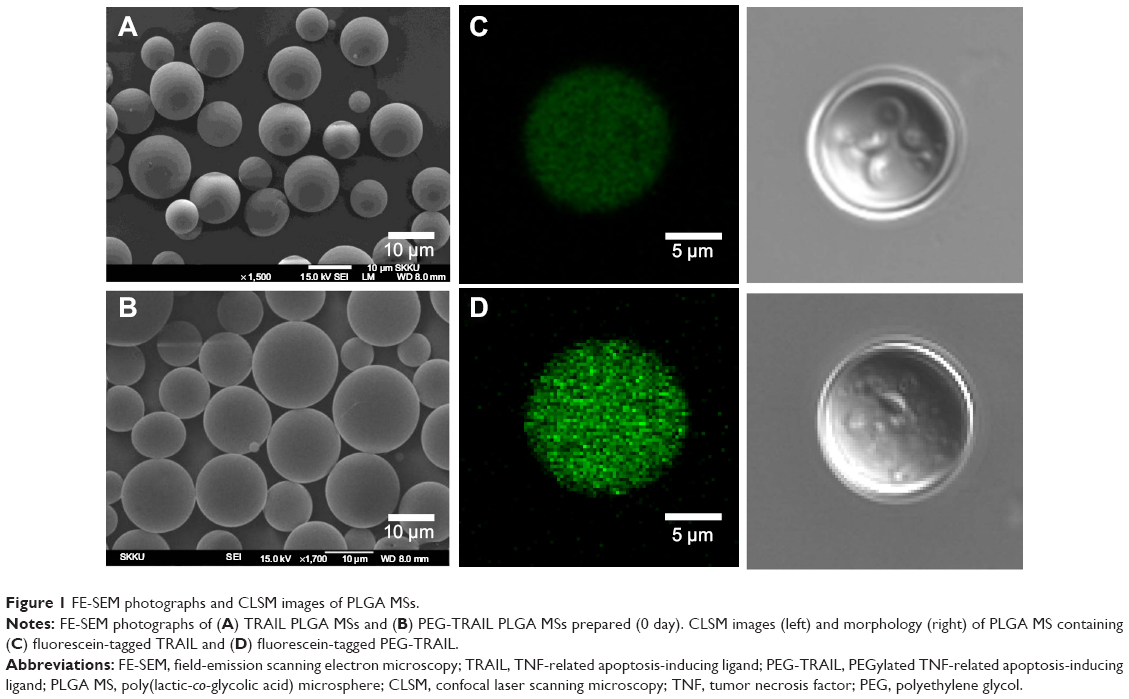 | Figure 1 FE-SEM photographs and CLSM images of PLGA MSs. |
Release profiles of PEG-TRAIL from PLGA MSs
Figure 2A shows the release profiles of TRAIL and PEG-TRAIL from PLGA MSs. The in vitro release results showed that PEG-TRAIL had continuous and gradual release from PLGA MSs vs native TRAIL. Approximately 72.3% of PEG-TRAIL was sustainably released from PLGA MSs for 7 days. On the contrary, ~50% of TRAIL was released from PLGA MSs within the first day, and no further significant release of TRAIL was observed in the same period. In addition, this gradual PEG-TRAIL release and erosion (size decrease) of PLGA MSs were demonstrated by CLSM and SEM, respectively (Figure 2A [inset] and B).
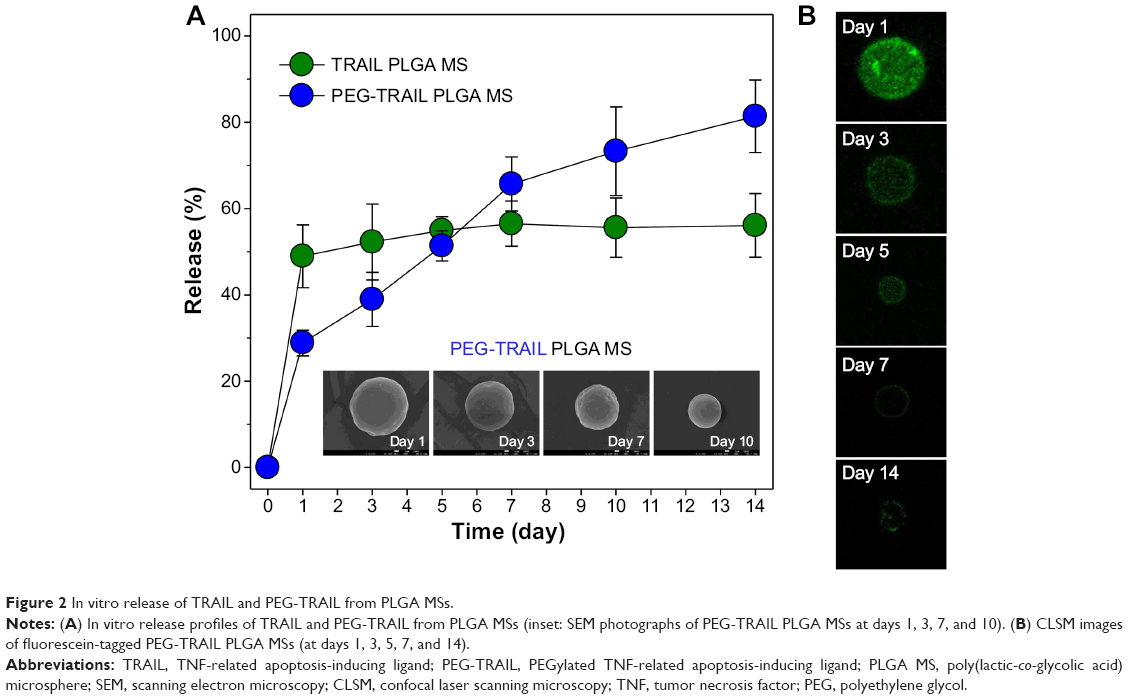 | Figure 2 In vitro release of TRAIL and PEG-TRAIL from PLGA MSs. |
CLSM visualization and viability of Mia Paca-2 cells treated with PEG-TRAIL PLGA MSs
Apoptosis induced by PEG-TRAIL released from PLGA MSs (10 mM PBS, pH 7.4) was determined using a TUNEL assay. As shown in Figure 3, Mia Paca-2 cells treated with PEG-TRAIL released on days 1, 3, and 7 showed significant green coloration, whereas Mia Paca-2 cells treated with TRAIL showed green coloration only on day 1 (Figure 3A). Viability study was performed to estimate the apoptotic activity and stability of the released protein drugs. The supernatant released from TRAIL PLGA MSs on days 3 and 7 exhibited little cytotoxicity to Mia Paca-2 cells, whereas the supernatant released from PEG-TRAIL PLGA MSs even at day 7 was found to be significantly cytotoxic (~70%) to the cells (Figure 3B).
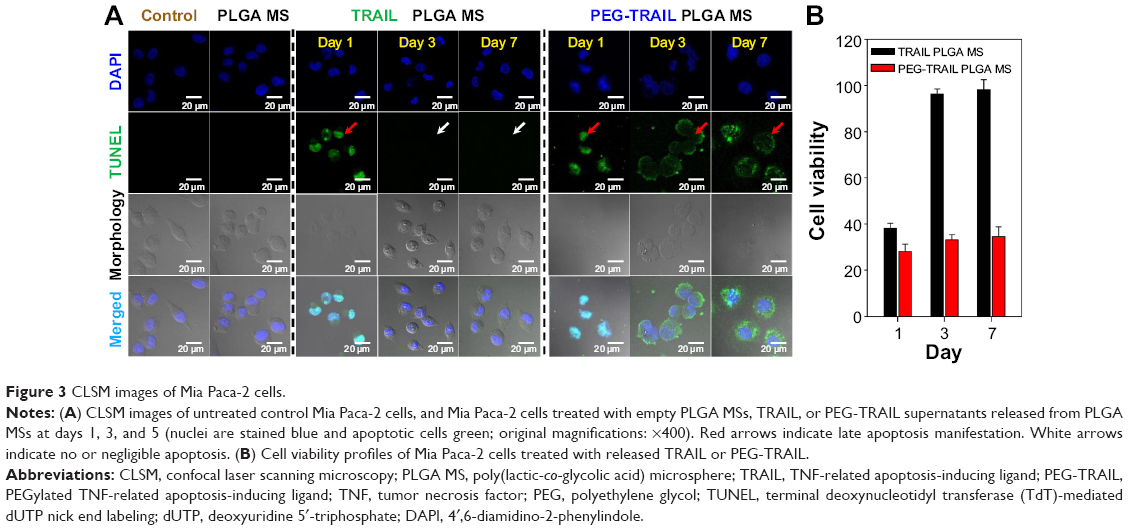 | Figure 3 CLSM images of Mia Paca-2 cells. |
In vivo antitumor effect of PEG-TRAIL PLGA MSs
Antitumor effects of TRAIL or PEG-TRAIL PLGA MSs were evaluated in Mia Paca-2 cell-xenografted mice (Figure 4A). At 4 weeks after inoculation, when the tumor volumes reached ~300 mm3, mice were administered TRAIL or PEG-TRAIL PLGA MSs (Figure 4B; in excised tumors) intratumorally weekly for 2 weeks (three administrations), and tumor volumes were calculated every 3 days. Mice were sacrificed at day 27 and tumor weights were measured (Figure 4C). As shown in Figure 5A, PEG-TRAIL PLGA MSs greatly suppressed tumor growth. Especially, tumor volumes of the mice treated with PEG-TRAIL PLGA MSs were much lower than those of mice treated with TRAIL PLGA MSs or blank PLGA MSs (706.5±204.3 mm3 vs 1481.5±390.4 mm3 and 1920.0±282.6 mm3, respectively) at 27 days (Figure 4A). In addition, the mean weight of tumors from mice treated with PEG-TRAIL PLGA MSs was 0.83±0.14 g at 27 days after the first administration, whereas those of tumors from mice treated with TRAIL or blank PLGA MSs were 1.52±0.24 g and 2.11±0.14 g, respectively (Figure 4C). As shown in Figure 6, hematoxylin and eosin–stained tumor tissue specimens from mice treated with PEG-TRAIL PLGA MSs contained significantly lower densities of tumor cells than specimens from mice treated with blank or TRAIL PLGA MSs.
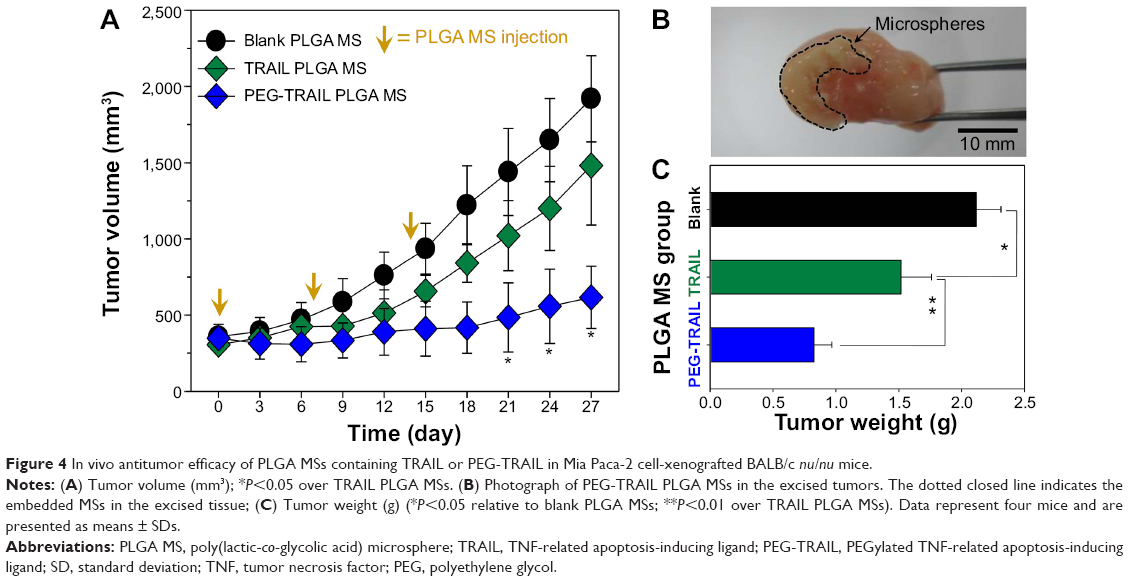 | Figure 4 In vivo antitumor efficacy of PLGA MSs containing TRAIL or PEG-TRAIL in Mia Paca-2 cell-xenografted BALB/c nu/nu mice. |
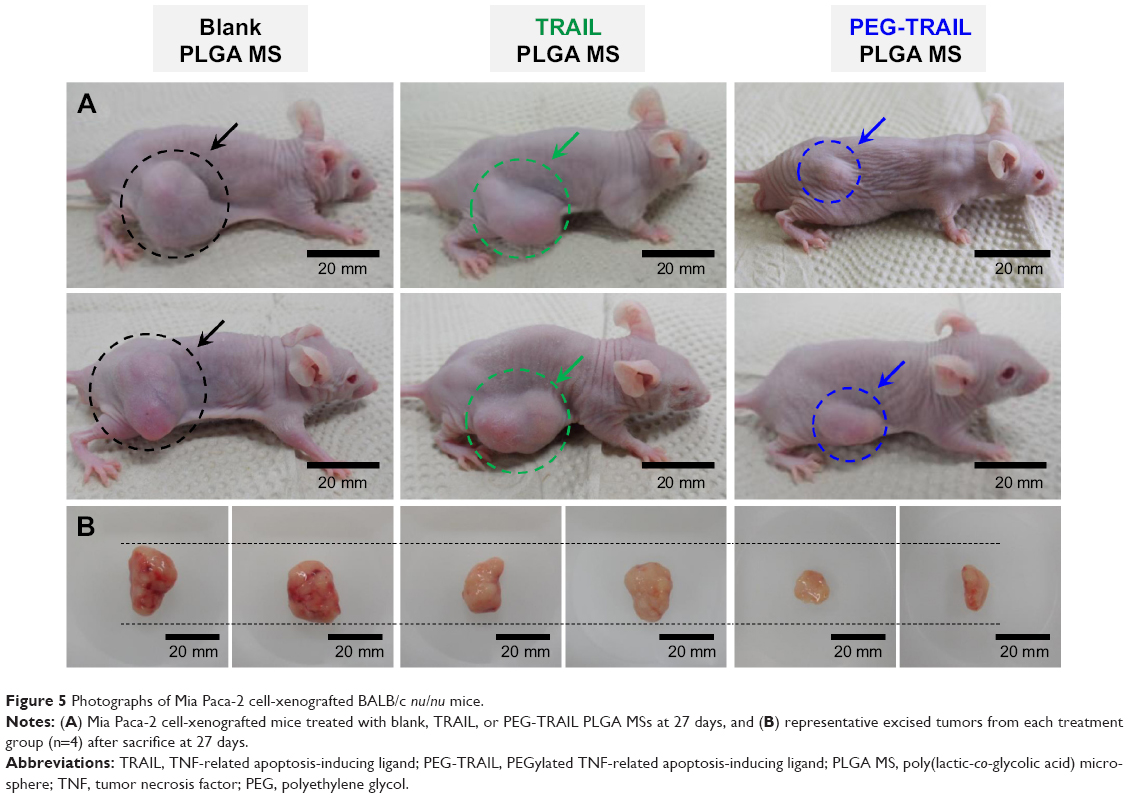 | Figure 5 Photographs of Mia Paca-2 cell-xenografted BALB/c nu/nu mice. |
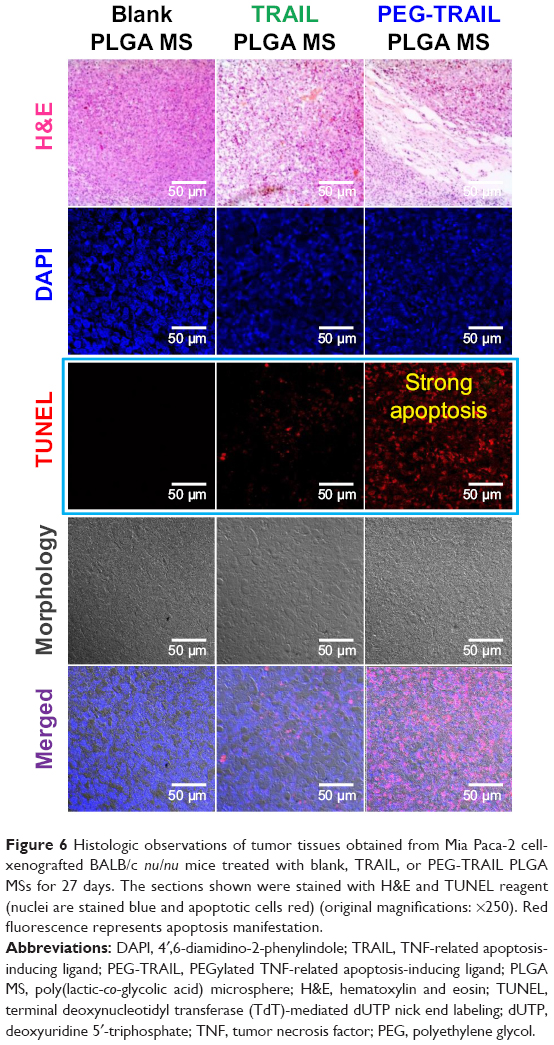 | Figure 6 Histologic observations of tumor tissues obtained from Mia Paca-2 cell-xenografted BALB/c nu/nu mice treated with blank, TRAIL, or PEG-TRAIL PLGA MSs for 27 days. The sections shown were stained with H&E and TUNEL reagent (nuclei are stained blue and apoptotic cells red) (original magnifications: ×250). Red fluorescence represents apoptosis manifestation. |
In vivo apoptosis induction by PEG-TRAIL PLGA MSs in Mia Paca-2 cell tumors
The results from TUNEL assays for tumor tissues showed that PEG-TRAIL PLGA MSs induced substantial apoptosis, and that TRAIL PLGA MSs induced a slightly lower level of apoptosis, as indicated by weaker red light (Figure 6).
Discussion
This study was performed to develop an effective antitumor PLGA MS containing PEGylated apoptotic TRAIL protein against pancreatic cancer. TRAIL protein has a complex trimeric structure and binds to the tripartite TRAIL receptors.11,12 The trimeric structure can be easily damaged by certain instability factors, and thus the protein tends to dissociate into a monomer structure, which has negligible apoptotic activity.23,26 Given this background, we sought to find an obvious pharmaceutical benefit by PEGylation of TRAIL for a sustained-release PLGA MS system in terms of enhanced efficacy in a pancreatic tumor model of mice.
Interestingly, PEGylated proteins have displayed much better release properties in PLGA MSs than non-PEGylated proteins in terms of controlled- or sustained-release profiles. A previous study has reported that PEGylated lysozyme was released gradually from PLGA MSs (eg, RG502) over 2 months, whereas >50% of native lysozyme was released within only 1 day.7 PEGylated interferons (Mw: 2 or 5 kDa) displayed greatly improved release pattern, as compared with native interferon that showed significant incomplete release (only 15% release over 3 weeks).8 Furthermore, PEGylated insulin-loaded PLGA MS displayed an initial release of <1% at day 1 and a continuous and complete release (>90%) over the next 2 weeks after several days’ lag period.9 In agreement with previous results, the PEGylation effect of TRAIL in terms of release from the PLGA MSs seemed clearly advantageous. PEG-TRAIL was released gradually over 2 weeks without a significant initial burst, whereas 50% of loading amount was released within the first day for native TRAIL with negligible additional release for the remaining 2 weeks. This improved release profile was dependent on the PEG attachment to TRAIL, which played a critical role in retarding the initial burst and decreasing random adsorption and aggregation of TRAIL in the PLGA matrix.
Protein instability has been regarded as one of the most significant causes for undermining 1) encapsulation yield, 2) continuous release, and 3) therapeutic effect in PLGA MSs.1,5,6 Many reports have shown that proteins can be unstable at the interface of organic solvents such as methylene chloride or ethylacetate, leading to protein unfolding and physical aggregation. Such destabilization processes eventually result in significantly reduced therapeutic effects of proteins in PLGA MSs. The TRAIL utilized in the present study is reported to be less soluble and unstable under a neutral condition because of the histidines in its N-terminus.26 Therefore, TRAIL could be unstable during fabrication and incubation due to the limited solubility and structural instability. Moreover, TRAIL is only biologically active when maintained in the trimeric form, which seriously decreases the pharmaceutical value of TRAIL-based delivery systems. However, the PLGA MSs containing PEG-TRAIL retained significant apoptotic activity, along with improved release property. We have previously demonstrated that PEG-TRAIL has significantly greater physicochemical stability than native TRAIL,27 and such increased stability by PEGylation might be responsible for well-retained in vitro bioactivity of PEG-TRAIL inside PLGA MSs. Actually, all of the released medium obtained from PEG-TRAIL PLGA MSs until 7 days was found to have significant apoptotic and cytotoxic activity (~70%) against pancreatic tumor cells. On the contrary, TRAIL PLGA MSs showed significant activity only at day 1. Mia Paca-2 cells treated with TRAIL released at days 3 and 7 from PLGA MSs exhibited no color in the TUNEL assay, indicating little or no apoptosis induction. This fact obviously demonstrates that PEG-TRAIL should be considered more valuable than native TRAIL, as a pharmaceutical partner for PLGA MSs.
The advantage of PEG-TRAIL PLGA MSs over native TRAIL PLGA MSs was also demonstrated in a Mia Paca-2 xenografted mice model. Particularly, PEG-TRAIL PLGA MSs markedly suppressed Mia Paca-2 cell-induced pancreatic tumors; tumor volumes after treatment for 27 days with PEG-TRAIL PLGA MSs were only ~twofold of those at the initial day, whereas the volumes of the tumors treated with TRAIL PLGA MSs or blank PLGA MSs were 4.2- and 5.5-fold of those at the beginning of treatments, respectively (Figure 6). In addition, this improved in vivo antitumor activity was clearly responsible for the well-preserved apoptotic activity of PEG-TRAIL.
Conclusion
Here, we described PEG-TRAIL-loaded PLGA MSs with remarkable antitumor activity against pancreatic cancer–bearing mice. The fabricated PLGA MSs provided a gradual and continuous PEG-TRAIL release for 14 days in vitro and reduced tumor sizes and weights owing to their well-retained apoptotic effects on Mia Paca-2 cells. This improvement seems to be due to the combination of PEGylation and MSs for the delivery and stabilization of TRAIL, as a dual delivery system (Figure 7). Furthermore, our PLGA MS system can be efficiently delivered to tumors in the pancreas by using a recently developed method of endoscopic ultrasonography-guided fine needle injection.28,29 In the context of local targeting efficiency to tumors, this would be more feasible and available for developing as an anticancer drug product because the size of the pancreas is much smaller than that of other organs, such as colon, lungs, and stomach. In this respect, we believe that the PLGA depot system of PEG-TRAIL would be a promising once-weekly antitumor treatment for pancreatic cancer.
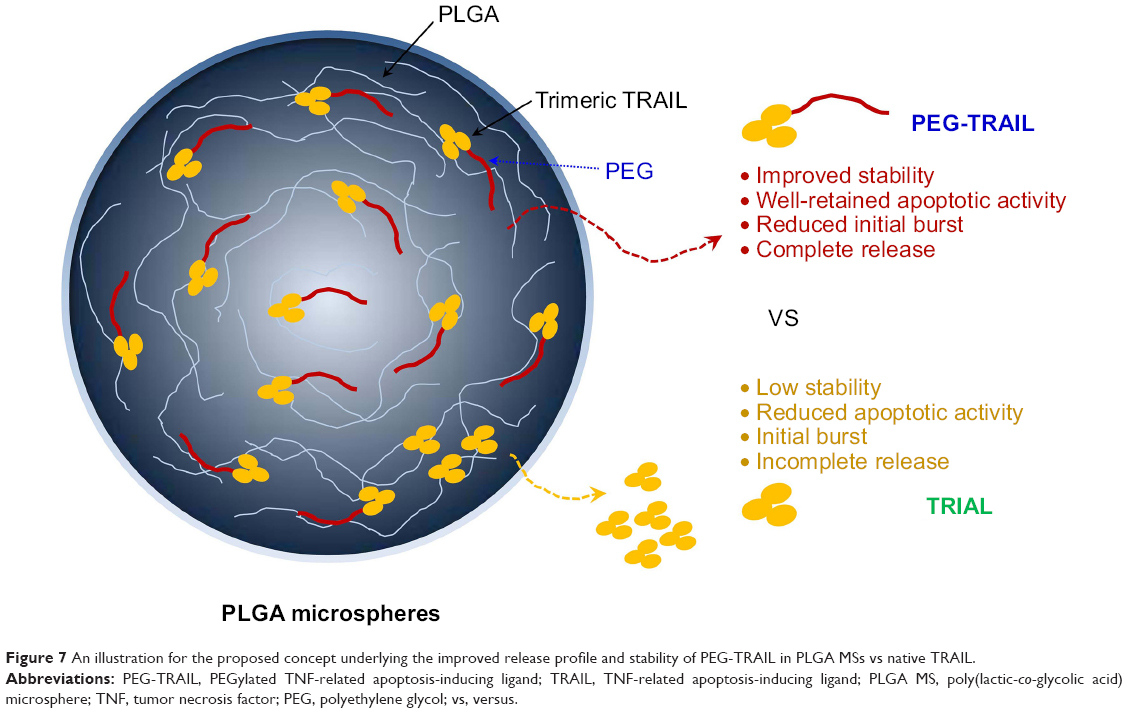 | Figure 7 An illustration for the proposed concept underlying the improved release profile and stability of PEG-TRAIL in PLGA MSs vs native TRAIL. |
Acknowledgment
This study was supported by the Korean Health Technology R&D Project, Korean Ministry of Health and Welfare (grant number HI12C0829).
Disclosure
The authors report no conflicts of interest in this work.
References
Taluja A, Youn YS, Bae YH. Novel approaches in microparticulate PLGA delivery systems encapsulating proteins. J Mater Chem. 2007;17:4002–4014. | ||
Gupta RK, Singh M, O’Hagan DT. Poly(lactide-co-glycolide) microparticles for the development of single-dose controlled-release vaccines. Adv Drug Deliv Rev. 1998;32(3):225–246. | ||
Van de Weert M, Hennink WE, Jiskoot W. Protein instability in poly(lactic-co-glycolic acid) microparticles. Pharm Res. 2000;17(10):1159–1167. | ||
Wischke C, Schwendeman SP. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int J Pharm. 2008;364(2):298–327. | ||
Pai SS, Tilton RD, Przybycien TM. Poly(ethylene glycol)-modified proteins: implications for poly(lactide-co-glycolide)-based microsphere delivery. AAPS J. 2009;11(1):88–98. | ||
Fu K, Klibanov AM, Langer R. Protein stability in controlled-release systems. Nat Biotechnol. 2000;18(1):24–25. | ||
Diwan M, Park TG. Pegylation enhances protein stability during encapsulation in PLGA microspheres. J Control Release. 2001;73(2–3):233–244. | ||
Diwan M, Park TG. Stabilization of recombinant interferon-alpha by pegylation for encapsulation in PLGA microspheres. Int J Pharm. 2003;252(1–2):111–122. | ||
Hinds KD, Campbell KM, Holland KM, Lewis DH, Piché CA, Schmidt PG. PEGylated insulin in PLGA microparticles. In vivo and in vitro analysis. J Control Release. 2005;104(3):447–460. | ||
Griffith TS, Lynch DH. TRAIL: a molecule with multiple receptors and control mechanisms. Curr Opin Immunol. 1998;10(5):559–563. | ||
LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003;10(1):66–75. | ||
French LE, Tschopp J. The TRAIL to selective tumor death. Nat Med. 1999;5(2):146–147. | ||
Ashkenazi A, Pai RC, Fong S, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104(2):155–162. | ||
Thomas RP, Farrow BJ, Kim S, May MJ, Hellmich MR, Evers BM. Selective targeting of the nuclear factor-kappaB pathway enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated pancreatic cancer cell death. Surgery. 2002;132(2):127–134. | ||
Ozawa F, Friess H, Kleeff J, et al. Effects and expression of TRAIL and its apoptosis-promoting receptors in human pancreatic cancer. Cancer Lett. 2001;163(1):71–81. | ||
Saito R, Bringas JR, Panner A, et al. Convection-enhanced delivery of tumor necrosis factor-related apoptosis-inducing ligand with systemic administration of temozolomide prolongs survival in an intracranial glioblastoma xenograft model. Cancer Res. 2004;64(19):6858–6862. | ||
Roth W, Isenmann S, Naumann U, et al. Locoregional Apo2L/TRAIL eradicates intracranial human malignant glioma xenografts in athymic mice in the absence of neurotoxicity. Biochem Biophys Res Commun. 1999;265(2):479–483. | ||
Kim TH, Youn YS, Jiang HH, Lee S, Chen X, Lee KC. PEGylated TNF-related apoptosis-inducing ligand (TRAIL) analogues: pharmacokinetics and antitumor effects. Bioconjug Chem. 2011;22(8):1631–1637. | ||
Chae SY, Kim TH, Park K, et al. Improved antitumor activity and tumor targeting of NH(2)-terminal-specific PEGylated tumor necrosis factor-related apoptosis-inducing ligand. Mol Cancer Ther. 2010;9(6):1719–1729. | ||
Youn YS, Na DH, Yoo SD, Song SC, Lee KC. Carbohydrate-specifically polyethylene glycol-modified ricin A-chain with improved therapeutic potential. Int J Biochem Cell Biol. 2005;37(7):1525–1533. | ||
Harris JM, Martin NE, Modi M. Pegylation: a novel process for modifying pharmacokinetics. Clin Pharmacokinet. 2001;40(7):539–551. | ||
Jiang HH, Kim TH, Lee S, Chen X, Youn YS, Lee KC. PEGylated TNF-related apoptosis-inducing ligand (TRAIL) for effective tumor combination therapy. Biomaterials. 2011;32(33):8529–8537. | ||
Kim TH, Jiang HH, Park CW, et al. PEGylated TNF-related apoptosis-inducing ligand (TRAIL)-loaded sustained release PLGA microspheres for enhanced stability and antitumor activity. J Control Release. 2011;150(1):63–69. | ||
Lim SM, Kim TH, Jiang HH, et al. Improved biological half-life and anti-tumor activity of TNF-related apoptosis-inducing ligand (TRAIL) using PEG-exposed nanoparticles. Biomaterials. 2011;32(13):3538–3546. | ||
Bae S, Ma K, Kim TH, et al. Doxorubicin-loaded human serum albumin nanoparticles surface-modified with TNF-related apoptosis-inducing ligand and transferrin for targeting multiple tumor types. Biomaterials. 2012;33(5):1536–1546. | ||
Youn YS, Shin MJ, Chae SY, Jin CH, Kim TH, Lee KC. Biological and physicochemical evaluation of the conformational stability of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Biotechnol Lett. 2007;29(5):713–721. | ||
Byeon HJ, Choi SH, Choi JS, et al. Four-arm PEG cross-linked hyaluronic acid hydrogels containing PEGylated apoptotic TRAIL protein for treating pancreatic cancer. Acta Biomater. 2014;10:142–150. | ||
Yan BM, Van Dam J. Endoscopic ultrasound-guided intratumoural therapy for pancreatic cancer. Can J Gastroenterol. 2008;22(4):405–410. | ||
Verna EC, Dhar V. Endoscopic ultrasound-guided fine needle injection for cancer therapy: the evolving role of therapeutic endoscopic ultrasound. Therap Adv Gastroenterol. 2008;1(2):103–109. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.