Back to Journals » OncoTargets and Therapy » Volume 10
Patients harboring ALK rearrangement adenocarcinoma after acquired resistance to crizotinib and transformation to small-cell lung cancer: a case report
Authors Zhu Y, Liao X, Wang W ![]() , Xu C
, Xu C ![]() , Zhuang W, Zhong L, Du K, Chen Y, Chen G, Fang M
, Zhuang W, Zhong L, Du K, Chen Y, Chen G, Fang M
Received 15 April 2017
Accepted for publication 6 June 2017
Published 27 June 2017 Volume 2017:10 Pages 3187—3192
DOI https://doi.org/10.2147/OTT.S139718
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ingrid Espinoza
You-cai Zhu,1 Xing-hui Liao,2 Wen-xian Wang,3 Chun-wei Xu,4 Wu Zhuang,5 Li-hua Zhong,4 Kai-qi Du,1 Yan-ping Chen,4 Gang Chen,4 Mei-yu Fang6
1Department of Chest Disease Diagnosis and Treatment Center, 2Department of Tumor Molecular Laboratory, Zhejiang Rongjun Hospital, Jiaxing, Zhejiang, 3Department of Chemotherapy, Zhejiang Cancer Hospital, Hangzhou, Zhejiang, 4Department of Pathology, Fujian Provincial Cancer Hospital, 5Department of Medical Thoracic Oncology, Fujian Provincial Cancer Hospital, Fujian Medical University Cancer Hospital, Fujian, Fuzhou, 6Department of Comprehensive Medical Oncology, Zhejiang Cancer Hospital, Hangzhou, Zhejiang, People’s Republic of China
Abstract: Anaplastic lymphoma kinase (ALK) rearrangement responds to ALK tyrosine kinase inhibitors (TKIs) in lung cancer. Many cases ultimately acquire resistance to crizotinib. Resistance, including ALK-dominant or ALK non-dominant, mechanisms have been described. Transformation to small-cell lung cancer is rare. Herein, we report a 49-year-old man diagnosed with adenocarcinoma, who was negative for EGFR and ALK genes as detected by reverse transcription polymerase chain reaction, and was treated with crizotinib. A new biopsy showed a small-cell lung cancer after disease progression. Then, next-generation sequencing (NGS) was carried out and detected a TP53 gene mutation, an ALK rearrangement, and no loss of the retinoblastoma gene (RB). Although a regimen for small-cell lung cancer may be one treatment option, a heterogeneous tumor may exist at the time of diagnosis and manifest during the course of disease.
Keywords: lung cancer, ALK, crizotinib, small-cell lung cancer
Introduction
Anaplastic lymphoma kinase (ALK) and echinoderm microtubule-associated protein-like 4 (EML4) gene rearrangements occur in 2%–7% of patients with non-small cell lung cancer (NSCLC).1 Crizotinib is the standard treatment for advanced ALK-positive NSCLC and has been shown to have impressive single-agent activity in this type of patient.2,3 While most patients respond to crizotinib, every patient will ultimately experience disease progression within 1–2 years.2 Drug resistance has not only become the major limitation of clinical efforts, but also is the most urgent issue in need of resolution for prolonging life in patients with NSCLC. It is encouraging that molecular acquired resistance mechanisms to tyrosine kinase inhibitors (TKIs) have been identified. With in-depth research, new therapeutic strategies to overcome resistance have been used to prolong survival of patients with advanced NSCLC. Mechanisms of acquired resistance to crizotinib that have been explored include on-target genetic alterations or off-target mechanisms of resistance;4 however, other complex resistance mechanisms, such as mechanisms of resistance to EGFR-TKIs, still exist.5 Histologic transformation into small-cell lung cancer (SCLC) has been reported in some ALK fusion adenocarcinomas; however, there are no established treatment strategies to manage such transformations.
Here, we report a patient who developed acquired resistance to crizotinib with transformation to SCLC. Moreover, we have summarized some published reports which have improved our understanding of the transformation to SCLC in ALK fusion adenocarcinoma.
Case report
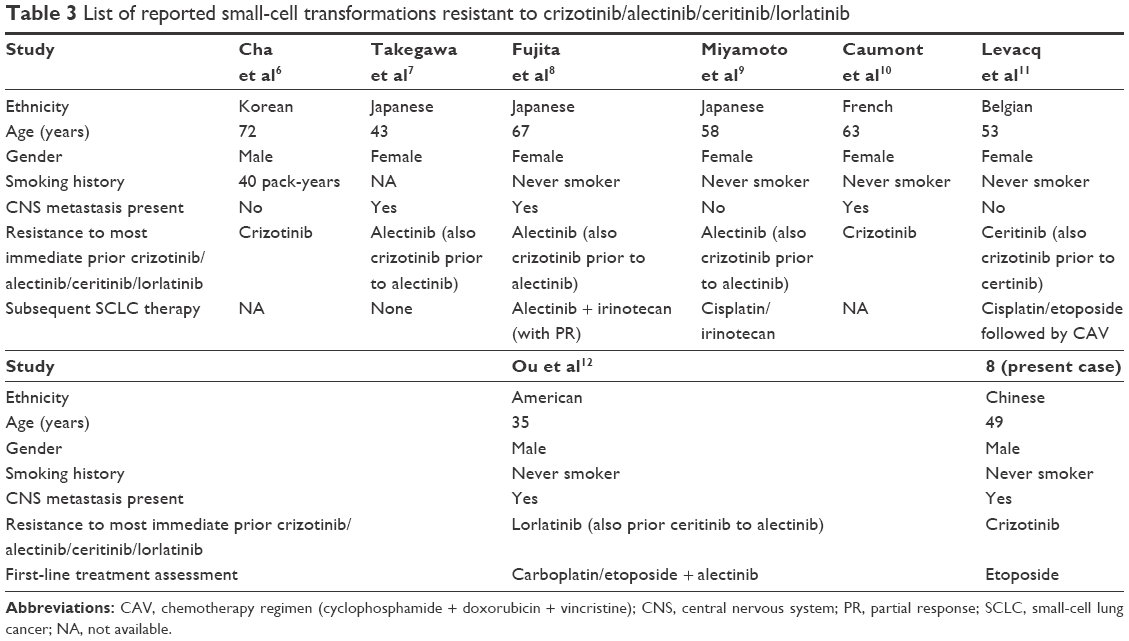
A 49-year-old man, a never smoker, presented to our hospital with a 1-month history of cough and blood-stained sputum in October 2012. CT scans revealed a mass in the right upper lung, right hilar lymph node enlargement, and multiple nodules in the right lung. Cerebral and lumbar magnetic resonance imaging (MRI) revealed a nodule in the right cerebellum and a centrum tumor at the L2 level (T4N1M1b stage IV). A pathologic diagnosis of adenocarcinoma cells was established based on a bronchoscopic biopsy (Figure 1). Immunohistochemical (IHC) analysis demonstrated positivity for TTF-1 and Napsin A, and negativity for cytokeratin (CK) 5/6 and P63 (Figure 1 and Table 1). Tumor tissue was shown to be wild-type of epidermal growth factor receptor variants by ARMS (AmoyDx, Xiamen, People’s Republic of China), and EML4-ALK fusion was shown by reverse transcription polymerase chain reaction (RT-PCR; AmoyDx; Figure 2). The patient received five cycles of first-line chemotherapy with cisplatin and paclitaxel from October 2012 to January 2013. The best tumor response was a partial response according to RECIST criteria, and the progression-free survival (PFS) was 7.0 months. In May 2013, the tumor progressed (right lower lobe nodules and brain metastases). The patient underwent crizotinib treatment (250 mg/bid, orally) from May 2013 to October 2014. He received whole-brain radiotherapy (2 Gy per fraction, 1 fraction per day ×20 days; total radiation dose 50 Gy). The curative effect of crizotinib treatment was stable disease (SD). After progression on crizotinib, the patient underwent multiple cycles of cytotoxic chemotherapy (gemcitabine, docetaxel, and bevacizumab; Table 2). A secondary biopsy of the enlarging mass in the right lung was performed. Histologic examination of the biopsy specimen revealed SCLC without adenocarcinoma components (Figure 3). IHC analysis demonstrated positivity for Syn and CD56, negativity for CgA, and a Ki-67 index of 98% (Figure 3). To search for new therapeutic strategies, additional gene detection was performed on the tissue sample by next-generation sequencing (Gene plus, Beijing, People’s Republic of China), which showed a TP53 gene mutation (p.R248W) and ALK rearrangement accompanied by ERBB3 p.P554Q, NOTCH1 p.P3S, IL7R p.P417R, and IL6ST p.A896V, but no loss of the retinoblastoma gene (RB). The next generation sequencing (NGS) assay was performed using the HiSEquation 4000 (Illumina, San Diego, CA, USA). Then, the patient underwent etoposide monotherapy. After two cycles, the effect was SD. The patient remains alive at the time of writing this article. We have listed small-cell transformations following treatment with ALK-TKIs, including crizotinib/alectinib/ceritinib/lorlatinib in Table 3. The authors confirm that written informed consent for publication of case details and any accompanying images has been provided by the patient.
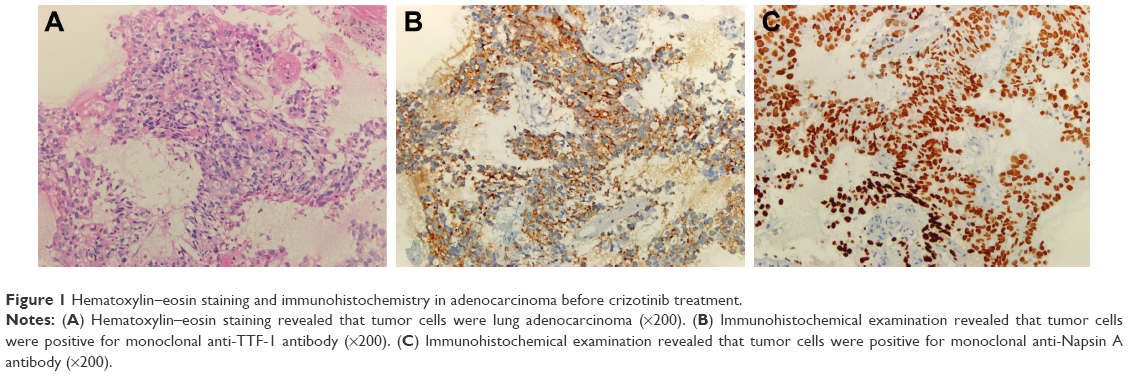 | Figure 1 Hematoxylin–eosin staining and immunohistochemistry in adenocarcinoma before crizotinib treatment. |
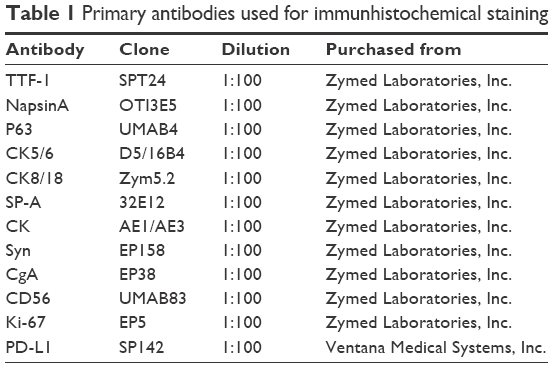 | Table 1 Primary antibodies used for immunhistochemical staining |
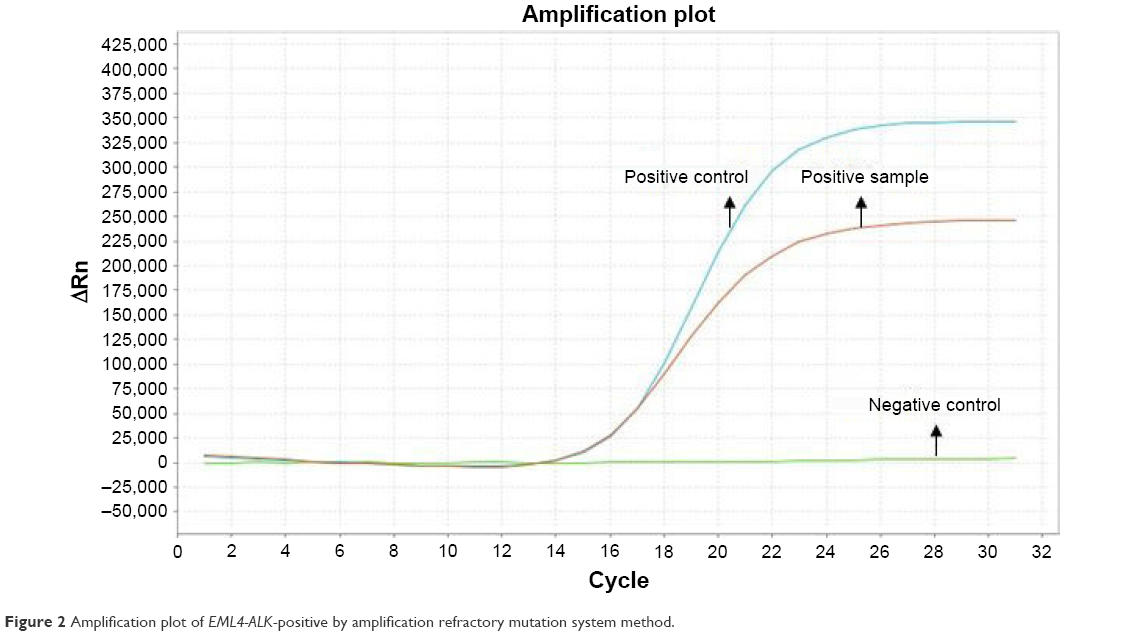 | Figure 2 Amplification plot of EML4-ALK-positive by amplification refractory mutation system method. |
 | Table 2 Details of the treatment process |
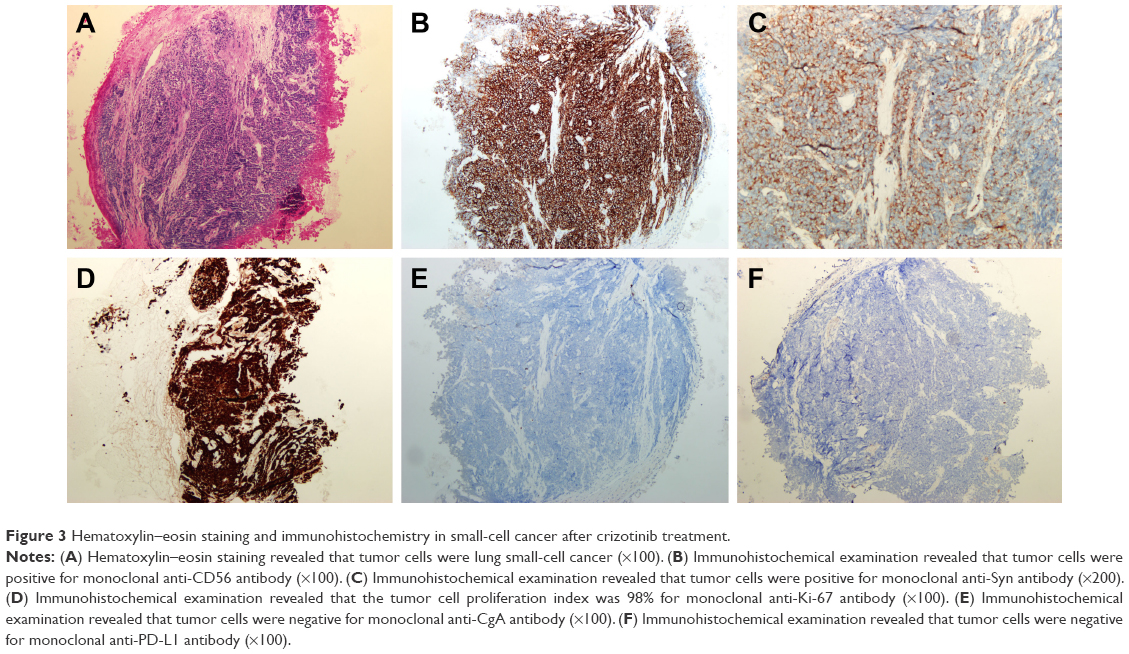 | Figure 3 Hematoxylin–eosin staining and immunohistochemistry in small-cell cancer after crizotinib treatment. |
 | Table 3 List of reported small-cell transformations resistant to crizotinib/alectinib/ceritinib/lorlatinib |
Discussion
With the rapid development of molecular diagnostic technology, targeted therapy is effective for patients with advanced NSCLC and associated driver genes; however, a majority of patients will eventually acquire resistance to the targeted drug and experience disease progression. Therefore, some ALK-positive patients also develop acquired resistance to ALK-TKIs. Known acquired mechanisms that cause resistance to ALK-TKIs have been described, and can be divided into two types (ALK dominant and ALK non-dominant).13 ALK-dominant mechanisms include ALK secondary mutations and ALK amplification, whereas ALK non-dominant mechanisms include bypassing downstream signaling, such as the epidermal growth factor receptor (EGFR), KRAS, KIT, MET, and insulin-like growth factor 1 receptor pathways. Approximately 25% of acquired mechanisms of resistance are mediated by unknown mechanisms.14 With respect to EGFR-TKIs, 3%–15% of the patients with EGFR-mutated lung cancer develop acquired resistance to EGFR-TKIs by histologic transformation to SCLC.15,16 Similarly, a mechanism underlying SCLC transformation in ALK-positive tumors has been reported in some cases.6–12
In the present case in which the re-biopsied SCLC tissue was examined, ALK rearrangement was still detected. In addition, chemotherapy and/or anti-angiogenic drug treatment-induced change in initial tumor morphology should, therefore, be considered. If the transformation had occurred before crizotinib treatment, a rapid growth of the primary lesion during crizotinib therapy would have been observed. Moreover, the primary lesion did not show an increase in size during albumin paclitaxel or docetaxel combination with bevacizumab therapy, and only limited agent activity on SCLC was reported with these agents.17 These findings imply that the transformation to SCLC during crizotinib treatment was the main cause of acquired resistance in this case. The pathophysiologic mechanism underlying transformation to SCLC following ALK-TKI treatment is not well understood. According to the mechanism of EGFR-TKIs, two possibilities have been stated, including a phenotype switch from NSCLC to SCLC, and SCLC and adenocarcinoma may coexist at baseline, with SCLC becoming dominant during disease progression after EGFR-TKI therapy.5,18 We thought the mechanism of transformation to SCLC in ALK-TKIs was similar to that with EGFR-TKIs. Although the pathologic features of the present case were re-evaluated, SCLC was not identified in the primary biopsy specimen. Because of the initial bronchial biopsy, there was limited availability of the tissue sample.
In contrast, although the cause of transformation into SCLC is unclear, inactivation of tumor suppressor genes (RB and TP53) seems important and constitutes an initial event in the tumorigenic process.16 Some reports have revealed the role of RB gene loss and/or TP53 mutation in ALK-positive NSCLC transformed into SCLC11,15,16; however, in our case, there was no loss of RB, but a TP53 gene mutation occurred. We reasoned that RB gene loss or TP53 gene mutation could participate in the transformation of adenocarcinoma to SCLC, but the two genes were not always concurrent as the transforming event. Furthermore, we demonstrated a NOTCH1 gene mutation, and this constituted the first reported gene involving transformation to SCLC. It has been reported that approximately 25% of patients with SCLC have a NOTCH1 gene family abnormality identified on next-generation sequencing.19 We hypothesize that, if inactivation of the RB1 or P53 or NOTCH1 gene is identified in the primary tumor tissue, the tumor may be at higher risk of SCLC transformation following target drug therapy.
The present case supports attempts to re-biopsy the tumor for selection of treatment after acquired resistance. There are no established treatment strategies to manage patients with transformation to SCLC. In agreement with reported cases, Fujita et al8 reported treatment with a combination of irinotecan and alectinib, resulting in an ongoing partial response to the primary lesion with a maintained partial response to the other lesions. Miyamoto et al9 showed that after two cycles of cisplatin–irinotecan treatment, there was a reduction in the size of mediastinal lymph nodes, whereas there was an increase in the size and number of nodes in both lung fields. Ou et al12 suggest that the patient switch to a third-generation ALK-TKI within 2 months of treatment and disease progression. The patient underwent etoposide monotherapy in our case. After two cycles, the effect was SD. Nevertheless, as the heterogeneity of response to different therapeutic strategies of the reported case suggests, the re-biopsy site only represents a partial pathology of the resistance, and the mechanism of resistance may differ from one site to another. Given the heterogeneity and the variety of acquired resistance mechanisms to target drugs, re-biopsy of only one site might not always be appropriate. We think detection of gene variations in blood samples with homogeneity in ctDNA by NGS may be a supplemental method with re-biopsy for analysis, in some cases. It is clinically challenging to choose the treatment mode when transformation to SCLC occurs. More research on optimal treatment methods are needed.
Conclusion
Oncologists should realize the possibility of transformation to SCLC after patients acquire resistance to ALK-TKI therapy. A re-biopsy should be performed to facilitate histologic and molecular analysis. Transformation to SCLC is an important consideration for choosing appropriate therapy due to the potential efficacy of standard SCLC treatments or a combination of next-generation AKL-TKIs.
Acknowledgment
This work was supported by the Zhejiang Province of China (2013KYB051), Zhejiang Administration of Traditional Chinese Medicine Foundation (2013ZQ005), Science and Technology Planning Project of Zhejiang Province (2015C33194), and the National Clinical Key Specialty Construction Program (2013).
Disclosure
The authors report no conflicts of interest in this work.
References
Ettinger DS, Wood DE, Akerley W, et al; National Comprehensive Cancer Network. Non-small cell lung cancer, version 6. 2015. J Natl Compr Canc Netw. 2015;13(5):515–524. | ||
Solomon BJ, Mok T, Kim DW, et al; PROFILE 1014 Investigators. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. | ||
Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. | ||
Matikas A, Kentepozidis N, Georgoulias V, Kotsakis A. Management of resistance to crizotinib in anaplastic lymphoma kinase-positive non-small-cell lung cancer. Clin Lung Cancer. 2016;17(6):474–482. | ||
Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. | ||
Cha YJ, Cho BC, Kim HR, Lee HJ, Shim HS. A case of ALK-rearranged adenocarcinoma with small cell carcinoma-like transformation and resistance to crizotinib. J Thorac Oncol. 2015;11(5):e55–e58. | ||
Takegawa N, Hayashi H, Iizuka N, et al. Transformation of ALK rearrangement-positive adenocarcinoma to small cell lung cancer in association with acquired resistance to alectinib. Ann Oncol. 2016;27(5):25–32. | ||
Fujita S, Masago K, Katakami N, Yatabe Y. Transformation to SCLC after treatment with the ALK inhibitor Alectinib. J Thorac Oncol. 2016;11(6):e67–e72. | ||
Miyamoto S, Ikushima S, Ono R, et al. Transformation to small-cell lung cancer as a mechanism of acquired resistance to crizotinib and alectinib. Jpn J Clin Oncol. 2016;46(2):170–173. | ||
Caumont C, Veillon R, Gros A, Laharanne E, Bégueret H, Merlio JP. Neuroendocrine phenotype as an acquired resistance mechanism in ALK-rearranged lung adenocarcinoma. Lung Cancer. 2016;92:15–18. | ||
Levacq D, D’Haene N, de Wind R, Remmelink M, Berghmans T. Histological transformation of ALK rearranged adenocarcinoma into small cell lung cancer: a new mechanism of resistance to ALK inhibitors. Lung Cancer. 2016;102:38–41. | ||
Ou SI, Lee TK, Young L, et al. Dual occurrence of ALK G1202R solvent front mutation and small cell lung cancer transformation as resistance mechanisms to second generation ALK inhibitors without prior exposure to crizotinib. Pitfall of solely relying on liquid re-biopsy? Lung Cancer. 2017;106:110–114. | ||
Camidge DR, Doebele RC. Treating ALK-positive lung cancer-early success and future challenges. Nat Rev Clin Oncol. 2012;9(5):268–277. | ||
Isozaki H, Takigawa N, Kiura K. Mechanisms of acquired resistance to ALK inhibitors and the rationale for treating ALK-positive lung cancer. Cancers (Basel). 2015;7(2):763–783. | ||
Norkowski E, Ghigna MR, Lacroix L, et al. Small-cell carcinoma in the setting of pulmonary adenocarcinoma: new insights in the era of molecular pathology. J Thorac Oncol. 2013;8(10):1265–1271. | ||
Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015;16(4):e165–e172. | ||
Kalemkerian GP, Akerley W, Bogner P, et al. Small cell lung cancer. J Natl Compr Canc Netw. 2013;11(1):78–98. | ||
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. | ||
Kikuchi H, Sakakibara-Konishi J, Furuta M, et al. Expression of Notch1 and Numb in small cell lung cancer. Oncotarget. 2017;8(6):10348–10358. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.