Back to Journals » International Journal of Nanomedicine » Volume 17
PD-1 Targeted Nanoparticles Inhibit Activated T Cells and Alleviate Autoimmunity via Suppression of Cellular Energy Metabolism Mediated by PKM2
Authors Liu Z, Xu J, Li H, Shu J, Su G, Zhou C, Yang P ![]()
Received 18 November 2021
Accepted for publication 31 March 2022
Published 13 April 2022 Volume 2022:17 Pages 1711—1724
DOI https://doi.org/10.2147/IJN.S349360
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Zhangluxi Liu, Jing Xu, Hongxi Li, Jia Shu, Guannan Su, Chunjiang Zhou, Peizeng Yang
The First Affiliated Hospital of Chongqing Medical University, Chongqing Key Laboratory of Ophthalmology, Chongqing Eye Institute, Chongqing Branch of National Clinical Research Center for Ocular Diseases, Chongqing, People’s Republic of China
Correspondence: Peizeng Yang, The First Affiliated Hospital of Chongqing Medical University, Chongqing Key Laboratory of Ophthalmology, Chongqing Eye Institute, Chongqing, People’s Republic of China, Tel/Fax +86-23-89012851, Email [email protected]
Background: Effector T cells, especially T helper 1 (Th1) cells and T helper 17 (Th17) cells, are involved in the pathogenesis of many autoimmune diseases such as uveitis. Under hyperactive immune conditions, these effector T cells pathologically maintain a high expression level of programmed cell death protein 1 (PD-1) receptors and distinctively engage aerobic glycolysis via cellular energy metabolism mediated by pyruvate kinase M2 (PKM2). Therefore, we proposed that the synergy of metabolic inhibition and receptor guidance might target and down-regulate these hyperactive effector T cells to achieve anti-immune effects.
Methods: PD-1 antibody and TEPP-46 were integrated by polyethylene glycol (PEG) modified poly (lactic-co-glycolic acid) (PLGA) as a nanoplatform (TPP). Characteristics of TPP were basically detected. The biosafety of TPP was evaluated in vitro and in vivo. The targeting effect of TPP was detected by laser scanning confocal microscopy and flow cytometry (FCM). Interleukin-2 (IL-2)/interleukin-17A (IL-17A)/interferon-gamma (IFN-γ) producing cells were detected by FCM. Experimental autoimmune uveoretinitis (EAU) was induced in C57BL/6J mice as the inflammatory model.
Results: TPP had homogeneous distribution, good stability in vitro, and high biosafety in vitro and in vivo. Encapsulated TEPP-46 showed a sustained release profile with burst, steady and slow release periods. Early activation and proliferation of effector T cells was inhibited by TPP treatment in vitro. Th1 and Th17 cells were suppressed by TPP in vitro and in vivo. EAU was alleviated in mice by systemic administration of TPP.
Conclusion: The novel nanoplatform TPP could suppress Th1 and Th17 cells and exhibited an anti-inflammatory effect on EAU, providing an alternative approach to ameliorate autoimmune diseases mediated by these cells.
Keywords: anti-inflammation, experimental autoimmune uveitis, sustained release, aerobic glycolysis, biomaterial, Th1 and Th17 cells
Graphical Abstract:

Introduction
The immune system is a defense network of cellular interactions covering the whole body, which recognizes non-self from self to repel attacking pathogens.1 In the system, immune homeostasis builds the foundation of well-organized immune events. When the complex homeostasis is impaired, the host will react to autoantigen and trigger inflammation in tissues.2,3 Thus, autoimmune diseases, such as uveitis can develop.4 Most uveitis entities are non-infectious and autoimmunity or auto-inflammation are assumed to be the main pathological mechanisms.5 In clinic, different types of uveitis show various symptoms and outcomes, among which the most detrimental prognosis in terms of quality of life is visual impairment and visual loss.6 Presently, the therapeutic regimens primarily depend on glucocorticoids, immunosuppressants and biological agents which may cause a number of side effects due to lack of specificity.7,8 Therefore, such clinical condition encourages researchers to figure out a novel treatment approach.
As a critical player in negative pathway for maintaining immune homeostasis, programmed-cell-death-protein-1 (PD-1) receptors are principally observed on activated T cells and B cells or effector T cells and B cells.9 PD-1 receptors can specifically bind to its natural ligands PD-L1/PD-L2 to eliminate the signals of immune stimulation, maintaining T cell anergy and preventing the cells from autoimmune initiation.10,11 Increasing evidence shows that the disturbance of PD-1 pathway plays a crucial role in pathogenesis of autoimmunity and auto-inflammation.12,13 Once the pathway is damaged, activated lymphocytes will escape from immune checkpoint.14 Thus the immune effects and high expression level of PD-1 receptor can be persistently observed in the activated cells.15 Finally, infiltration of inflammatory cells into tissues and autoimmune destruction can develop. Consequently, using PD-1 receptors as a target to suppress certain lymphocyte populations might become an effective therapeutic approach in autoimmunity.
A large number of studies have shown that effector T cells are the main pathogenic factors in non-infectious uveitis, including Th1 and Th17 cells.16,17 Notably, activated effector T cells have a special metabolic profile, referred to aerobic glycolysis, which differs from most lymphocytes supplied with energy by oxidative phosphorylation (OXPHOS) and fatty-acid oxidation (FAO).18 The distinctive metabolic pattern of activated effector T cells is similar to that of high-consumption tumor cells because of their universal high metabolism levels for effector functions or proliferation.18–20 In the metabolic pathway of aerobic glycolysis, PKM2 acts as the decisive rate-limiting enzyme.21–23 It affects intracellular signaling related to glycolysis.24,25 Generally, PKM2 is a monomer/dimer in cells with high anabolic levels.26,27 Its activity is regulated by complex allosterism, offering an opportunity to control the activation and pathogenicity of effector T cells by pharmacological regulation of PKM2 configuration. The specific allosteric activator of PKM2, termed TEPP-46, can tetrammerize PKM2, whereby nuclear translocation of PKM2 and aerobic glycolysis efficiency of effector T cells can be limited.28 Consequently, the development of effector T cells can be inhibited, including Th1 and Th17 cells, which are considered to critically influence the progress of inflammatory infiltration in non-infectious uveitis.
Taken together, the strategy that combines targeting PD-1 receptors and inhibiting glycolysis metabolism might specifically suppress the activity of effector T cells and creatively bring a novel idea to alleviate autoimmune diseases. To realize the synergistic therapy, we proposed to utilize biomaterials to assemble both PD-1 antibody and TEPP-46. Nowadays, biomaterials are able to accomplish the complex functional demands of different biological fields, including tumor treatment, medical imaging, regulation of immune system, etc.29,30 For example, some microRNA and cytokines have been utilized in conjunction with biomaterials for immunoregulation.31,32 Polyethylene glycol (PEG) modified poly (lactic-co-glycolic acid) (PLGA) is a promising biomedical polymer used to assemble multi-functional platforms. It has been approved by the US Food and Drug Administration (FDA) for its good bio-compatibility, biodegradability, and extremely low cytotoxicity.33 Furthermore, PEGylated PLGA (PLGA-PEG) is mostly synthesized as hollow nanofibers or nanoparticles with porous shell, which endows the composition remarkable characteristics of drug delivery and controlled-release.34,35
In this study, a novel nanoparticle was elaborately designed. Firstly, PKM2 activator TEPP-46 was encapsulated by PEGylated PLGA as the suppressive agent to inhibit aerobic glycolysis. Secondly, PD-1 antibody, as the targeted probe, was modified on the surface of nanoparticles by covalent combination with polymer terminal. Accordingly, we successfully formulated the functional nanoparticles and investigated its performance and biosafety. Of note, we demonstrated that the nanoparticles could specifically target PD-1+ cells and reduce inflammatory cells in vitro as well as alleviate inflammation of murine EAU model in vivo. The therapeutic regimen with nanoparticles had an evident anti-inflammatory and anti-immunity function.
Materials and Methods
Materials
PLGA-COOH (50:50, 30kDa) including a premixed content of ~3.4kDa polyethylene glycol (PEG) was purchased from Xi’anRuiXi Co., Ltd (Xi’an, China). Polyvinyl alcohol (PVA, 26 kDa), 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), 2- (N-morpholino) ethanesulfonic acid (MES monohydrate), N- (3-dimethyl-ami-nopropyl)-N’-ethylcarbodiimide hydrochloride (EDC) and N-hydroxy-succinimide (NHS) were purchased from Sigma-Aldrich Co., Ltd (St Louis, MO, USA). Dimethyl sulfoxide (DMSO) was purchased from Solarbio Ltd (Beijing, China). Tepp-46 was obtained from MedChemExpress (New Jersey, USA). PD-1 antibody was purchased from Biolegend (114114, San Diego, USA). Human interphotoreceptor-retinoid-binding protein (IRBP651-670, LAQGAYRTAVDLESLASQLT) peptide was produced by Sangon Biochem Ltd (Shanghai, China). 4’,6-diamidino-2-phenylindole (DAPI) was purchased from Boster Biological Technology Co., Ltd (California, USA). Cell counting kit-8 (CCK-8) was purchased from Dojindo (Kyushu, Japan).
Synthesis of Nanoparticles
The nanoparticles were formulated with a double emulsion solvent evaporation method. First, 50 mg PLGA-PEG was completely dissolved in 2 mL dichloromethane. Then 200 µL TEPP-46 solution (20 mg/mL in DMSO) was added. And 200 µL double distilled water was added. The obtained liquid was emulsified for 2 min (5 s on, 5 s off) by an ultrasonic probe. After 10 mL 4% PVA solution was added in resulting emulsion, the mixed compound was emulsified for another 5 min (5 s on, 5 s off). The final compound was stirred for 4 h by a magnetic stirrer to volatilize organic solvent. Next, the obtained suspension was centrifuged and the sediment was re-suspended by phosphate buffer saline (PBS). The same operation to the sediment was repeated three times. Next, nanoparticles loaded with TEPP-46 were prepared (TP). PLGA-PEG NPs were formulated using the same process except adding TEPP-46.
Then a carbodiimide method was used to connect PD-1 antibody with TP.36,37 Prepared TP were resuspended in MES buffer (0.1 M, pH=6.0) with EDC/NHS for 1 h. As mentioned previously, operations of centrifugation and resuspension to the sediment were repeated three times using MES buffer (0.1 M, pH=8.0) and then PD-1 antibody was added. The chemical reaction between PD-1 antibody and PLGA-PEG-COOH lasted for 1.5 h. Until now, nanoparticles both carrying TEPP-46 and PD-1 antibody (TPP) were well-prepared. Additionally, DiI was added to dichloromethane before emulsification when the nanoparticles were required for fluorescence.
Performance of TPP
The morphology of TPP was detected by scanning electron microscopy (SEM) with energy dispersive X-ray detector (EDX) and transmission electron microscopy (TEM). The pore structure of TPP was analyzed using N2 adsorption/desorption techniques based on Brunauer, Emmett & Teller (BET) theory. The connection of PD-1 antibody was observed by fluorescence microscopy and quantified by flow cytometry (FCM) using a FITC-conjugated goat anti-mouse IgG (GeneCopoeia, L146B, USA). The particle size distribution of TPP and zeta potential of PLGA-PEG NPs, TP and TPP were measured by dynamic light scattering (DLS). The stability of TPP was analyzed by measuring its particle size and zeta potential on Day 1, Day 5 and Day 10 after preparation in PBS at 37°C.
To determine whether TEPP-46 had been successfully loaded in PLGA-PEG, the structure of TPP was damaged by pure DMSO. The absorption spectrum of free TEPP-46 and dissolved TPP were measured respectively using Nanophotometer 80 (NP80). Different concentrations of free TEPP-46 were also detected by NP80 to figure out its standard absorption curve. The percentage of loaded TEPP-46 was then calculated by the following equations: Entrapment efficiency% (EE)= (T-U)/T, Loading capacity % (LC)= (T-U)/TNP (T: total TEPP-46 content; U: unloaded TEPP-46; TNP: total nanoparticles).
TEPP-46 Release
To evaluate the drug release function of TPP, 4 mg TEPP-46 was used to prepare TPP. The prepared TPP was dispersed in 10 mL PBS followed by a 96-hour study of drug release. The samples were harvested at 1 h, 3 h, 6 h, 12 h, 24 h, 48 h, 72 h, and 96 h respectively and measured by NP80. The amount of efficiently loaded and released TEPP-46 was also calculated according to the standard absorption curve.
Preparation for PD-1+ Lymphocytes
The anesthetized C57BL/6J mice were sacrificed by euthanasia and suspension of spleen cells was obtained in a sterile environment. After centrifugation in lymphocyte separation solution (TBDScience, China), the spleen lymphocytes were separated from the middle layer and lymphocytes were removed by red blood cell lysis buffer (Absin, China). Harvested lymphocytes were cultured in RPMI 1640 complete medium containing 6.25 µg/mL concanavalin A (ConA, Sigma, USA) for 72 h to prepare PD-1+ lymphocytes.
Murine EAU Model
Six-week-old female C57BL/6J mice were purchased from Animal Experiment Center of Chongqing Medical University. Each mouse was subcutaneously injected at neck, groin, axilla and dorsum with the agent mixing 500 µg IRBP peptide with Complete Freund’s adjuvant (CFA). Meanwhile, the mice were intraperitoneally injected with 1 µg pertussis toxin (KKCL1350, Sigma, USA). Then the immunized mice were kept at room temperature for 2 weeks to develop EAU. All experiments with mice were approved by the Animal Care and Use Committee of The First Affiliated Hospital of Chongqing Medical University and followed the Laboratory Animal-Guideline for Ethical Review of Animal Welfare (GB/T 35892–2018) issued by Standardization Administration of China.
The Biosafety of TPP in vitro
To detect the cytological biocompatibility of TPP, human renal tubular epithelial cells (HK-2, 89111512, Procell Life Technology Co., Ltd, China) were plated in 96-well plates and co-cultured with TPP at gradient concentrations (0.05 mg/mL, 0.10 mg/mL, 0.20 mg/mL, 0.40 mg/mL and 0.80 mg/mL) at 37°C. After being co-cultured for 24 h, CCK-8 assay was used to detect the cell viability.
The hematological biocompatibility of TPP was evaluated by hemolysis test. C57BL/6J mice were anesthetized and their blood was collected in tubes containing ethylene diamine tetraacetic acid-K2 (EDTA-K2). After dilution, the blood samples were respectively added into 10 mL normal saline (negative control), 10 mL double distilled water (positive control) and 10 mL suspension of TPP in PBS. After water bathing for 30 min at 37°C, all groups were centrifuged (750 g, 5 min). The absorbance of the supernatant was measured by NP80 (wavelength 415nm). The hemolysis ratio (HR) was calculated according to the following formula: HR=100%×(AT-AN)/(AP-AN) (AT: Absorption value of TPP group at 415 nm; AN: Absorption value of negative control at 415 nm; AP: Absorption value of positive control group at 415 nm).
The Biosafety of TPP in vivo
Six-week-old female C57BL/6J mice were randomly divided into control group and TPP group. Mice were injected via the tail vein with PBS in control group and TPP in TPP group. After 24 h, mice were sacrificed by euthanasia and the blood sample of each mouse was collected to detect chemistry analytes and blood counts. Another ten mice were divided and injected in the same way. Mice were sacrificed on the 7th day by euthanasia after injection of agents. The hearts, livers, spleens, lungs and kidneys of mice were collected for hematoxylin and eosin (H&E) staining.
Targeting Ability of TPP
C57BL/6J mouse lymphocytes stimulated by ConA were plated in 24-well plates (1×105 cells per cell) and divided into targeted group and non-targeted group. Cells of targeted group were treated with TPP while cells of non-targeted group with equal amount of TP for 2 h. The cells were respectively collected, stained by DAPI, and centrifuged to remove free nanoparticles from culture solution. HK-2 cells were used as a negative control (PD-1− cells) with the same treatments. Laser scanning confocal microscopy (LSCM) and fluorescence microscopy were used to observe the combination between nanoparticles and cells at 400 magnification or 200 magnification. Meanwhile, flow cytometry (FCM) was used to quantify the binding efficiency (excitation wavelength: 540 nm; emission wavelength: 590 nm).
Suppression Functions of TPP to the Immune Activity in vitro
Spleen lymphocytes from C57BL/6J mice were pre-stained with CellTraceViolet (CTV, Invitrogen, USA) and washed with PBS to remove free dye. Stained lymphocytes were plated in 24-well plates (1×106 cells per well) and divided into three groups for different treatment agents, namely PBS group (control), TPP group and mixture of free TEPP-46, free PD-1 antibody and empty nanoparticles (Mixture) group. TPP group and Mixture group always had a concentration of TEPP-46 as 25 µM. The lymphocytes were co-cultured with ConA and corresponding agents simultaneously for 72 h. Before detection by FCM, a group of newly CTV stained lymphocytes was prepared as parent control.
For cytokines’ staining, the lymphocytes were also plated, grouped and treated according to the method stated previously except for adding CTV. Then the lymphocytes were stimulated by ionomycin, PMA and brefeldin A (Biolegend, USA) for 6 h. Fixation/Permeabilization Kit (Becton, Dickinson and Company, USA) was used to fix and permeate the lymphocytes. Anti-mouse PE-conjugated IL-2 (Biolegend, 503807, USA), PE-conjugated IL-17A (Invitrogen, 12–7177-81, USA), FITC-conjugated IFN-γ (Invitrogen, 11–7311-82, USA) were used to mark different cytokines of lymphocytes for 1 h. At last, the lymphocytes were detected by FCM.
TPP Alleviates EAU
C57BL/6J mice were randomly divided into three groups as described previously and immunized by IRBP as described previously. Corresponding agents of different groups were injected via the tail vein on the 7th day after immunization. On day 14, clinical grading of EAU in mice was scored according to Caspi’s criteria. The mice were sacrificed by euthanasia and the eyes were collected for H&E staining and histopathologically scored. The severity of EAU was scored on a scale of 0 (no disease) to 4 (severe disease) by an ophthalmologist blinded to related experiments. The spleens of mice were collected to isolate lymphocytes. IL-17A and IFN-γ were also stained as previously described and analyzed by FCM to investigate the percentage of Th1 and Th17 cells.
Statistical Analyses
Statistical analysis in this study was performed by GraphPad Prism 7 software. All data were shown as mean±standard deviation (SD). One-way ANOVA was used to determine the statistical significance among groups and Student’s t-test was applied to analyze data between two independent groups. A two-tailed p-value less than 0.05 was considered statistically significant (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, NS not significant).
Results
Synthesis and Characteristics of TPP
The nanoparticles loaded with TEPP-46 and PD-1 antibody (TPP) were successfully synthesized with the use of the double emulsion solvent evaporation method and carbodiimide method. TEPP-46 was encapsulated in PLGA-PEG and PD-1 antibody modified the surface of nanoparticles. To investigate whether the functions of TPP could satisfy our expectation, its characteristics were carefully detected. To be specific, TPP was spherical, solid and homogeneous with porous surface observed by SEM and TEM (Figure 1A). The elements of carbon, oxygen and sulphur were detected on the surface of TPP (Supplementary Figure 1). The N2 adsorption-desorption isotherm and the corresponding Barrett-Joyner-Halenda (BJH) pore size distribution curve are shown in Supplementary Figure 2. According to the classification of physical adsorption isotherms defined by the International Union of Pure and Applied Chemistry (IUPAC), the isotherm of TPP was attributed to type IV, which was a porous structure (Supplementary Figure 2A) and the BJH desorption average pore diameter was 36.99 nm (Supplementary Figure 2B). The diameter of TPP was 218.7±13.9 nm (Figure 1B) and its zeta potential was −17.4±0.7 mV (Figure 1C) measured by DLS analysis. Meanwhile, the polydispersity index (PDI) showed a narrow distribution of most TPP in particle size (0.06±0.02), further indicating a good homogeneity of TPP. The zeta potential of PLGA-PEG NPs and TP respectively were −27.23±9.8 mV and −26.1±8.7 mV (Supplementary Figure 3). The encapsulated TEPP-46 had no influence on the zeta potential of nanoparticles while the conjugation of PD-1 antibody decreased the value of zeta potential. However, the data had no statistical significance. In addition, TPP was dispersed in PBS for 10 days and detected by DLS analysis on day 1, day 5 and day 10 to investigate the stability of TPP. As shown in Figure 2A, the diameter and PDI did not change significantly within the 10 days, demonstrating satisfactory stability of TPP which laid a foundation for the further utilization of TPP in vitro and in vivo. The modification with PD-1 antibody on TPP was visualized by using fluorescence microscopy (Figure 1D). DiI labeled PLGA-PEG and FITC labeled PD-1 antibody were observed to be overlapping, indicating that PD-1 antibody was connected to TPP. The binding efficiency was verified to be 80.5% by FCM, as shown in Figure 1D.
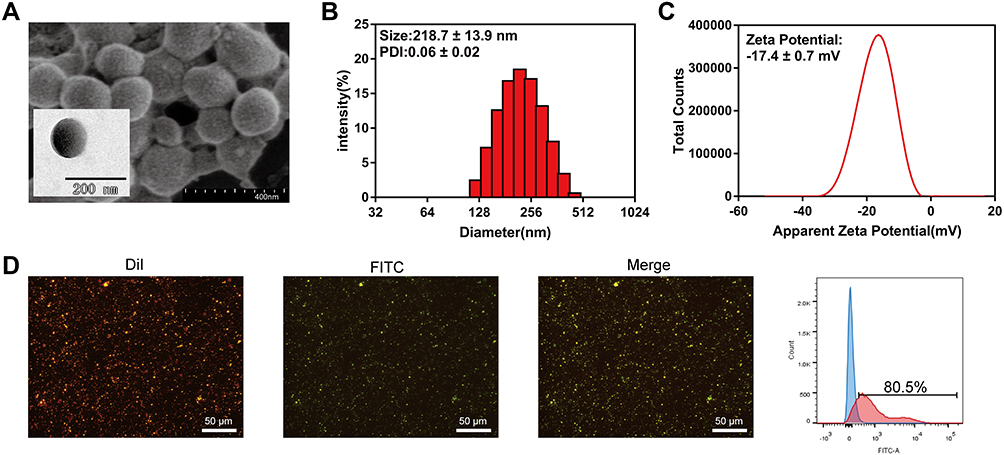 |
Figure 1 Characteristics of TPP. (A) SEM and TEM images of TPP. (B) Diameter and PDI of TPP. (C) Zeta potential of TPP. (D) PD-1 combination (DiI labeled PLGA-PEG, FITC labeled PD-1 antibody) was observed by fluorescence microscopy (× 400) and the binding efficiency was analyzed by FCM. |
 |
Figure 2 The stability and drug release of TPP. (A) Particle sizes and PDI of TPP on day 1, day 5, day 10. (B) Ultraviolet absorption spectrum of TPP, PLGA-PEG and TEPP-46. The absorption peaks of TPP and free TEPP-46 appeared at 272 nm and 305 nm. (C) Standard curve of TEPP-46 related to concentration and absorbance intensity. (D) Release pattern of TEPP-46 released from TPP within 96 h. |
To investigate the drug-loading and drug-releasing ability of TPP, the ultraviolet absorption spectrum of free TEPP-46 and TPP were detected. As shown in Figure 2B, the absorption peaks of free TEPP-46 appeared at 272 nm and 305 nm. The curve of TPP was almost overlapping with the curve of PLGA-PEG except for two peaks at 272 nm and 305 nm which was the same as free TEPP-46, indicating that TEPP-46 was reliably loaded in TPP. According to the standard absorption curve of TEPP-46 (Figure 2C), the entrapment efficiency and the loading capacity were figured out as 57.38±2.084% and 4.48±0.20%.
To analyze the release behavior of TPP, the amount of released TEPP-46 was detected at different time points over a 96-hour period study. In brief, the whole release period was a sustained process. To be specific, an initial burst effect was observed during the first twelve hours, followed by a steady release period of approximately 60 hours, and finally a slow release period (Figure 2D). Fifty percent of TEPP-46 was released during the burst period and 34% during the steady period. Remaining TEPP-46 was released during the slow period. The sustained-release effect suggested that TPP had the potential to continue its therapeutic effects throughout the treatment process.
The Biosafety of TPP
To investigate the biocompatibility of TPP, the cytological and hematological toxicity were tested. As shown in Supplementary Figure 4A, there was no significant difference in the viability of HK-2 cells among groups at different concentrations of TPP, suggesting good cytological biocompatibility of TPP. The absorption spectrum of supernatant in hemolysis test is shown in Supplementary Figure 4B and the hemolysis ratio of TPP was 3.6%, demonstrating high hematological biocompatibility of TPP. To investigate the biosafety of TPP in murine model, the blood and serum samples of mice were tested 24 h after TPP or PBS treatment. Results are shown in Table 1. There was no statistical difference in liver function indexes (AST, ALT) and kidney function indexes (BUN, CRE) between TPP group and PBS group, demonstrating no hepatic and renal toxicity in TPP treatment. Moreover, TPP did not significantly impair the amount of various blood cells (Table 1), indicating the absence of cytotoxicity. To further testify the compatibility of TPP with murine tissue, the hearts, livers, spleens, lungs and kidneys of mice were collected for H&E staining 7 days after TPP or PBS treatment. No significant histological change was observed in H&E staining of all organs (Figure 3).
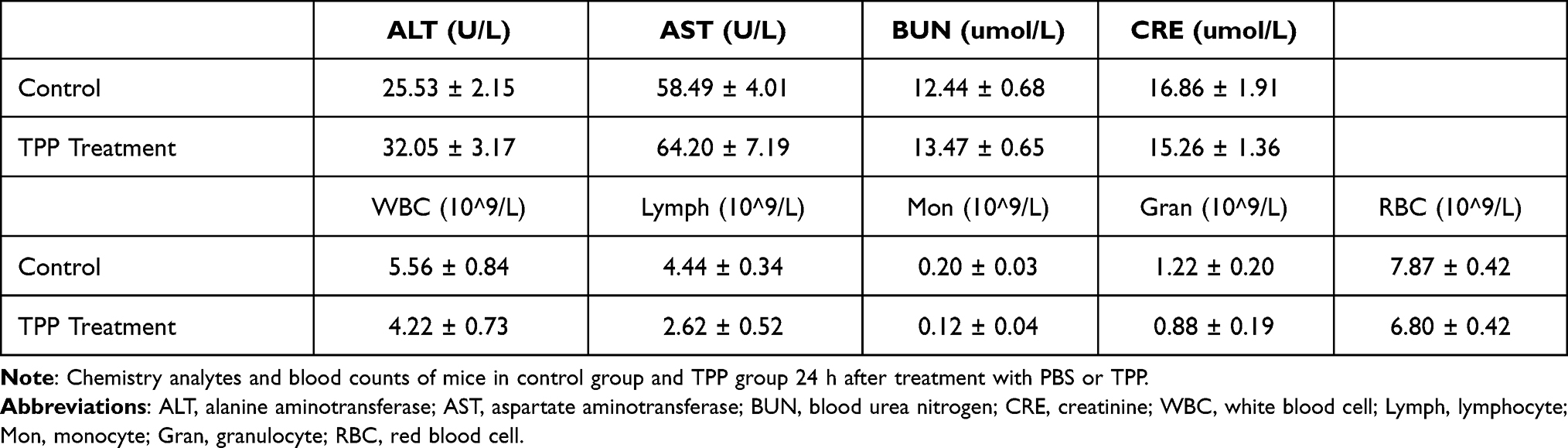 |
Table 1 Chemistry Analytes and Blood Counts of Mice |
 |
Figure 3 The compatibility of TPP with murine tissue. H&E staining (×200) of hearts, livers, spleens, lungs and kidneys of mice in control group and TPP group 7 days after treatment with PBS or TPP. |
Targeting Ability of TPP
To verify the targeting ability of TPP to PD-1+ lymphocytes, activated lymphocytes were respectively treated with non-targeted TP and targeted TPP for 2 h and visualized by LSCM. There were a large number of nanoparticles accumulating around cells treated with targeted TPP, while few nanoparticles were observed around cells treated with non-targeted TP, as shown in Figure 4A. Furthermore, the binding efficiency of TPP to lymphocytes was quantitatively detected by FCM and is shown in Figure 4B, which was significantly higher in targeted group than that in non-targeted group. Both results demonstrated that TPP could target PD-1+ lymphocytes more efficiently via the modification by PD-1 antibody. Besides, neither TP nor TPP could specifically be combined with HK-2 cells (Supplementary Figure 5), demonstrating that TPP had no targeting effect on PD-1− cells.
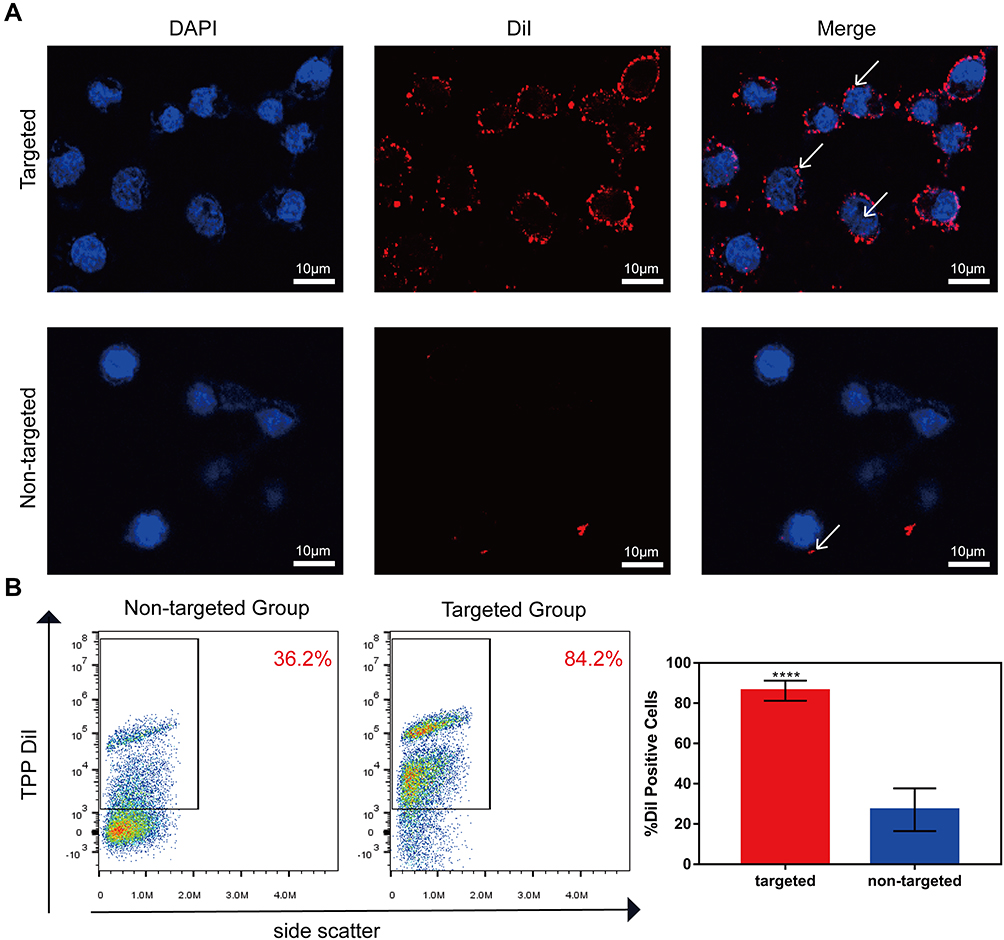 |
Figure 4 Targeting ability of TPP. (A) LSCM images of the combination of stimulated lymphocytes and TPP (targeted group) or TP (non-targeted group) (×400). The nucleus was stained blue by DAPI and TPP was stained red by DiI. (B) FCM analysis of the binding efficiency between stimulated lymphocytes and TPP (targeted group) or TP (non-targeted group). (****p<0.0001). |
Suppression Functions of TPP to the Immune Activity in vitro
To investigate whether the designed TPP can suppress the immune activity in vitro, the spleen lymphocytes were isolated from mice immunized by IRBP and without any treatment and plated as three groups, namely PBS group (control), TPP group and mixture of free TEPP-46, free PD-1 antibody and empty nanoparticles (mixture) group. Spleen lymphocytes were stimulated with ConA and corresponding agents for each group. FCM results showed that, the fluorescence distribution of CTV shifted to the left in PBS group and mixture group compared to that in TPP group, suggesting decreased fluorescence intensity of lymphocytes in PBS group and mixture group, and the quantitative mean fluorescence intensity of TPP group was significantly stronger than that of PBS group and mixture group (Figure 5B). As the fluorescence intensity of tested cells decreased with cell proliferation, it can be concluded from these results that TPP treatment evidently inhibited the proliferation capability of lymphocytes. A remarkable decrease of IL-2-producing cells was meanwhile observed in TPP-treated lymphocytes (Figure 5A), suggesting an impairment in early T cell activation in TPP treatment. In accordance, the percentage of IFN-γ/IL-17A-producing cells was also significantly reduced by TPP treatment compared to that of PBS or mixture agents (Figure 6A and B), indicating a considerable suppression ability of TPP to such crucial pro-inflammatory cells in EAU model.
 |
Figure 5 Suppressive effect of TPP on early activation and proliferation of stimulated lymphocytes. (A) The percentage of IL-2 producing cells in TPP group, mixture group and PBS group was detected by FCM. (B) The proliferation of lymphocytes in TPP group, mixture group and PBS group was detected by FCM using CelltraceViolet. (*p<0.05, **p<0.01,***p<0.001, ****p<0.0001). Abbreviation: NS, not significant. |
 |
Figure 6 Inhibitory effect of TPP on Th1 and Th17 cells in vitro. (A) The percentage of IL-17 producing cells in TPP group, mixture group and PBS group was detected by FCM. (B) The percentage of IFN-γ producing cells in TPP group, mixture group and PBS group was detected by FCM. (*p<0.05, **p<0.01). Abbreviation: NS, not significant. |
TPP Alleviates EAU
To determine the therapeutic effect of TPP in vivo, we induced the EAU model in mice to evaluate the anti-inflammatory effect of TPP treatment. Groups were set as previously described. Clinical remission of uveitis was characterized by a decreased clinical score as determined by conjunctival hyperemia, ciliary injection, posterior synechiae, aqueous flare and cells. Meanwhile, retinal structural destruction, inflammatory cells infiltrating the retina and vasculitis in choroid or retina referred to the histological assessment of uveal inflammation. As shown in Figure 7A and B, TPP-treated mice had lowest clinical scores and histological scores among the three groups, demonstrating the effective alleviating function of TPP treatment in EAU model. In addition, the IFN-γ/IL-17A-producing cells in the spleen were also significantly inhibited by TPP treatment (Figure 8A and B), which represented the favorable inhibition of pathogenic inflammatory Th1/Th17 cells.
 |
Figure 7 TPP alleviated the inflammatory activity of EAU. (A) Clinical analysis of mice in TPP group, mixture group and PBS group. (B) H&E staining (×100) of eyes for histopathological analysis of mice in TPP group, mixture group and PBS group. (*p<0.05, **p<0.01). Abbreviation: NS, not significant. |
 |
Figure 8 Inhibitory effect of TPP on Th1 and Th17 cells in vivo. (A) The percentage of IL-17 producing cells in TPP group, mixture group and PBS group was detected by FCM. (B) The percentage of IFN-γ producing cells in TPP group, mixture group and PBS group was detected by FCM. (*p<0.05, **p<0.01, ***p<0.001). Abbreviation: NS, not significant. |
Discussion
In this study, we reported a novel therapeutic strategy based on TPP, a specific drug delivery platform. It targeted active lymphocytes by recognition of PD-1 receptors and suppressed the energy metabolism of effector T cells, collectively achieving an anti-inflammation effect. We showed that TPP successfully encapsulated TEPP-46 and found a distinctive three-period release profile, namely burst, steady and slow period. The sustained-release characteristics favored maintaining a high drug concentration within the eye and the blood circulation, thereby delaying the expiration of treatment. Therefore, it would benefit patients in terms of reduced administration frequency. The assembled TPP turned hydrophobic TEPP-46 to part of a hydrophilic system on account of the amphiphilicity of PLGA-PEG,38,39 which promoted the humoral compatibility of TEPP-46 and elevated its bioavailability. PEG modification further enhanced the hydrophilicity of the system, lowered the immunogenicity and prolonged the cycle period, preventing TPP from rapid blood clearance. Compared with non-targeted group, TPP could bind more efficiently to stimulated lymphocytes due to the surface modification of PD-1 antibody. With systemic administration, TPP might be more likely to spread toward inflamed retina, exerting a local cumulative effect within the eye. Overall, the bioavailability and utilization efficiency of TEPP-46 were both improved through the integration of nanoplatform.
IL-2, also known as T cell growth factor, is a cytokine primarily produced by activated T cells, especially CD4+ T cells. It maintains the growth and proliferation of T cells and promotes the activation and differentiation of T cells as well as other lymphocytes, such as natural killer (NK) cells and macrophage.40 In this study, the production of IL-2 was significantly reduced with TPP treatment, suggesting its inhibitory effect on the initial activation of T cells. In addition, IL-17A and IFN-γ were also reduced after treatment with TPP both in vitro and in vivo, indicating its suppression on Th1 and Th17 cells. The proliferation, early activation and activity of these aberrant lymphocytes were collectively inhibited by TPP treatment and therefore the inflammation mediated by them was consequently suppressed.
Non-infectious uveitis refers to a group of clinically heterogenic inflammatory diseases presumably mediated by Th1 and Th17 cells.41,42 To date, comprehensive treatment of local or systemic administration of corticosteroids and immunosuppressive agents has remained to be the mainstay for these autoimmune uveitis entities.43 Long-term use of such treatment regimen could cause obvious and even severe side effects as well as probable drug resistance.44 Nowadays many researchers resort to biomaterials for more effective and safe immunosuppression strategies. With the application of biomaterials, the dosage of certain agents can often be cut down to minimal level. This favors reduced side effects and drug tolerance to a great extent by such accurate and effective delivery method. Moreover, biomaterials can effectively reduce the toxicity of agents, protect them from degradation or help to penetrate biological barriers in vivo.
Although some studies have achieved beneficial results, the targeted regulation of dysfunctional pathogenic lymphocytes remains unsatisfactory. In this case, we focused our study on the activated effector T cells especially pro-inflammatory Th1 and Th17, two cell populations critical for the development and progression of non-infectious uveitis. PD-1 receptors are involved in the immunosuppressive regulation pathway. Its expression is an evident indicator of the ongoing immunization events, marking the active condition of Th1 and Th17 cells. Recent studies have revealed an unconventional metabolism pathway in these active effector T cells, termed aerobic glycolysis, differing from other lymphocytes.22 This metabolism pattern can be particularly impacted by allosteric activator TEPP-46. Within the nanoplatform system, encapsulated TEPP-46 could selectively inhibit the energy metabolism of activated effector T cells, following the targeted guidance of PD-1 antibody. Therefore, the combination of TEPP-46 with PD-1 antibody could give birth to a synergetic inhibitory effect on inhibition of activated Th1 and Th17 cells. Our study elucidated that TPP could effectively inhibit the EAU activity in association with significantly down-regulated Th1 and Th17 cells.
There are some limitations in our study. First, laboratory preparation methods of TPP are insufficient to meet the demand of mass production, which restricts its further utilization in clinic. Second, the pathological mechanism of uveitis entities is much more complicated than in an animal model and there are some discrepancies in cellular mechanisms between different classifications of uveitis. Certain underlying mechanisms and specific animal models require further investigation.
Conclusion
In conclusion, we synthesized a novel nanoplatform TPP to targeted suppress effector T cells, including Th1 and Th17 cells, through the synergetic effect of PD-1 antibody and TEPP-46. TPP was demonstrated to have good stability, high biocompatibility and biosafety. It was able to target active lymphocytes and significantly suppress their early activation, proliferation in vitro. It also effectively inhibited the immune activity of Th1 and Th17 cells and ameliorate EAU in mice. Our study provided a promising therapeutic alternative for auto-inflammatory and autoimmune disease.
Acknowledgments
Thanks for the support of Natural Science Foundation Major International (Regional) Joint Research Project (81720108009), National Natural Science Foundation Key Program (81930023), Chongqing Outstanding Science Project (2019), Chongqing Chief Medical Scientist Project (2018), Chongqing Key Laboratory of Ophthalmology (CSTC, 2008CA5003), Chongqing Science & Technology Platform and Base Construction Program (cstc2014pt-sy10002).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jones EL, Laidlaw SM, Dustin LB. TRIM21/Ro52 - Roles in innate immunity and autoimmune disease. Front Immunol. 2021;12:738473.
2. Bentley ER, Little SR. Local delivery strategies to restore immune homeostasis in the context of inflammation. Adv Drug Deliv Rev. 2021;178:113971.
3. Raverdeau M, Christofi M, Malara A, et al. Retinoic acid-induced autoantigen-specific type 1 regulatory T cells suppress autoimmunity. EMBO Rep. 2019;20(5):e47121.
4. Horai R, Caspi RR. Microbiome and Autoimmune Uveitis. Front Immunol. 2019;10:232.
5. Zhong Z, Su G, Kijlstra A, Yang P. Activation of the interleukin-23/interleukin-17 signalling pathway in autoinflammatory and autoimmune uveitis. Prog Retin Eye Res. 2021;80:100866.
6. Bertrand PJ, Jamilloux Y, Kodjikian L, et al. Quality of life in patients with uveitis: data from the ULISSE study (Uveitis: cLInical and medico-economic evaluation of a Standardised Strategy for the Etiological diagnosis). Br J Ophthalmol. 2021;105(7):935–940.
7. Jabs DA, Rosenbaum JT, Foster CS, et al. Guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders: recommendations of an expert panel. Am J Ophthalmol. 2000;130(4):492–513.
8. Shen Z, Huang W, Liu J, Tian J, Wang S, Rui K. Effects of Mesenchymal Stem Cell-Derived Exosomes on Autoimmune Diseases. Front Immunol. 2021;12:749192.
9. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242.
10. keir ME, Liang SC, Guleria I, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203(4):883–895.
11. Fife BT, Pauken KE, Eagar TN, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10(11):1185–1192.
12. Zhang Y, Liu Z, Tian M, et al. The altered PD-1/PD-L1 pathway delivers the ‘one-two punch’ effects to promote the Treg/Th17 imbalance in pre-eclampsia. Cell Mol Immunol. 2018;15(7):710–723.
13. Ansari MJ, Salama AD, Chitnis T, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 2003;198(1):63–69.
14. Geisler AN, Phillips GS, Barrios DM, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol. 2020;83(5):1255–1268.
15. Zhao P, Wang P, Dong S, et al. Depletion of PD-1-positive cells ameliorates autoimmune disease. Nat Biomed Eng. 2019;3(4):292–305.
16. Liu X, Diedrichs-Möhring M, Wildner G. The Role of IFN-alpha in Experimental and Clinical Uveitis. Ocul Immunol Inflamm. 2019;27(1):23–33.
17. Wu S, Ma R, Zhong Y, et al. Deficiency of IL-27 Signaling Exacerbates Experimental Autoimmune Uveitis with Elevated Uveitogenic Th1 and Th17 Responses. Int J Mol Sci. 2021;22(14):7517.
18. Geltink RIK, Kyle RL, Pearce EL. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol. 2018;36:461–488.
19. Li YH, Li XF, Liu JT, et al. PKM2, a potential target for regulating cancer. Gene. 2018;668:48–53.
20. Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186(6):3299–3303.
21. Lunt SY, Muralidhar V, Hosios AM, et al. Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation. Mol Cell. 2015;57(1):95–107.
22. Angiari S, Runtsch MC, Sutton CE, et al. Pharmacological Activation of Pyruvate Kinase M2 Inhibits CD4+ T Cell Pathogenicity and Suppresses Autoimmunity. Cell Metab. 2020;31(2):391–405.e8.
23. Seki SM, Posyniak K, McCloud R, et al. Modulation of PKM activity affects the differentiation of TH17 cells. Sci Signal. 2020;13(655):eaay9217.
24. Dhanesha N, Patel RB, Doddapattar P, et al. PKM2 promotes neutrophil activation and cerebral thrombo-inflammation: therapeutic implications for ischemic stroke. Blood. 2021;1:453.
25. Zhu S, Guo Y, Zhang X, et al. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. 2021;503:240–248.
26. Dai H, Zeng W, Luo H. C-MET-dependent signal transduction mediates retinoblastoma growth by regulating PKM2 nuclear translocation. Cell Biochem Funct. 2020;38(2):204–212.
27. Wong N, Ojo D, Yan J, Tang D. PKM2 contributes to cancer metabolism. Cancer Lett. 2015;356(2Pt A):184–191.
28. Anastasiou D, Yu Y, Israelsen WJ, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8(10):839–847.
29. Jin S, Gao J, Yang R, et al. A baicalin-loaded coaxial nanofiber scaffold regulated inflammation and osteoclast differentiation for vascularized bone regeneration. Bioact Mater. 2021;8:559–572.
30. Feng X, Xu W, Li Z, Song W, Ding J, Chen X. Immunomodulatory Nanosystems. Adv Sci. 2019;6(17):1900101.
31. Pan W, Zhu S, Dai D, et al. MiR-125a targets effector programs to stabilize Treg-mediated immune homeostasis. Nat Commun. 2015;6:7096.
32. McHugh MD, Park J, Uhrich R, Gao W, Horwitz DA, Fahmy TM. Paracrine co-delivery of TGF-β and IL-2 using CD4-targeted nanoparticles for induction and maintenance of regulatory T cells. Biomaterials. 2015;59:172–181.
33. Jiang G, Huang Z, Yuan Y, Tao K, Feng W. Intracellular delivery of anti-BCR/ABL antibody by PLGA nanoparticles suppresses the oncogenesis of chronic myeloid leukemia cells. J Hematol Oncol. 2021;14(1):139.
34. Sarkar Siddique JC, Chow L. Gold Nanoparticles for Drug Delivery and Cancer Therapy. Applied Science. 2020;10(11):3824.
35. Sarkar Siddique JC, Chow L. Application of Nanomaterials in Biomedical Imaging and Cancer Therapy. Nanomaterials. 2020;10(9):1700.
36. Sadeghi AR, Nokhasteh S, Molavi AM, et al. Surface modification of electrospun PLGA scaffold with collagen for bioengineered skin substitutes. Mater Sci Eng C Mater Biol Appl. 2016;68:995.
37. Wang Q, Sui G, Wu X, et al. A sequential targeting nanoplatform for anaplastic thyroid carcinoma theranostics. Acta Biomater. 2020;102:367–383.
38. Zhang F, Song Q, Huang X, et al. A Novel High Mechanical Property PLGA Composite Matrix Loaded with Nanodiamond-Phospholipid Compound for Bone Tissue Engineering. ACS Appl Mater Interfaces. 2016;8(2):1087–1097.
39. Lee S, Han D, Kang HG, et al. Intravenous sustained-release nifedipine ameliorates nonalcoholic fatty liver disease by restoring autophagic clearance. Biomaterials. 2019;197:1–11.
40. Abbas AK, Trotta E, Simeonov R, Marson A, Bluestone JA. Revisiting IL-2: biology and therapeutic prospects. Sci Immunol. 2018;3(25):eaat1482.
41. Guedes MC, Borrego LM, Proença RD. Roles of interleukin-17 in uveitis. Indian J Ophthalmol. 2016;64(9):628–634.
42. Luger D, Silver PB, Tang J, et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector T category. J Exp Med. 2008;205(4):799–810.
43. Shahab MA, Mir TA, Zafar S. Optimising drug therapy for non-infectious uveitis. Int Ophthalmol. 2019;39(7):1633–1650.
44. Buchman AL. Side effects of corticosteroid therapy. J Clin Gastroenterol. 2001;33(4):289–294.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.