Back to Journals » International Medical Case Reports Journal » Volume 8
Pediatric primary optic nerve sheath meningioma
Authors Vanikieti K, Preechawat P, Poonyathalang A ![]()
Received 13 February 2015
Accepted for publication 24 March 2015
Published 4 August 2015 Volume 2015:8 Pages 159—163
DOI https://doi.org/10.2147/IMCRJ.S82795
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Scott Fraser
Kavin Vanikieti, Pisit Preechawat, Anuchit Poonyathalang
Department of Ophthalmology, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Abstract: Primary optic nerve sheath meningioma (PONSM) is extremely rare among children. We report two cases of pediatric PONSM. The first case was a 12-year-old boy who presented with gradual visual loss of his right eye and was found to be associated with neurofibromatosis type 2. The second case was a 10-year-old boy who presented with gradual proptosis of his left eye with normal visual acuity. Severe visual loss is a common clinical manifestation of pediatric PONSM. Although the visual acuity in the second case was normal, his vision rapidly deteriorated to 20/200. In both cases, the diagnosis of PONSM was confirmed by magnetic resonance imaging, and a successful tumor growth control was achieved after stereotactic radiotherapy was implemented.
Keywords: meningioma, optic nerve, children, pediatric, sheath, orbit
Introduction
Primary optic nerve sheath meningioma (PONSM) is a proliferation of meningothelial cells within the nerve sheath of the orbital or intracanalicular portion of the optic nerve. The classic diagnostic triad includes slowly progressive, painless monocular visual loss, optic atrophy, and optociliary shunt vessels. Although PONSM accounts for one-third of primary optic nerve tumors, it is not common in patients younger than 20 years of age, and represents less than 5% of all cases of PONSM.1 The authors report the clinical presentation, treatment, and visual outcome in two cases of pediatric PONSM.
Case reports
Case 1
A 12-year-old boy presented with painless, gradual loss of his right vision over 2 months. His past history and family history were unremarkable. Ocular examination revealed visual acuity of counting fingers in his right eye and 20/20 in his left eye. The right eye showed 2 mm of proptosis and marked limitation of extraocular movement in all directions (Figure 1). Anterior segment examination was unremarkable in both eyes. Neither Lisch nodules nor cataract was detected. There was a right relative afferent pupillary defect. Fundus examination showed generalized, marked optic disc edema with optociliary shunt vessels in the right eye (Figure 2A). Systemic evaluation revealed multiple plexiform neurofibromas in both hands (Figure 3) and one café-au-lait spot on his trunk. Magnetic resonance imaging (MRI) of orbit and brain revealed diffuse enlargement and contrast enhancement along the length of the right intraorbital optic nerve sheath consistent with an optic nerve sheath meningioma (Figure 4). There were an en plaque meningioma along left anterior temporal convexity, a falcine meningioma, and multiple schwannomas of bilateral cranial nerves V, VI, VII, VIII, IX, and XI, and spinal nerve roots (Figure 5). The child was diagnosed with definite neurofibromatosis type 2 (NF-2). He received fractionated stereotactic radiotherapy; a total dose of 50 Gy was given in 1.8 Gy daily fractions. The child’s vision remained stable over 6 months of follow-up, and no significant, short-term side effects of radiation were observed.
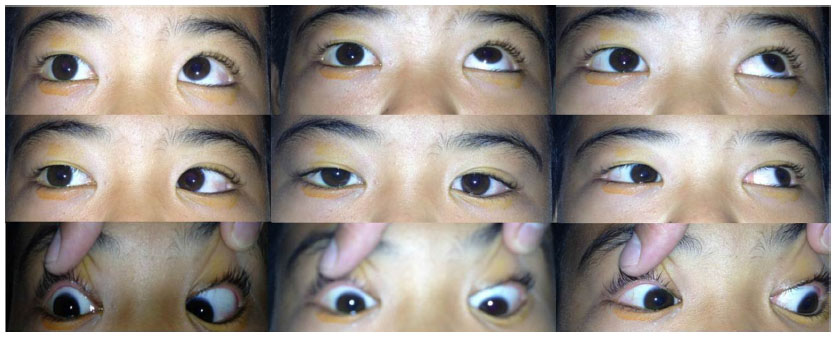 | Figure 1 Marked limitation of extraocular movement in all directions of the right eye. |
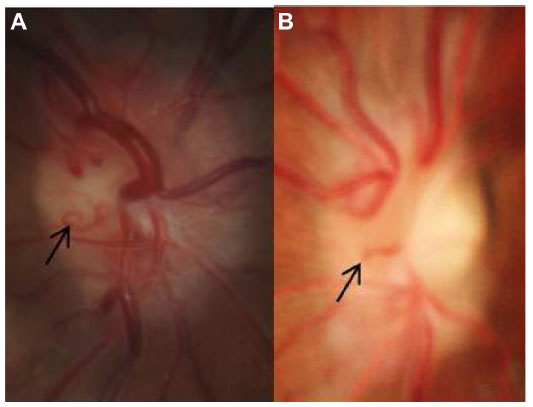 | Figure 2 Fundus photograph showing generalized optic disc edema with optociliary shunt vessels (black arrows) in case 1 (A) and case 2 (B). |
 | Figure 3 Multiple plexiform neurofibromas in both hands of case 1. |
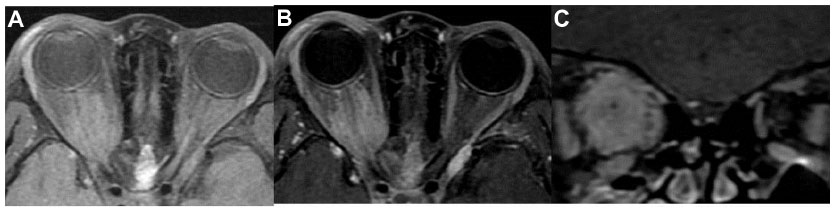 | Figure 4 MRI of orbit (case 1). (A) Axial T1-weighted, (B) axial T1-weighted with gadolinium injection, and (C) coronal T1-weighted with gadolinium injection showing diffuse enlargement and contrast enhancement of the right intraorbital optic nerve sheath. |
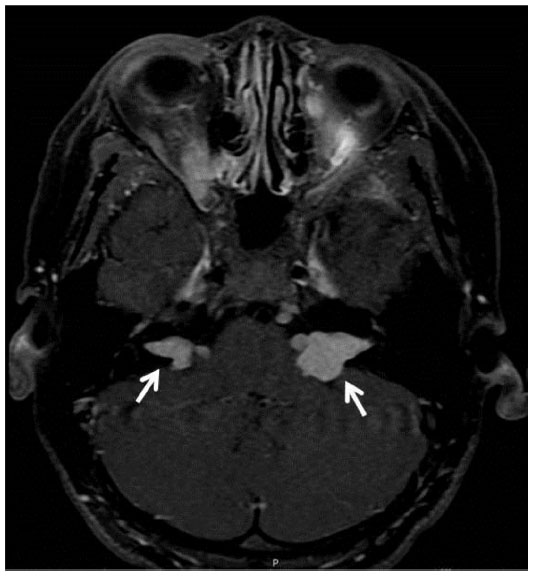 | Figure 5 Bilateral vestibular schwannomas (white arrows) at bilateral cerebellopontine angle extending to bilateral expanded internal acoustic canal (case 1). |
Case 2
A 10-year-old boy presented with gradually progressive and painless proptosis in his left eye for 2 years. His past history and family history were unremarkable. Ocular examination revealed visual acuity of 20/20 in his right eye and 20/25 in his left eye. The left eye showed 4 mm of proptosis and marked limitation of extraocular movement in all directions. Anterior segment examination was unremarkable in both eyes. Neither Lisch nodules nor cataract was detected. There was a left relative afferent pupillary defect. Fundus examination showed generalized, marked optic disc edema with optociliary shunt vessels in the left eye (Figure 2B). General physical examination was unremarkable. MRI of orbit and brain revealed a large, enhancing intraconal mass at the left orbit along the optic nerve sheath extending from the posterior globe to the orbital apex and surrounding the non-enhancing optic nerve (Figure 6). There were no other brain abnormalities. Incisional biopsy via transconjunctival approach was performed in order to confirm the diagnosis. The pathological result was consistent with optic nerve sheath meningioma. Conservative treatment was recommended at that time. Six months later, his left vision rapidly deteriorated to 20/200. The child subsequently received stereotactic radiotherapy; a total dose of 50 Gy was given in 1.8 Gy daily fractions. Over 15 years of follow-up, his left vision had gradually decreased to 5/200, and the right vision remained stable. Left fundus examination showed generalized optic atrophy, gliosis, and optociliary shunt vessels. There was no evidence of radiographic progression of the tumor.
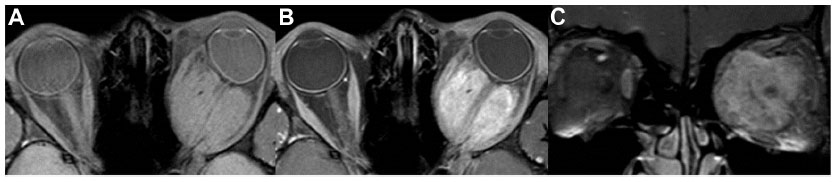 | Figure 6 MRI of orbit (case 2). |
Discussion
Unlike adult patients, pediatric PONSM is very rare and has some distinct clinical features (Table 1) including more frequent association with neurofibromatosis, worse presenting visual acuity, and higher incidence of intracranial extension. Visual loss is the most common (80%) initial presenting symptom in both groups as well as lacking tumor-related mortality.1,2 The sex distribution of PONSM appears to be similar in adults and children with a female predominance. In contrary, both cases in our report were male. Most cases of pediatric PONSM in previous reports presented with visual acuity of 20/200 or worse.1,3–5 Our first case similarly presented with visual acuity of counting fingers. Although the second case had good acuity at the initial presentation, his vision rapidly deteriorated to 20/200 within 6 months. This finding supports the aggressive behavior of pediatric PONSM and high risk of progressive visual decline.1,6 Observation without treatment should be followed with caution. Proptosis is the second most common (68%) presenting symptom.1 It usually follows the visual loss but can be the initial manifestation alone as shown in our second case. The optic disc findings may be either edema or atrophic depending on the duration of optic nerve compression. The presence of optociliary shunt vessels previously was found in 28% of patients.1 The optic disc findings in our two cases initially showed edema and subsequently became atrophic with persistent shunt vessels. The association of meningioma with NF-2 has been well established. The incidence of NF-2 in pediatric PONSM is greater than adult PONSM.1 In one large study, there were 35% of pediatric PONSM patients having concomitant diagnosis of NF-2.1 One of our cases (50%) was associated with NF-2, supported by the presence of bilateral vestibular schwannomas.3 PONSM in childhood should be considered as one of the initial manifestations of NF-2. MRI is sufficient to confirm the diagnosis of PONSM, thus obviating the need for tissue biopsy.6 However, according to the rarity of this tumor in children and its aggressive nature, we decided to perform the incisional biopsy in our second case. Although stereotactic radiotherapy is the treatment of choice for adult PONSM,6 the application of this treatment in pediatric cases is controversial. The radiation risk particularly to the cortical and hypothalamic–pituitary system, especially in the developing child, is a major concern. There have been only a few cases reported with short-term follow-up.7–9 Pitz et al reported a case of a 13-year-old boy who had pre-radiation visual acuity of counting fingers and vision subsequently improved to 20/200 with no progression of tumor at 37-month follow-up.7 Arvold et al reported a case of an 11-year-old girl who underwent stereotactic radiotherapy and had stable visual outcome and tumor control at 14-month follow-up.8 The youngest patient who received stereotactic radiotherapy was 9 years old, but the radiation side effects were difficult to determine due to only 1-month follow-up.9 Interestingly, we present a long-term result, 15-year follow-up, after radiation in the second case. Radiotherapy effectively stabilized the child’s vision and tumor progression. No significant side effects of radiation including local dermatologic changes, radiation retinopathy, or secondary intracranial malignancy were observed. Other optional treatments include close observation throughout the course of the disease and surgical resection of the tumor from the sheath, though the latter may injure pial vessels, which may compromise the blood flow of the intraorbital optic nerve.1 Although young patients tend to develop intracranial extension of the tumor, we did not find the extension of the tumor in our cases at any point of the disease.
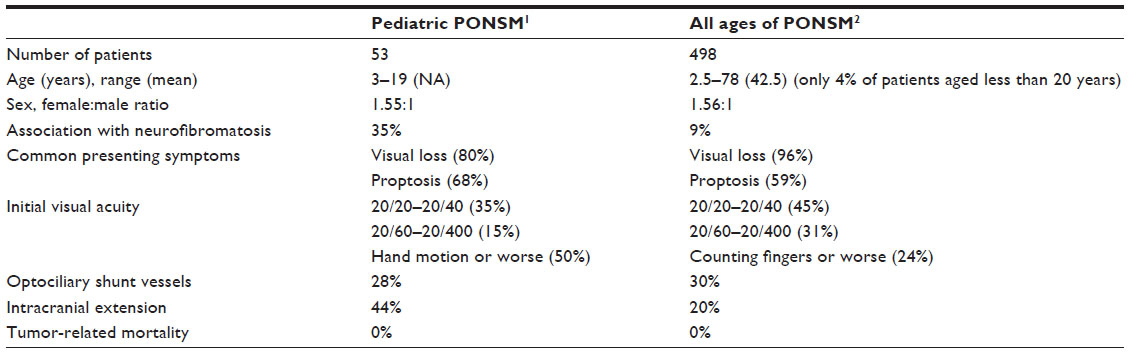 | Table 1 Comparison of demographic and clinical features between pediatric PONSM and all ages of PONSM |
Conclusion
PONSM should be included in the differential diagnosis of primary optic nerve tumor in children in spite of its rarity. Once the diagnosis of pediatric PONSM is made, the search for evidence of NF-2 is crucial. Pediatric PONSM seems to have aggressive clinical behavior with rapid decline and poor visual outcome. Stereotactic radiotherapy in selected cases is shown to be effective in long-term tumor growth control and has no serious complications.
Disclosure
The authors report no conflicts of interest in this work.
References
Harold Lee HB, Garrity JA, Cameron JD, Strianese D, Bonavolonta G, Patrinely JR. Primary optic nerve sheath meningioma in children. Surv Ophthalmol. 2008;53:543–558. | |
Dutton JJ. Optic nerve sheath meningiomas. Surv Ophthalmol. 1992;37:167–183. | |
Dailey RA. Optic nerve sheath meningiomas of childhood. Ophthalmol Clin North Am. 1991;4:519–529. | |
Kuroda R, Nakatani J, Yorimae A, Nakao Y, Ohtori T. Clinical experience of intraorbital optic nerve sheath meningioma – report of eight cases. Neurol Med Chir (Tokyo). 1990;30:468–475. | |
Karp LA, Zimmerman LE, Borit A, Spencer W. Primary intraorbital meningiomas. Arch Ophthalmol. 1974;91:24–28. | |
Eddleman CS, Liu JK. Optic nerve sheath meningioma: current diagnosis and treatment. Neurosurg Focus. 2007;23:E4. | |
Pitz S, Becker G, Schiefer U, et al. Stereotactic fractionated irradiation of optic nerve sheath meningioma: a new treatment alternative. Br J Ophthalmol. 2002;86:1265–1268. | |
Arvold ND, Lessell S, Bussiere M, et al. Visual outcome and tumor control after conformal radiotherapy for patients with optic nerve sheath meningioma. Int J Radiat Oncol Biol Phys. 2009;75:1166–1172. | |
Baumert BG, Villà S, Studer G, Mirimanoff RO, Davis JB, Landau K. Early improvements in vision after fractionated stereotactic radiotherapy for primary optic nerve sheath meningioma. Radiother Oncol. 2004;72:169–174. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.