Back to Journals » OncoTargets and Therapy » Volume 7
PD-1 as an emerging therapeutic target in renal cell carcinoma: current evidence
Authors Tykodi S
Received 10 March 2014
Accepted for publication 13 May 2014
Published 25 July 2014 Volume 2014:7 Pages 1349—1359
DOI https://doi.org/10.2147/OTT.S48443
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Scott S Tykodi
Department of Medicine, Division of Medical Oncology, University of Washington, Seattle, WA, USA
Abstract: Renal cell carcinoma (RCC) is the most common primary malignant tumor of the kidney in adults, representing approximately 4% of all adult cancers in the United States. Metastatic RCC is poorly responsive to conventional cytotoxic chemotherapies but can be sensitive to T-cell-directed immunotherapies such as interferon-α or interleukin-2. Despite recent progress in the application of antiangiogenic “targeted therapies” for metastatic RCC, high-dose interleukin-2 remains an appropriate first-line therapy for select patients and is associated with durable complete remissions in a small fraction of treated patients. Thus, advanced RCC provides a unique opportunity to investigate the requirements for effective antitumor immunotherapy. Accumulating evidence suggests that resistance mechanisms exploited by RCC and other tumor types may play a dominant role in limiting the effectiveness of tumor-reactive adaptive immune responses. Expression of the inhibitory coreceptor programmed cell death-1 (PD-1) on tumor-infiltrating lymphocytes within RCC tumors, as well as the expression of the PD-1 ligand (PD-L1) on RCC tumor cells, are strong negative prognostic markers for disease-specific death in RCC patients. Monoclonal antibodies targeting either PD-1 or PD-L1 have now entered clinic trials and have demonstrated promising antitumor effects for refractory metastatic RCC. This review summarizes the results of published and reported studies of PD-1- and PD-L1-targeted therapies enrolling patients with advanced RCC, focusing on key safety, toxicity, and efficacy end points. Prospects for advanced phase clinical testing and novel therapy combinations with PD-1- and PD-L1-targeted agents are discussed.
Keywords: renal cell carcinoma, immune checkpoint, immunotherapy, T-lymphocyte, PD-1, PD-L1
Introduction
Cumulative evidence suggests that cellular immune responses may play an important role in modulating tumor progression in cancer patients. Specific examples include the documentation of rare spontaneous regressions of renal cell carcinomas (RCCs), melanomas, and other tumors;1identification of naturally occurring T-cells recognizing tumor-associated antigens in tumor patients;2and the positive association of T-cell infiltration into tumors with favorable disease outcome.3Such observations have encouraged the development and clinical testing of therapeutic strategies designed to augment tumor-specific T-cell responses in cancer patients, including vaccines, cytokines, and cellular therapies. Pioneering clinical studies undertaken by investigators at the National Cancer Institute Surgery Branch demonstrate that autologous tumor-infiltrating lymphocytes (TILs) expanded in vitro can be transferred to melanoma patients following lymphodepleting conditioning therapy, resulting in objective tumor regression in approximately 50% of treated patients. These results represent one of the most compelling demonstrations of T-cell-mediated antitumor effects targeting solid tumors.4
To date, however, cancer vaccines or cellular therapy products developed for advanced RCC have proven to be disappointing and ineffective for most patients.5–7 Immunotherapy with systemically administered cytokines, including interferon-α (IFN-α) or interleukin-2 (IL-2), has been incrementally more successful as these agents have gained US Food and Drug Administration (FDA) approval for the treatment of advanced RCC. Nevertheless, significant clinical benefit is observed for only a small minority of cytokine-treated patients.8
More recently, preclinical studies and results from early phase clinical trials now suggest that resistance mechanisms exploited by tumors may play a dominant role in limiting the effectiveness of T-cell-mediated cancer therapies.9 Thus, an emerging and highly promising approach to cancer immunotherapy for advanced RCC and other cancers is antibody-mediated blockade of inhibitory coreceptors expressed on T-lymphocytes, or so-called immune checkpoints.
Immune checkpoint blockade for cancer therapy
T-lymphocytes express stimulatory as well as inhibitory coreceptors that are now recognized to play critical roles in regulating the initiation and termination of adaptive immune responses. Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4; CD152) is expressed on activated effector T-cells, and represents the first well-characterized and therapeutically targeted immune checkpoint receptor. CTLA-4 is structurally related to the stimulatory coreceptor CD28 and competes for binding to common ligands B7.1 (CD80) and B7.2 (CD86) expressed on antigen-presenting cells (Figure 1).10 The essential role of CTLA-4 in downregulating T-cell activation and maintaining lymphocyte homeostasis was demonstrated in ctla-4-knockout mice that developed a lymphoproliferative disease associated with multiorgan lymphocytic infiltration, causing death by 3–4 weeks of age.11,12
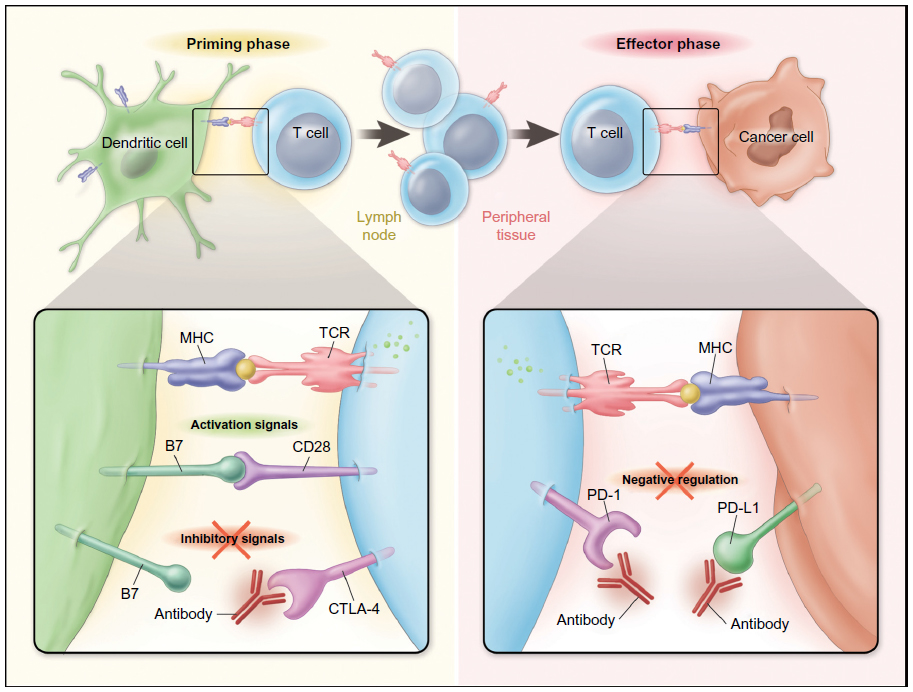 | Figure 1 Stimulatory and Inhibitory coreceptors regulate T-cell responses to tumor antigens. |
Despite severe immunopathology developing in ctla-4-knockout mice, preclinical testing of monoclonal antibody (mAb) blockade of CTLA-4 as tumor immunotherapy both as monotherapy and in association with tumor vaccines demonstrated antitumor effects without apparent toxicity.13,14These preclinical data encouraged the development and clinical testing of two fully humanized mAbs specific for human CTLA-4, ipilimumab and tremelimumab. In early phase clinical testing, both antibodies produced objective responses in patients with melanoma, despite differences in dose and schedule for administration.15,16 In a pivotal phase III clinical trial, ipilimumab given with or without a gp100 peptide vaccine versus the gp100 vaccine alone demonstrated a median survival benefit for the ipilimumab-treated patients (10.0 months versus [vs] 6.4 months; P<0.001).17 Interestingly, a minority of ipilimumab-treated patients showed prolonged disease control even after completion of active treatment, suggesting sustained functional modulation of the T-cell compartment in these patients.17,18 Ipilimumab received FDA approval for the treatment of advanced melanoma in 2011, validating the therapeutic potential of immune checkpoint blockade. This study also established benchmark data for autoimmune toxicities associated with ipilimumab-mediated disruption of a regulatory immune pathway. Immune-related adverse events occurred in 60% of ipilimumab-treated patients (10%–15% grade 3/4) and included patient deaths secondary to immune-related toxicity.17 Severe immune-related toxicities often required active management with corticosteroids or other immune-suppressive treatments.19
Programmed cell death-1 (PD-1; CD279) is another immune checkpoint receptor emerging as a target for antitumor immunotherapy.9 PD-1 is expressed on activated effector T-cells as well as natural killer cells and b cells. High-level PD-1 expression is found on TILs and also on T-cells in the setting of chronic viral infections.20 Thus, PD-1 expression on effector T-cells is associated with constitutive antigen exposure and is a marker of T-cell unresponsiveness or exhaustion.
Unlike CTLA-4, the ligands for PD-1 are not restricted to antigen-presenting cells and the hematopoietic compartment. Known ligands for PD-1 include B7 family member proteins programmed cell death-1 ligand 1 (PD-L1; also known as B7-H1 or CD274) and programmed cell death-1 ligand 2 (PD-L2; also known as B7-DC or CD273). PD-L1 is expressed by multiple tissues, including T- and B-cells, dendritic cells, and macrophage. PD-L1 is also expressed by many different solid tumor types, suggesting that inhibitory receptor–ligand interactions inhibiting antitumor T-cell immunity are occurring in the tumor microenvironment (Figure 1).21 Tumor expression of PD-L1 has been associated with deregulated oncogene signaling,22,23 tumor hypoxia,24 and local inflammatory signals, including IFN-γ.25 A model of tumor upregulation of PD-L1 expression in response to TILs and local cytokine release as a mechanism to evade endogenous immune responses has been termed “adaptive resistance” and may represent the most common mechanism for PD-L1 expression in a wide variety of tumor histologies.25 PD-L2 has a more restricted expression pattern than PD-L1, primarily expressed by hematopoietic cells and also some lymphomas, but with far less frequent expression in solid tumors.9
As with CTLA-4, the functional consequence of absent PD-1 or PD-L1 expression has been evaluated in both PD-1 (pdcd1) and PD-L1 knockout mice generally showing less severe immune dysfunction. Disruption of PD-1 resulted in strain-specific autoimmune syndromes, including arthritis, glomerulonephritis, or dilated cardiomyopathy, whereas disruption of PD-L1 was not associated with a phenotype of spontaneous autoimmunity, suggesting substantial redundancy in mechanisms controlling peripheral T-cell tolerance.26–28 The more subtle immunopathology observed with PD-1 or PD-L1 knockout anticipated that autoimmune side effects associated with therapeutic PD-1 pathway blockade might be less severe than for CTLA-4. Preclinical studies of PD-1:PD-L1 inhibition by gene disruption of PD-1 or antibody blockade of PD-L1 have observed augmented T-cell-mediated antitumor effects.29 The evolving insight gained from preclinical studies supporting a key role of PD-1:PD-L1 interactions in immune regulation and tumor resistance to adaptive immune responses, along with evidence for antitumor activity of anti-CTLA-4 mAbs in clinical trials, has provided strong encouragement for development and testing of PD-1- and PD-L1-specific agents. Great enthusiasm for this approach is reflected by the entry of nine different PD-1/PD-L1-directed agents into clinical trials as of this writing (Table 1).
PD-L1 has been shown to bind to B7.1 in addition to PD-1, an interaction that may also deliver an inhibitory signal to T-cells. Thus, therapeutic mAb blockade of PD-1 versus PD-L1 will result in nonidentical disruption of signaling interactions. PD-1 blockade would not interfere with a PD-L1:B7.1 interaction. Conversely, PD-L1 blockade would leave PD-1:PD-L2 interactions intact.9
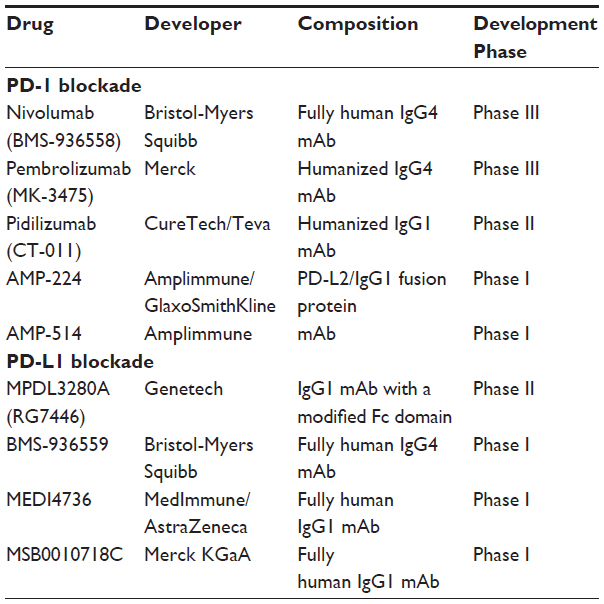 | Table 1 PD-1- or PD-L1-blocking agents in clinical trails |
Regulatory T-cells (Tregs) inhibit cellular immune responses and help to maintain self-tolerance. Tregs express constitutive high levels of several immune checkpoint receptors, including CTLA-4, PD-1, and others. Rather than exerting inhibitory effects, these receptors appear to enhance Treg activity or proliferation. Thus, separate from their influence on effector T-cells, therapeutic antibodies blocking checkpoint receptors may suppress the activity of Tregs as an additional mechanism contributing to overall enhanced antitumor immunity.9
Advanced RCC is a target for T-cell-directed immunotherapy
RCC is the most common primary malignant tumor of the kidney in adults, representing approximately 4% of all adult cancers.30 RCC encompasses a heterogeneous group of tumors subdivided largely by their histologic appearance. The most common histologic types of RCC include clear cell (75% of tumors), papillary (10%), and chromophobe (5%) tumors. Additional rare subtypes of RCC are recognized, including medullary, collecting duct, and RCC associated with Xp11 translocation. A pathologic diagnosis of unclassified RCC still accounts for 4%–6% of all tumors.31 The high frequency of clear cell histology and predisposing genetic lesion for this RCC subtype have skewed clinical research toward clear cell tumors.
Clear cell RCC tumors in both hereditary and sporadic forms are associated with loss of function of the von Hippel-Lindau (VHL) tumor suppressor gene on the short arm of chromosome 3 (3p25.3) as a result of deletion, mutation, or epigenetic silencing.32 The loss of VHL expression results in the deregulation of hypoxia-inducible factor-1 and -2 transcription factors and constitutive expression of a number of hypoxia-responsive gene products that control angiogenesis, cell cycle, and energy homeostasis. Insight into the abnormal molecular biology common to most clear cell RCC tumors has encouraged the clinical development of novel targeted therapies for this disease directed at signaling pathways affected by VHL inactivation. Since 2005, seven new antiangiogenic drugs have been approved by the FDA for the treatment of advanced RCC. These include oral tyrosine kinase inhibitors (sorafenib, sunitinib, pazopanib, axitinib) that disrupt vascular endothelial growth factor (VEGF) receptor-mediated signaling, the VEGF-specific mAb bevacizumab, and inhibitors of mammalian target of rapamycin (temsirolimus and everolimus). Targeted therapies have been rapidly adopted as first- and second-line treatments for metastatic clear cell RCC. However, they are limited by the development of tumor resistance and disease progression that has been uniformly observed in treated patients.33–39
From the 1980s until the introduction of targeted therapies, the treatment of advanced RCC was unique among metastatic carcinomas. Most patients received immunotherapy with the cytokines IFN-α or IL-2 as standard front-line therapy. IFN-α was shown in randomized trials to improve survival compared with medroxyprogesterone acetate or vinblastine, despite a response rate of only 14%–16%,40,41 which reflected the intrinsic resistance of RCC to cytotoxic chemotherapies and hormonal therapies.42 A Cochrane review of pooled data further supported a survival benefit for IFN-α versus controls (hazard ratio of 0.74).43 More recently, IFN-α in combination with bevacizumab has shown superior efficacy to IFN-α monotherapy measured by response rate and progression-free survival, thereby maintaining a role for IFN-α in contemporary treatment of RCC.36,44
High-dose (HD) IL-2 received FDA approval for the treatment of metastatic RCC in 1992 based on pooled results of 255 patients treated as part of seven phase II clinical trials conducted at 21 institutions. The overall objective response rate (ORR) was 14% with 5% complete responses (CRs).45 Most remarkable was the durability of CRs following IL-2 therapy. For example, the long-term follow-up of 259 RCC patients treated by HD IL-2 at the National Cancer Institute Surgery Branch noted recurrent disease in only four of 23 patients (17%) who achieved a CR with a median duration of ongoing CR >11 years at the time of reporting.46 Due to the unique potential to achieve unmaintained CRs, HD IL-2 has remained a first-line treatment option for select RCC patients, despite the limitations of severe toxicities and the requirement for patient referral to cancer centers specializing in IL-2 administration. The uniquely good outcomes for a small fraction of RCC patients achieving CR after HD IL-2 provides strong motivation for the development and testing of novel T-cell-directed immunotherapeutics for advanced RCC with the hope for potent and durable tumor responses associated with less toxicity than with HD IL-2.
PD-1 pathway targets associated with metastatic RCC
The expression of PD-1 pathway proteins has been extensively studied in RCC tumors and shown to closely associate with RCC progression and poor disease-specific outcomes. In the largest reported series of 306 clear cell RCC tumors, Thompson et al47 analyzed PD-L1 expression by immunostaining formalin-fixed paraffin-embedded tissue samples. Tumor cell membranous expression of PD-L1 (with a threshold of ≥5% of tumor cells stained with mAb 5H1) was seen in 24% of samples and strongly associated with cancer-specific death (risk ratio [RR] 3.92; P<0.001). This association also remained significant in multivariate modeling (RR 2.0, P=0.003) (Figure 2). In a similar analysis, Thompson et al48 also analyzed PD-L1 expression detected by immunostaining in a series of 196 RCC tumors with fresh frozen tissue available. These analyses demonstrated high PD-L1 expression by tumor cells (>10%), PD-L1 expression on infiltrating lymphocytes (>50%), or the composite of both parameters was strongly associated with cancer progression and death from RCC. The unadjusted RR for cancer-specific death in the 44% of tumors scored PD-L1 positive was 4.53 (P<0.001). More recently, tumor cell expression of PD-L1 has been addressed in non-clear cell RCC histologies. Using a threshold of ≥5% of tumor cells stained positive with mAb 405.9A11, PD-L1 expression was observed in two of 36 (5%) chromophobe tumors, five of 50 (10%) papillary tumors, and three of seven (43%) translocation tumors.49
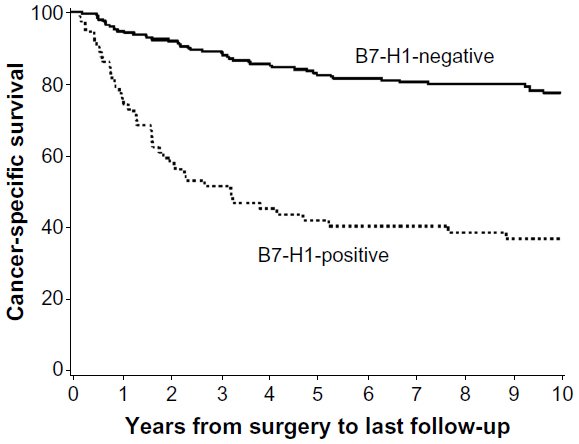 | Figure 2 Programmed cell death-ligand 1 (PD-L1; or B7-H1) expression on clear cell renal cell carcinoma tumor is associated with cancer-specific death. |
PD-1 expression by tumor-infiltrating lymphocytes has also been analyzed in both fresh-frozen or formalin-fixed paraffin-embedded clear cell RCC tissue. High PD-1 expression was associated with distant metastatic relapse and inferior relapse-free and overall survival versus the PD-1-negative patient subset.50,51
As observed in other tumor histologies, increased numbers of (FOXP3+CD4+CD25high) Tregs are seen in peripheral blood and TIL populations in RCC patients and also are associated with adverse patient outcomes.52 In addition, high numbers of intratumoral Tregs have been associated with poor survival for RCC patients treated with low- or intermediate-dose IL-2.53
Clinical outcomes with PD-1 pathway-blocking antibodies in metastatic RCC
The recognition of advanced RCC as a tumor responsive to immunotherapy, together with evidence for a PD-1/PD-L1-mediated immune escape mechanism operating in RCC tumors, has provided a compelling rationale for inclusion of patients with advanced RCC in early clinical studies of blocking antibodies specific for PD-1 or PD-L1. Results of published and reported studies of PD-1- or PD-L1-targeted therapies enrolling patients with advanced RCC are discussed in the following sections, focusing on key safety, toxicity, and efficacy end points.
Anti-PD-1
Nivolumab
Nivolumab (BMS-936558, MDX-1106, ONO-4538. [Bristol-Myers Squibb; New York, NY, USA]) is a fully human IgG4 mAb that binds human PD-1 with high affinity and blocks PD-1 binding to its known ligands PD-L1 and PD-L2. In a first-in-human dose escalation phase I study, 39 patients with five different tumor histologies, including one patient with clear cell RCC, received nivolumab on an intermittent dosing regimen (Table 2).54 The RCC patient received three doses of nivolumab at 10 mg/kg over the first 5 months on the study with a resulting partial response (PR). The patient had ongoing tumor regression and was judged to be in CR 2.6 years after the start of therapy, which continued to the last reported follow-up at a time point 4 years from the final treatment dose of nivolumab.55
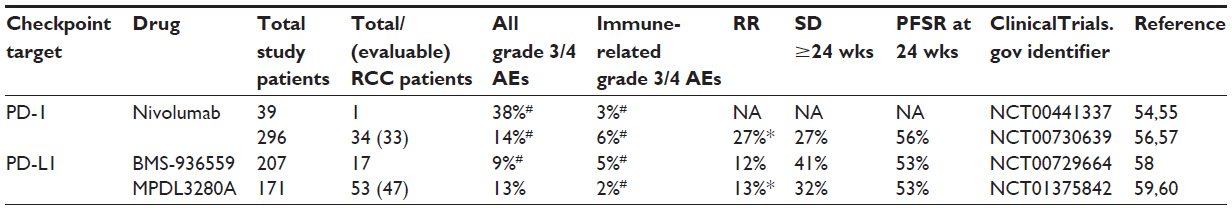 | Table 2 Safety and efficacy data from pilot studies with blocking monoclonal antibodies targeting PD-1 and PD-L1 |
In a second phase I trial with nivolumab, biweekly administration was investigated in a dose escalation format in 296 patients with clear cell RCC (N=34), melanoma (N=104), non-small cell lung cancer (N=122), castration-resistant prostate cancer (N=17), or colorectal cancer (N=19) at doses of 0.1 mg/kg, 0.3 mg/kg, 1.0 mg/kg, 3.0 mg/kg, or 10.0 mg/kg. A maximum tolerated dose (MTD) was not reached. For all enrolled patients, grade 3/4 treatment-related adverse events were observed in 41 of 296 patients (14%; Table 2). Grade 3/4 treatment-related adverse events of special interest reflecting a potential immune-mediated causality occurred in 18 of 296 patients (6%; Table 2) and included pneumonitis, vitiligo, colitis, hepatitis, hypophysitis, and thyroiditis. There were three drug-related deaths (1%) due to pneumonitis; however, none in the group of treated RCC patients.56 Within a cohort of 236 patients evaluable for treatment efficacy, 33 patients had metastatic RCC and were treated with 1.0 mg/kg (N=17) or 10.0 mg/kg (N=16) dose levels of nivolumab. Objective responses measured by Response Evaluation Criteria In Solid Tumors (RECIST 1.0) criteria57 were observed in four of 17 patients (24%) treated at the 1.0 mg/kg dose level and five of 16 patients (31%) treated at the 10.0 mg/kg dose level for an overall response rate of 27% (Table 2). Of ten responding patients with follow-up >1 year, five of the ten (50%) maintained an ongoing response for ≥1 year. The median duration of response was 12.9 months for both doses.56 An additional nine patients had stable disease for ≥24 weeks (27%). The progression-free survival rate at 24 weeks for the entire RCC cohort of 33 patients was 56% (Table 2). This study also observed objective responses for tumor types other than RCC, including melanoma (28%) and non-small cell lung cancer (18%).58
A randomized, blinded, dose-ranging phase II study with nivolumab in advanced clear cell RCC (NCT01354431) completed enrollment in the third quarter of 2011. Eligible patients had received prior treatment with at least one antiangiogenic drug in the metastatic setting and not more than three prior lines of therapy in total. Patients were randomized to one of three treatment arms receiving 0.3 mg/kg, 2.0 mg/kg, or 10.0 mg/kg nivolumab on an every-3-week schedule until disease progression or intolerable toxicity. Results from this trial have not yet been reported.
A phase III randomized trial of nivolumab versus everolimus in patients with advanced clear cell RCC (NCT01668784) recently completed enrollment of 822 patients in February 2014. Eligible patients must have received one or two prior antiangiogenic regimens and not more than three prior lines of therapy. Patients were randomized 1:1 to receive everolimus versus nivolumab at 3 mg/kg every 2 weeks until disease progression or intolerable toxicity. If a positive outcome is observed in this pivotal phase III study, a development timeline leading to FDA approval of nivolumab in 2015 is anticipated.
Anti-PD-L1
BMS-936559
BMS-936559 (MDX-1105; Bristol-Myers Squibb) is a fully human immunoglobulin G4 (IgG4) mAb that binds human PD-L1 with high affinity and blocks PD-L1 binding to both PD-1 and B7.1. In a first-in-human phase I trial with BMS-936559, biweekly administration was investigated in a dose escalation format in 207 patients with clear cell RCC (N=17), non-small cell lung cancer (N=75), melanoma (N=55), colorectal cancer (N=18), ovarian cancer (N=17), pancreatic cancer (N=14), gastric cancer (N=7), or breast cancer (N=4) at doses of 0.1 mg/kg, 0.3 mg/kg, 1.0 mg/kg, 3.0 mg/kg, or 10.0 mg/kg. As for nivolumab, an MTD for the mAb was not reached. For all enrolled patients, grade 3/4 treatment-related adverse events were observed in 19 of 207 patients (9%; Table 2). Grade 3/4 treatment-related adverse events of special interest reflecting a potential immune-mediated causality occurred in ten of 207 patients (5%; Table 2) and included adrenal insufficiency, hepatitis, pulmonary sarcoidosis, diabetes mellitus, myasthenia gravis, and endophthalmitis. There were no drug-related deaths. Within a cohort of 160 patients evaluable for treatment efficacy, 17 patients had metastatic RCC and were treated with 10.0 mg/kg BMS-936559. Objective responses measured by RECIST 1.0 criteria were observed in two of 17 patients (12%; Table 2) lasting 4 months and 17 months. An additional seven patients had stable disease for ≥24 weeks (41%). The progression-free survival rate at 24 weeks for the RCC cohort of 17 patients was 53% (Table 2). In addition to RCC, objective responses were also observed in patients with melanoma (17%), non-small cell lung cancer (10%), and ovarian cancer (6%).59
MPDL3280A
MPDL3280A (Genetech; South San Francisco, CA, USA) is a human IgG1 mAb with an engineered fragment crystallizable (Fc) domain designed to inhibit antibody-dependent cell-mediated cytotoxicity, thereby avoiding a cytotoxic effect against activated T-cells expressing PD-L1. MPDL3280A blocks PD-L1 binding to both PD-1 and B7.1. In a first-in-human phase I trial, MPDL3280A was administered every 3 weeks in a dose escalation format to 171 patients with advanced solid tumors that included RCC (N=55), non-small cell lung cancer (N=52), and melanoma (N=44) and 20 additional patients with eleven different tumor histologies.60 Dosing of MPDL3280A spanned a range from 0.01 mg/kg to 20 mg/kg with 162/171 patients treated with ≥3 mg/kg. An MTD was not reached. For all enrolled patients, grade 3/4 treatment-related adverse events were observed in 22 of 171 patients (13%; Table 2). Grade 3/4 immune-related adverse events occurred in four patients (2%; Table 2) and included hepatitis, colitis, and hyperglycemia. There were no drug-related deaths. There were 47 RCC patients within a cohort of 140 patients evaluable for efficacy. Objective responses measured by RECIST 1.161 criteria were observed in six patients (13%, Table 2). Enrollment allowed for both clear cell and non-clear cell RCC histologies, and there was one responder from the non-clear cell cohort (one of six non-clear cell tumors). An additional 32% of RCC patients had stable disease for ≥24 weeks, and the progression-free survival rate at 24 weeks for the RCC cohort was 53% (Table 2). In addition to RCC, objective responses were observed in patients with melanoma (29%), non-small cell lung cancer (22%), and individual patients with colorectal or gastric carcinoma.62
On target activity of PD-1 pathway-specific agents
The heterogeneity observed between RCC tumors and other tumor histologies for PD-L1 expression measured by immunohistochemistry analyses suggested that pretreatment PD-L1 expression by tumors might represent a useful marker predicting response to PD-1 or PD-L1-specific blocking mAb therapy. This association has been evaluated as part of the phase I studies of both the anti-PD-1 antibody nivolumab as well as the anti-PD-L1 antibody MPDL3280A. As part of the phase I testing of nivolumab, pretreatment tumor tissue for 42 patients was available for analysis by immunohistochemistry for intratumoral expression of PD-L1 (using the 5H1 mAb). For all histologies, pretreatment tumor was positive for PD-L1 expression (defined as cell surface staining on ≥5% of tumor cells) in 25 of 42 tumors (60%). There were nine objective responses associated with the 25 PD-L1-positive tumors (36% ORR). Conversely, none of the 17 patients with PD-L1-negative tumors had an objective response (P=0.006). Five RCC patients were included in the analysis of intratumoral PD-L1 expression. Four of five RCC tumors were positive for PD-L1 expression (80%), and two of the four PD-L1-positive RCC tumors were associated with objective responses (one CR, one PR). The patient with the PD-L1-negative tumor was a nonresponder.58
The association of pretreatment tumor expression of PD-L1 was also evaluated as part of phase I testing of the PD-L1-specific mAb MPDL3280A. A proprietary immunohistochemistry assay was developed by the study sponsor (Genetech/Roche), which defined PD-L1+ based on a positive PD-L1 stain on infiltrating immune cells. For the RCC cohort, tumor tissue from 31 of 47 patients was available for analysis and showed a PD-L1+ phenotype for ten of 31 (32%) tumors. There were two objective responses in the PD-L1+ cohort (one CR, one PR; 20% ORR) and six additional patients had stable disease (60%). In the PD-L1− cohort, there were two responding patients with PR (10%) and eleven of 21 patients (52%) had stable disease.61 This study also conducted expression profiling of immune-related genes from 96 of 171 pretreatment tumor samples showing an association of T-cell-related genes, including interferon-γ, CD8α, granzyme-A, and EOMES, with tumor response.63 These observations were consistent with previous gene expression profiling studies of melanoma tumors associating an inflammatory tumor microenvironment phenotype with a more favorable response to subsequent immunotherapy.64
A trend favoring a higher response rate for PD-L1-expressing tumors treated with anti-PD-1 or anti-PD-L1 has been seen for RCC and for other tumor histologies sensitive to PD-1/PD-L1 blockade, including melanoma.65 However, observations that include responses of PD-L1-negative tumors,61 the potential inducibility of PD-L1 expression within tumors,63 intratumoral heterogeneity of PD-L1 expression, and discordance in PD-L1 expression between primary tumor and metastases66 create considerable uncertainty about the ultimate role that PD-L1 phenotype may play in guiding patient selection for anti-PD-1/PD-L1 therapies. Standardization of reagents and scoring cut points for PD-L1 expression, along with substantially larger datasets associating PD-L1 phenotype for RCC tumors with treatment outcomes as part of phase II and phase III studies with nivolumab and other agents in clinical development, are anticipated to provide greater insight into the role pretreatment PD-L1 tumor phenotype may serve as a clinically useful biomarker.
The antitumor activity mediated by PD-1 or PD-L1 blockade is presumed to represent augmented tumor-reactive T-cell immunity. Anecdotal observations from paired pre- and post-treatment biopsies of tumor lesions regressing following PD-1 or PD-L1 blockade reveal increased numbers of infiltrating CD8+ T-cells and associated upregulation of PD-L1 expression in the tumor microenvironment.54,63,67 In vitro studies have also demonstrated augmented expansion and enhanced effector function of tumor-antigen specific T-cells stimulated in the presence of blocking antibodies targeting PD-1 or PD-L1.68,69
Future perspectives
The initial response rates observed with nivolumab and PD-L1-blocking antibodies in heavily pretreated RCC and other tumor types have generated considerable enthusiasm for the further development of PD-1 pathway-targeted agents. However, unresolved questions remain regarding clinical outcomes for PD-1 pathway-specific agents in RCC. Outcomes from larger numbers of patients are needed to determine whether anti-PD-1 or anti-PD-L1 agents have the potential to result in complete remissions in a subset of RCC patients. In addition, it remains uncertain whether tumor responses can be maintained if active treatment with PD-1 or PD-L1-specific antibody is suspended. Insight into the durability of anti-PD-1 and anti-PD-L1-mediated responses may be forthcoming from longer follow-up of RCC patients treated on phase I studies of nivolumab, BMS-936559, or MPDL3280A. Each of these trials allowed a maximum duration of initial therapy of 1 year (MPDL3280A) or 2 years (nivolumab, BMS-936559).58–60 Therefore, patients without intolerable treatment-associated side effects and with ongoing stable disease or partial remissions who received the maximum duration of therapy will then transition to surveillance to detect new disease progression. Finally, the signaling interactions blocked by PD-1 versus PD-L1 mAbs are nonidentical. However, substantially more data will be needed to determine whether the relative activity or toxicity of blocking PD-1 versus PD-L1 is meaningfully different. Thus far, results from initial studies, including the range of tumor types responding to these agents and the disease control rate represented by progression-free survival rate at 24 weeks, reflect a high degree of similarity.
Rapid progress is being made in the clinical development of PD-1 and PD-L1-blocking mAbs as monotherapy for advanced RCC, culminating in a phase III trial with the lead agent in development, nivolumab, which has completed enrollment. The early phase clinical development of PD-1- and PD-L1-targeted compounds is now focusing on combination approaches in a search for enhanced antitumor activity with acceptable toxicity (Table 3). Recent intriguing observations regarding the antigen targets recognized by tumor-specific CD8+ T-cells in melanoma patients is worth comment, and could influence the future direction of development of PD-1 pathway-targeted therapies. Using next-generation exome sequencing coupled with epitope prediction algorithms, CD8+cells specific for mutated tumor proteins were identified within a TIL sample collected from a melanoma patient responding to ipilimumab and within the in vitro expanded TIL products from three melanoma patients responding to an adoptive cellular therapy treatment protocol.70,71 These observations have fueled speculation that mutation-specific T-cells could represent key elements of effective antitumor immunity. Although the contribution of mutation-specific cytotoxic T-lymphocytes (CTLs) in RCC patients remains conjecture at this point, it is interesting to note that seminal work on RCC-associated tumor antigen identification previously discovered tumor-specific mutations as the antigen targets for RCC-specific CD8+ CTL clones.72,73 Strategies intending to capture and expand mutation-specific T-cell responses unique to individual tumors in combination with PD-1 pathway-targeted therapy could include the use of cellular therapy products expanded from TIL or tumor vaccines that incorporate autologous tumor into the immunogen – in preference to vaccines targeting broadly shared, nonmutated antigens. Such a study combining the anti-PD-1 mAb CT-011 with an autologous tumor/dendritic cell fusion vaccine is ongoing (Table 3).
A large (and expanding) array of receptors known to modulate T-cell function extends well beyond CTLA-4 and PD-1, suggesting a great potential for combination therapy targeting multiple immune checkpoint receptors to result in additive antitumor activity.9,20 Indeed, the combination of ipilimumab and nivolumab has already been evaluated in a pilot study in patients with melanoma. Preliminary observations for this combination suggest a higher response rate and deeper tumor regressions than for either antibody as monotherapy.74 Combinations of checkpoint-blocking mAbs targeting PD-1 or PD-L1 along with CTLA-4, blockade of PD-1 plus LAG-3 (another inhibitory receptor expressed on activated CD4+ and CD8+ T-cells),9 or dual blockade of both PD-1 with PD-L1 are currently undergoing evaluation in early phase studies for RCC and other tumors (Table 3). Further development of this general approach is limited primarily by the availability of clinical grade reagents targeting human checkpoint receptors or their ligands.
Trials of PD-1- and PD-L1-blocking mAbs are also moving forward in combination with established therapies for advanced RCC. Studies investigating anti-PD-1 in combination with cytokine immunotherapies IFN-α or HD IL-2 are underway or in development. Preclinical studies associating therapies targeting the VEGF signaling pathway with enhanced antitumor T-cell immunity75–77 have created substantial interest in the potential for combining immune checkpoint-blocking agents with antiangiogenic therapies. Despite unanticipated renal toxicity observed with the combination of the anti-CTLA-4 mAb tremelimumab plus sunitinib,78 pilot studies of anti-PD-1 mAbs with sunitinib or pazopanib and the pairing or MPDL3280A with bevacizumab are moving forward (Table 3).
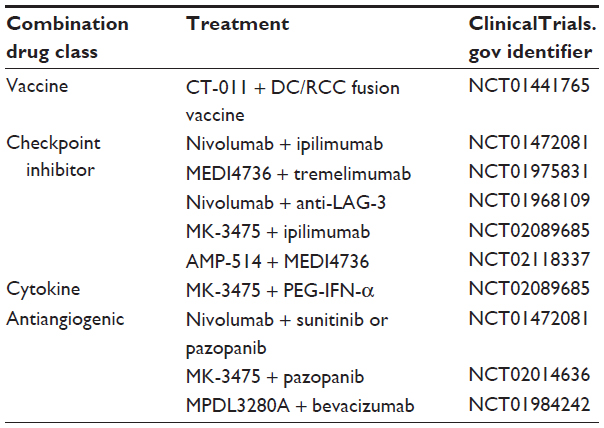 | Table 3 Selected combination trials for renal cell carcinoma incorporating blocking monoclonal antibodies targeting PD-1 or PD-L1 |
Conclusion
The cytokine era of immunotherapy for advanced RCC was followed by a decade of clinical research dominated by antiangiogenic therapies targeting the VEGF and mammalian target of rapamycin signaling pathways, establishing targeted therapies as the current standard of care for most patients. Emerging data with blocking antibodies targeting PD-1 or PD-L1, demonstrating spontaneous and durable regressions for a subset of treatment-refractory RCC tumors, suggest that the PD-1 pathway represents a dominant control point for the regulation of tumor-reactive T-cell responses. The compelling early phase clinical data appear certain to usher in a new era of clinical research for RCC dominated by intense clinical study of PD-1- and PD-L1-blocking agents. Fierce competition between available agents to establish efficacy in treatment-refractory RCC patients, as first-line therapy, and as part of novel combination therapy is envisaged.
Disclosure
The author has received funding for clinical trials on behalf of his institution from Bristol-Myers Squibb. The author reports no other conflicts of interest in this work.
References
Papac RJ. Spontaneous regression of cancer. Cancer Treat Rev. 1996; 22(6):395–423. | |
Nagorsen D, Scheibenbogen C, Marincola FM, Letsch A, Keilholz U. Natural T cell immunity against cancer. Clin Cancer Res. 2003;9(12):4296–4303. | |
Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. | |
Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8(4):299–308. | |
Amato RJ, Hawkins RE, Kaufman HL, et al. Vaccination of metastatic renal cancer patients with MVA–5T4: a randomized, double-blind, placebo-controlled phase III study. Clin Cancer Res. 2010;16(22):5539–5547. | |
Figlin RA, Thompson JA, Bukowski RM, et al. Multicenter, randomized, phase III trial of CD8(+) tumor-infiltrating lymphocytes in combination with recombinant interleukin–2 in metastatic renal cell carcinoma. J Clin Oncol. 1999;17(8):2521–2529. | |
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–915. | |
Yang JC, Childs R. Immunotherapy for renal cell cancer. J Clin Oncol. 2006;24(35):5576–5583. | |
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | |
Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med. 2012;366(26):2517–2519. | |
Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270(5238):985–988. | |
Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–547. | |
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. | |
van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190(3):355–366. | |
Phan GQ, Yang JC, Sherry RM, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003;100(14):8372–8377. | |
Ribas A, Camacho LH, Lopez-Berestein G, et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol. 2005;23(35):8968–8977. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | |
Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase ii and phase III trials of ipilimumab in metastatic or locally advanced, unresectable melanoma. European J Ca. 2013;49(Suppl 3):abstr LBA24. | |
Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691–2697. | |
Odorizzi PM, Wherry EJ. Inhibitory receptors on lymphocytes: insights from infections. J Immunol. 2012;188(7):2957–2965. | |
Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy – inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res. 15 2012;18(24):6580–6587. | |
Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–88. | |
Yamamoto R, Nishikori M, Tashima M, et al. B7-H1 expression is regulated by MEK/ERK signaling pathway in anaplastic large cell lymphoma and Hodgkin lymphoma. Cancer Sci. 2009;100(11):2093–2100. | |
Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 1 2014;74(3):665–674. | |
Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra137. | |
Latchman YE, Liang SC, Wu Y, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101(29):10691–10696. | |
Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11(2):141–151. | |
Nishimura H, Okazaki T, Tanaka Y, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291(5502):319–322. | |
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99(19):12293–12297. | |
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | |
Lopez-Beltran A, Carrasco JC, Cheng L, Scarpelli M, Kirkali Z, Montironi R. 2009 update on the classification of renal epithelial tumors in adults. Int J Urol. 2009;16(5):432–443. | |
Nickerson ML, Jaeger E, Shi Y, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14(15):4726–4734. | |
Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–134. | |
Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115–124. | |
Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28(6):1061–1068. | |
Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370(9605):2103–2111. | |
Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356(22):2271–2281. | |
Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372(9637):449–456. | |
Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011;378(9807):1931–1939. | |
Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet. 1999;353(9146):14–17. | |
Pyrhonen S, Salminen E, Ruutu M, et al. Prospective randomized trial of interferon alfa-2a plus vinblastine versus vinblastine alone in patients with advanced renal cell cancer. J Clin Oncol. 1999;17(9):2859–2867. | |
Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet. 2009;373(9669):1119–1132. | |
Coppin C, Porzsolt F, Awa A, Kumpf J, Coldman A, Wilt T. Immunotherapy for advanced renal cell carcinoma. Cochrane Database Syst Rev. 2005(1):CD001425. | |
Rini BI, Halabi S, Rosenberg JE, et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J Clin Oncol. 2008;26(33):5422–5428. | |
Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, Louie AC. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. 1995;13(3):688–696. | |
Klapper JA, Downey SG, Smith FO, et al. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma : a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer. 2008;113(2):293–301. | |
Thompson RH, Kuntz SM, Leibovich BC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 2006;66(7):3381–3385. | |
Thompson RH, Gillett MD, Cheville JC, et al. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci U S A. 2004;101(49):17174–17179. | |
Fay AP, Callea M, Gray KP, et al. PD-L1 expression in non-clear cell renal cell carcinoma. J Clin Oncol. 2014;32(Suppl 4):424. | |
Thompson RH, Dong H, Lohse CM, et al. PD-1 is expressed by tumor-infiltrating immune cells and is associated with poor outcome for patients with renal cell carcinoma. Clin Cancer Res. 2007;13(6):1757–1761. | |
Kang MJ, Kim KM, Bae JS, et al. Tumor-infiltrating PD1-positive lymphocytes and FoxP3-positive regulatory T cells predict distant metastatic relapse and survival of clear cell renal cell carcinoma. Transl Oncol. 2013;6(3):282–289. | |
Griffiths RW, Elkord E, Gilham DE, et al. Frequency of regulatory T cells in renal cell carcinoma patients and investigation of correlation with survival. Cancer Immunol Immunother. 2007;56(11):1743–1753. | |
Jensen HK, Donskov F, Nordsmark M, Marcussen N, von der Maase H. Increased intratumoral FOXP3-positive regulatory immune cells during interleukin-2 treatment in metastatic renal cell carcinoma. Clin Cancer Res. 2009;15(3):1052–1058. | |
Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. | |
Lipson EJ, Sharfman WH, Drake CG, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013;19(2):462–468. | |
Drake CG, McDermott DF, Sznol M, et al. Survival, safety, and response duration results of nivolumab (Anti-PD-1; BMS-936558; ONO-4538) in a phase I trial in patients with previoulsy treated metastatic renal cell carcinoma 9mRCC): long-term patient follow-up. J Clin Oncol. 2013;31(Suppl):4514. | |
Therasse, P, SG Arbuck, EA Eisenhauer et al. New Guidelines to Evaluate the Response to Treatment in Solid Tumors. J Natl Cancer Inst. 2000;92(3):205–216. | |
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. | |
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. | |
Herbst RS, Gordon MS, Fine GD, et al. A study of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic tumors. J Clin Oncol. 2013;31(Suppl):3000. | |
Eisenhauer, EA, P Therasse, J Bogaerts et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45(2):228–247. | |
Cho DC, Sosman JA, Sznol M, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with metastatic renal cell carcinoma (mRCC). J Clin Oncol. 2013;31(Suppl):4505. | |
Powderly JD, Koeppen H, Hodi FS, et al. Biomarkers and associations with the clinical activity of PD-L1 blockade in a MPDL3280A study. J Clin Oncol. 2013;31(Suppl):3001. | |
Gajewski TF, Fuertes M, Spaapen R, Zheng Y, Kline J. Molecular profiling to identify relevant immune resistance mechanisms in the tumor microenvironment. Curr Opin Immunol. 2011;23(2):286–292. | |
Grosso J, Horak CE, Inzunza D, et al. Association of tumor PD-L1 expression and immune biomarkers with clinical activity in patients (pts) with advanced solid tumors treated with nivolumab (anti-PD-1; BMS-936558; ONO-4538). J Clin Oncol. 2013;31(Suppl):3016. | |
Callea M, Genega EM, Gupta M, et al. PD-L1 expression in primary clear cell renal cell carcinomas (ccRCC) and their metastases. J Clin Oncol. 2014;32(Suppl 4):467. | |
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. | |
Blank C, Kuball J, Voelkl S, et al. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int J Cancer. 2006;119(2):317–327. | |
Wong RM, Scotland RR, Lau RL, et al. Programmed death-1 blockade enhances expansion and functional capacity of human melanoma antigen-specific CTLs. Int Immunol. 2007;19(10):1223–1234. | |
van Rooij N, van Buuren MM, Philips D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31(32):e439–e442. | |
Robbins PF, Lu YC, El-Gamil M, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19(6):747–752. | |
Brandle D, Brasseur F, Weynants P, Boon T, Van den Eynde B. A mutated HLA-A2 molecule recognized by autologous cytotoxic T lymphocytes on a human renal cell carcinoma. J Exp Med. 1996;183(6):2501–2508. | |
Gaudin C, Kremer F, Angevin E, Scott V, Triebel F. A hsp70-2 mutation recognized by CTL on a human renal cell carcinoma. J Immunol. 1999;162(3):1730–1738. | |
Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. | |
Jinushi M, Dranoff G. Triggering tumor immunity through angiogenesis targeting. Clin Cancer Res. 2007;13(13):3762–3764. | |
Finke JH, Rini B, Ireland J, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14(20):6674–6682. | |
Ko JS, Zea AH, Rini BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–2157. | |
Rini BI, Stein M, Shannon P, et al. Phase 1 dose-escalation trial of tremelimumab plus sunitinib in patients with metastatic renal cell carcinoma. Cancer. 2011;117(4):758–767. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.