Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Patient considerations in the management of ulcerative colitis – role of vedolizumab
Authors Kothari M, Mudireddy P, Swaminath A
Received 18 January 2015
Accepted for publication 10 June 2015
Published 19 August 2015 Volume 2015:11 Pages 1235—1242
DOI https://doi.org/10.2147/TCRM.S65650
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Garry Walsh
Video abstract presented by Megha Kothari
Views: 1382
Megha Kothari, Prashant Mudireddy, Arun Swaminath
Division of Gastroenterology and Hepatology, Lenox Hill Hospital-NSLIJ, New York, NY, USA
Abstract: Ulcerative colitis (UC) is a subtype of inflammatory bowel disease which causes inflammation of the large intestine and affects approximately 7.6–24.6 per 100,000 persons. The therapeutic goal for UC patients is inducing remission, maintaining remission, and ideally, obtaining mucosal healing. Vedolizumab, approved by the US Food and Drug Administration in May 2014 for the treatment of moderate-to-severe UC and Crohn’s disease, is a newly developed anti-integrin therapy. This review focuses on the preclinical development of vedolizumab and data from early trials, and details the results of the landmark trails that led to its approval in the USA with a specific focus on the management of UC. Additionally, data on safety and the current UC management protocols are also discussed.
Keywords: inflammation, mucosal healing, remission, immunomodulator drugs, infliximab, ulcerative colitis
Introduction
The incidence of ulcerative colitis (UC) ranges from 1.2 to 20.3 cases per 100,000 person-years, and prevalence ranges from 7.6 to 24.6 per 100,000 persons.1 Approximately 50% of patients present with proctosigmoiditis, 30% with left-sided colitis, and 20% with pancolitis, and approximately 50% progress to more extensive disease over the first 5 years of disease.2 UC is characterized by recurring episodes of inflammation of the mucosal layer and is limited to the colon.
The therapeutic goal for UC patients is not only achieving symptomatic relief (induction of remission) but also to obtain mucosal healing. Thereafter, the goal is to prevent disease flares (maintenance of remission).
The treatment options for UC consist of sulfasalazine, 5-aminosalicylic acid (5-ASA), corticosteroids, immunomodulator drugs (azathioprine, 6-mercaptopurine), calcineurin inhibitors (cyclosporine and tacrolimus), anti-tumor necrosis factor (TNF)-alpha antibodies (infliximab [IFX], adalimumab, golimumab), and an integrin antagonist (vedolizumab). Introduction of anti-TNF agents in the last decade has revolutionized the care of UC patients and management of moderate-to-severe UC. The ACT 1 and ACT 2 trials demonstrated the efficacy of IFX in the induction and maintenance of moderate-to-severe UC. Treatment with IFX has shown nearly a 60%–70% response rate in UC, but up to 40% are primary nonresponders, and 10% per year develop secondary nonresponse to IFX therapy.3–5 While primary nonresponse may be due to proinflammatory pathways that bypass TNF-alpha, secondary nonresponse has been correlated with the development of antidrug antibodies, accelerated drug clearance, albumin concentration, and degree of systemic inflammation.6 Additionally, response to a second anti-TNF is lower than in anti-TNF-naïve patients.4
The efficacy and safety of vedolizumab in the induction and maintenance of UC were established in the GEMINI 1 study.7 This antibody, unlike natalizumab, does not penetrate the blood–brain barrier and therefore avoids immunomodulation of T-cell trafficking to the central nervous system. Vedolizumab received US Food and Drug Administration approval in May 2014 for the treatment of moderate-to-severe Crohn’s disease (CD) and UC.8 The most recent treatment algorithms highlighted by the American College of Gastroenterology, last published in 2010, predate the development of vedolizumab. The European Crohn’s and Colitis Organization’s ulcerative management guidelines (https://www.ecco-ibd.eu/images/6_Publication/6_3_ECCO%20Guidelines/2012_UC_Cosensus_Update_2_Current_Management.pdf) date to 2012 and do acknowledge vedolizumab but do not specifically describe the role of vedolizumab. In the most recent guidelines published by the American Gastroenterological Association, The Toronto Consensus, with regard to the nonhospitalized UC patient, vedolizumab is recommended for those with primary failure to anti-TNF therapy and as an equivalent option to a second anti-TNF agent in those with secondary failure to anti-TNF.9 Others have suggested that for induction and maintenance of moderate-to-severe UC, anti-TNF agents and anti-integrin agents are similarly positioned.10
Review of pharmacology, mode of action, and pharmacokinetics of vedolizumab
Vedolizumab (Entyvio), produced by Takeda Pharmaceuticals, evolved as is a humanized monoclonal antibody (IgG1), composed of two light chains of the kappa subclass and two IgG1 heavy chains, whose action is to selectively bind to integrin α4β7, and modulates gut lymphocyte trafficking.
Vedolizumab blocks the interaction of α4β7 integrin, a cell surface glycoprotein variably found on T-lymphocytes, with cell adhesion modules (MAdCAM-1, VCAM-1, and fibronectin) expressed on gut endothelial cells and thereby inhibits the infiltration of T-lymphocytes into inflamed gastrointestinal tissue.3,8,11 This allows vedolizumab to provide more specific anti-inflammatory activity while avoiding central nervous system toxicity which has limited the acceptance of the first anti-integrin therapy approved for treatment of CD, natalizumab. In a study of 14 healthy subjects, vedolizumab did not affect the CD4+ lymphocyte cell counts, CD8+ lymphocyte cell counts, or the CD4+:CD8+ ratios in the cerebrospinal fluid.12
In a placebo-controlled study, healthy volunteers received vedolizumab or placebo followed by intramuscular vaccination with hepatitis B vaccine and with oral cholera vaccine. Those treated with vedolizumab did not have lower rates of protective immunity to hepatitis B but did have lower cholera titers, which speaks to the gut specificity of the effect of vedolizumab.13
Phase II studies of pharmacokinetics suggest that vedolizumab exhibits dose-proportional pharmacokinetics, and at a minimal dose of 2 mg/kg, there is maximal saturation of α4β7 receptors found on peripheral serum lymphocytes. The mean elimination half-life of vedolizumab is 15–22 days, with detectable steady levels of the drug at one infusion every 8 weeks (q8wk), after initial induction at 0 week, 2 weeks, and 6 weeks.6 The clearance of vedolizumab conformed to a standard linear model over a broad range of body weights, so a fixed dose has been recommended.
Pharmacokinetics of vedolizumab in patients with renal or hepatic insufficiency has not been studied.
The refinement process which led to vedolizumab
Vedolizumab was the end result of an evolutionary process that included precursor antibodies, LDP-02, MLN-02, and MLN-0002.
The first Phase I/II trial of α4β7 integrin inhibitor (LDP-02) was a double-blind, placebo-controlled study of 29 patients with moderate-to-severe UC. The trial assessed the clinical and endoscopic effect of an infusion of single escalating dose (0.15 mg/kg subcutaneous, 0.15 mg/kg intravenous [IV], 0.5 mg/kg IV, 2.0 mg/kg IV) of LDP-02 at 30 days. An IV dose of 0.5 mg/kg was found to completely saturate antibody receptors. Complete endoscopic (Baron grade =0) and clinical remission (Mayo score =0) was found in 40% of patients at an optimal dose of 0.5 mg/kg. Headache was the only reported side effect in this study.14 A drug modification was sought subsequently to develop a more humanized antibody, as described below.
A Phase II trial using α4β7 antibody derived from the NS0 mouse myeloma cell line (MLN-02, more humanized light chain than the light chain of LDP-02) was a multicenter, double-blind, placebo-controlled trial in 181 patients with active UC. Patients were randomized to three groups (placebo, 0.5 mg/kg, and 2.0 mg/kg), and each patient received two infusions (on days 1 and 29). Patients were followed for 6 weeks with sigmoidoscopy performed at baseline and at weeks 4 and 6. The primary outcome was clinical remission at week 6 (defined as Mayo score 0–1 without rectal bleeding and Baron grade 0–1). The results demonstrated clinical remission in 14% of patients in the placebo group, 32% in the 2.0 mg/kg group, and 33% in the 0.5 mg/kg group (P=0.03). The secondary outcome was clinical improvement by at least 3 points on the Mayo score. This was seen in 33% of the patients in the placebo group, 66% in the 0.5 mg/kg group, and 53% in the 2.0 mg/kg group. Endoscopic remission was seen in 8% of patients in the placebo group, 12% in the 2.0 mg/kg group, and 28% in the 0.5 mg/kg group (P=0.007).15 While these results were encouraging, 44% of exposed patients developed detectable human antihuman antibodies (HAHAs) at week 8. Among the patients who developed immunogenicity/HAHAs, there was faster clearance of the drug, more rapid loss of α4β7 receptor saturation, and subsequently a decrease in clinical response.7
Additional work led to the development of a less immunogenic antibody, MLN-0002, using a Chinese hamster ovary cell-based system.7,16 Only 11% of patients developed HAHAs by week 8 with exposure to MLN-0002.17 In 2012, Parikh et al performed a Phase II trial using the new formulation of the antibody, MLN-0002. This was performed using escalating doses of MLN-0002 (2 mg/kg, 6 mg/kg, and 10 mg/kg) versus placebo in patients with UC and CD. Patients were included if they had clinical and endoscopic UC for >2 years, Mayo score of 2–7, and were on stable doses of other inflammatory bowel disease (IBD) medications. Forty-seven patients from eleven centers were included in the study, 2 mg/kg (n=13), 6 mg/kg (n=14), and 10 mg/kg (n=11), and placebo (n=9). Patients received four infusions, one each on days 1, 15, 29, and 85, and were followed for 253 days. Primary outcomes were to study the clinical pharmacology and safety of vedolizumab, and the secondary outcome was to study clinical remission and response. Fecal calprotectin was used as a surrogate marker for mucosal inflammation as documented by the partial Mayo score (PMS).16
Secondary outcome of clinical response at the end of induction (day 43) was seen in 51% of patients in the MLN-0002 group and 13% in the placebo group. From days 29–253, the all-severity (PMS 0–9) vedolizumab responders were >50% versus 22%–33% in the placebo group, while the percentage of responders was highest in the 6 mg/kg group. Over 50% of patients exposed to vedolizumab continued to show clinical response over the full length of the trial. Fecal calprotectin decreased from 405 μg/g to 54 μg/g in the vedolizumab group and from 310 μg/g to 192 μg/g in the placebo-treated group.16
MLN-0002 was moved to Phase III clinical trials as vedolizumab, and we detail the seminal clinical study of UC in the next section.
Role of vedolizumab in induction of remission in UC
GEMINI 1, a Phase III study of vedolizumab in UC patients, was published in the New England Journal of Medicine in 2013 by Feagan et al. This was a randomized, double-blind, placebo-controlled trial evaluating the efficacy of vedolizumab in inducing and maintaining remission in patients with moderate-to-severe UC. Inclusion criteria were age of 18–80 years, Mayo score of 6–16, and endoscopic subscore of at least 2 despite previous treatments with steroids, thiopurines, and/or anti-TNFs. Patients were allowed to take current medications at stable doses (ie, mesalamine, prednisone ≤30 mg/day, and/or immunosuppressive agents) which was permitted through week 6. Patients were ineligible if they had anti-TNFs within 60 days, cyclosporine, or thalidomide within 30 days. The primary outcome of induction therapy was clinical response at week 6 (reduction in Mayo score of ≥3 points and ≥30% from baseline plus a decrease in rectal bleeding subscore of ≥1 point or absolute rectal bleeding subscore of ≤1 point). Secondary outcomes were clinical remission (Mayo score ≥2 and no subscore higher than 1) and mucosal healing (endoscopic subscore of 0 or 1) at week 6.7
For induction therapy (cohort 1), patients were randomized in 3:2 ratio to receive IV vedolizumab 300 mg (225 patients) or placebo (149 patients) at days 1 and 15 (with or without use of glucocorticoids, with previous anti-TNF exposure limited to 50%). In addition, another 521 patients (cohort 2) received open-label vedolizumab for induction therapy at days 1 and 15.5 In cohort 1, 47% of patients receiving vedolizumab versus 25% of patients receiving placebo had a clinical response, 16.9% versus 5.4% of patients achieved clinical remission, and 40.1% versus 24.8% achieved mucosal healing at week 6 follow-up. In cohort 2, 44% had clinical response, 19% had clinical remission, and 36.7% had mucosal healing, similar to cohort 1, suggesting that no significant bias toward treatment was shown in the open-label group (Table 1; data from cohort 1 only).7
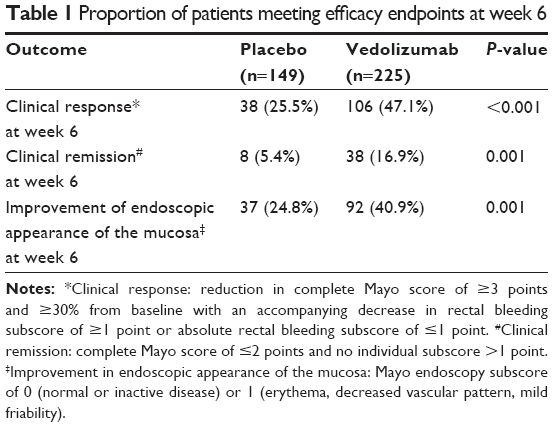 | Table 1 Proportion of patients meeting efficacy endpoints at week 6 |
A Cochrane analysis of four trials (606 patients) published recently confirmed the efficacy of vedolizumab (included different versions of the antibody eventually named vedolizumab; LDP-02, MLN-02, and MLN-0002) in induction of remission in UC compared to placebo. Seventy-seven percentage (293/382) of patients who received one of these formulations failed to achieve clinical remission compared to 92% (205/224) in the placebo group (relative risk [RR] 0.86, 95% confidence interval [CI] 0.80–0.91) after 4–6 weeks of treatment. After week 6 (pooled analysis of three studies), 48% of patients in treatment group failed to show clinical response compared to 72% in placebo group (RR 0.68, 95% CI 0.59–0.78). Sixty-eight percentage of patients in the treatment group failed to achieve endoscopic remission versus 81% of the placebo patients (RR 0.82, 95% CI 0.75–0.91). These moderate- to high-quality studies suggest that vedolizumab is superior to placebo in induction of clinical remission, clinical response, and endoscopic remission in patients with UC.17
Role of vedolizumab in maintenance of remission in UC
In GEMINI 1 study, only patients who had clinical response at week 6 in cohort 1 and cohort 2 were randomly assigned in a double-blind fashion (1:1:1) to one of the following regimens beginning at week 6: vedolizumab 300 mg q8wk (122 patients), vedolizumab 300 mg every 4 weeks (q4wk; 125 patients), or placebo q4wk (126 patients). Patients who did not have a clinical response at 6 weeks received open-label vedolizumab q4wk for up to 52 weeks. Primary outcome was clinical remission at week 52, and secondary measures were durable clinical response, durable clinical remission, mucosal healing, and steroid-free remission at 52 weeks. Concomitant ASA and steroids were permitted through week 52; however, concomitant immunomodulators were only permitted outside the USA.7
At week 52, 41.8% of patients receiving vedolizumab q8wk and 44.8% of patients receiving vedolizumab q4wk were in clinical remission, compared to 15.9% from the placebo group (P<0.001). All of the secondary outcomes were significantly higher among the vedolizumab groups compared to the placebo group. Additionally, concurrent treatment with steroids and/or thiopurines and previous therapy with anti-TNF did not change efficacy of vedolizumab. There was no significant difference between the two vedolizumab regimens (Table 2).7
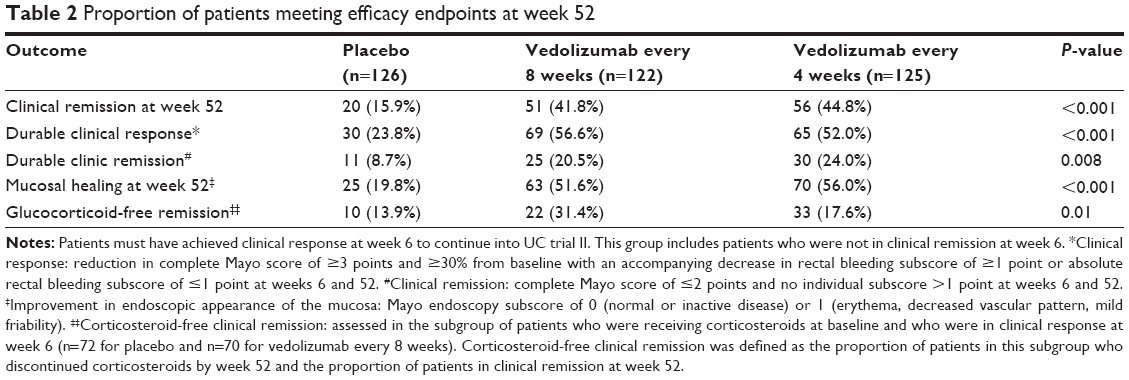 | Table 2 Proportion of patients meeting efficacy endpoints at week 52 |
While these results may appear comparable to earlier biologic therapies for UC, it should be clarified that the final numbers were enriched by only allowing drug responders into the maintenance arm. Hence, the interpretation of the maintenance results should be read as “41.8% of those enrolled into the maintenance trail at q8wk dosing for vedolizumab, which was 47% of the randomized induction population, achieved clinical response at week 52.”
What happened to those who did not meet clinical response at week 6 and were administered open-label vedolizumab q4wk? Currently, the data are available only in abstract form and are combined with “early terminators” and those who completed the 52-week maintenance trial and then dose escalated to q4wk as part of the GEMINI LTS study, to be completed in 2016. Two analyses were presented, a prespecified analysis involving the efficacy population (EP; GEMINI 1 completers) and a post hoc analysis involving observed cases (OCs; EP patients who had baseline and ≥1 partial Mayo score post-baseline measurements).
Of the GEMINI completers who enrolled in GEMINI LTS, 66% were in clinical remission at week 52, and 73% were in clinical remission at week 104. Also, 79% had clinical response at week 52, and 80% had clinical response at week 104. At week 104, 65.3% (EP) and 79.5% (OCs) of patients with prior biological failure had remission, and 73.7% (EP) and 89.7% (OCs) had a response.18 In the GEMINI 1 early terminators group, 31% and 19% had clinical response, and 3% and 13% had clinical remission at week 52 and week 108, respectively.19 While this data suggest that a lack of response to induction therapy can extrapolate to poor long-term response to vedolizumab, it should be noted that the sample size in the early terminators group was extremely small (only eight patients received vedolizumab q4wk). However, given the heterogeneous populations enrolled in this registry, the interpretation of these data remains vague, though encouraging.
Additionally, another abstract from open-label findings suggests that patients who lost response to dosing at q8wk may have improvements in mean disease activity scores with an increase in dosing frequency to q4wk without an apparent increased risk of adverse events (AEs).18
Safety and tolerability
Vedolizumab is a very well-tolerated medication. The rates of AEs in the vedolizumab group were similar to the placebo group. A Cochrane meta-analysis of two studies found that 79% of patients in vedolizumab group and 80% of patients in the placebo group experienced at least one AE (RR 0.99, 95% CI 0.93–1.07). The pooled analysis of these two studies also showed that the withdrawals due to AEs were significantly lower in the vedolizumab group (6%) compared to placebo (11%) (RR 0.55, 95% CI 0.35–0.87).2 The most common infusion-related side effects include viral upper respiratory tract infection symptoms, back pain, rash, and itching, which occurred in 4% of those receiving vedolizumab versus 3% in the placebo group (Table 3).7 One case of anaphylaxis (1/1,434) was reported by a CD patient during the second infusion which was managed with discontinuation of infusion and treatment with antihistamine and IV hydrocortisone. Serious infections were more common in CD patients than UC patients, and anal abscesses were the most frequently reported serious adverse reaction in CD patients.7 There was no increased rate of serious infections or significant changes in laboratory values (ie, liver function test or hematological test) in the treatment group.
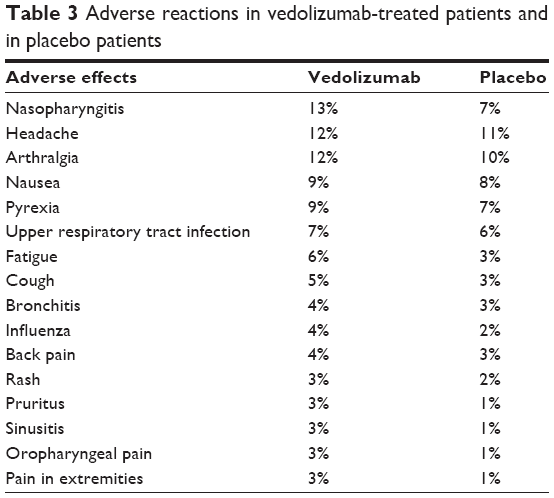 | Table 3 Adverse reactions in vedolizumab-treated patients and in placebo patients |
Additionally, data regarding the safety of vedolizumab come from a recent study by Loftus et al using the GEMINI 1 and 2 database. They showed that the infectious AE (ie, nasopharyngitis, urinary tract infection) and infection serious AE (ie, abscess, sepsis) profiles were similar among patients (both UC and CD) who received vedolizumab monotherapy and those who received vedolizumab with concomitant steroids and/or immunomodulators. Compared with placebo, vedolizumab generally led to similar rates of infection, with the exception of nasopharyngitis which was less common in placebo group.7
Other integrin inhibitors, such as natalizumab (only approved for CD), lack specificity for immunosuppression and can subsequently affect immunomodulation in the brain. Their immunosuppressive effect on the brain can lead to progressive multifocal leukoencephalopathy (PML) in rare cases. PML is a rare and fatal demyelinating disease of the brain caused by reactivation of JC virus; this occurs due to the nonspecific α4 inhibitory effects of natalizumab.8
As of June 2013, 3,129 patients have been treated with vedolizumab. It is estimated that six to seven cases would have been seen in the vedolizumab group, if the risk was similar to those receiving natalizumab (>1 case in 500 patients).2 However, no cases have been reported.2,5 Govani et al described the rule of three to assess with a 95% CI the upper bound of likelihood (=3/n) for PML to occur on anti-adhesion molecules. As 3,129 patients have been treated with vedolizumab with zero events of PML, we can estimate that the risk of PML is zero, with a 95% CI from 0% to 0.1% or 10/10,000.20 Given that the risk of PML in natalizumab-treated patients was increased after 2 years of exposure, we can estimate the risk of PML in those exposed to vedolizumab for >2 years. Sands et al reported that 995 patients have been treated with vedolizumab for >2 years. Therefore, the estimated risk of PML after 2 years of vedolizumab treatment is zero, with a 95% CI from 0% to 0.3% or 30/10,000 patients exposed for 2 years.2 If the sample size increases without any PML events, we can more accurately estimate a near 0% risk of developing PML.
Malignancies were reported among 0.4% of patients treated with vedolizumab, including patients with colon cancer (n=2), transitional cell carcinoma (n=1), breast cancer (n=1), carcinoid tumor of the appendix (n=1), and squamous cell carcinoma (n=1). Malignancy was reported in only one of 297 (0.3%) patients treated with placebo (squamous cell carcinoma).7 While expert consensus recommends avoiding anti-TNF exposure for 5 years after treatment of a malignancy,21 it is not yet clear if vedolizumab should abide by the same rule of thumb.
Long-term complications such as serious malignancies and infections are also associated with the use of anti-TNF agents and immunomodulators. A meta-analysis suggested that patients receiving immunomodulators for the treatment of IBD have a lymphoma risk of 0.012% (approximately fourfold higher; 3/100,000 average population → 12/100,000 patients). Patients receiving anti-TNF agents have a 0.059% (approximately threefold higher) risk of non-Hodgkin’s lymphoma (NHL), with scattered case reports of a rare NHL (hepatosplenic T-cell lymphoma) with the use of azathioprine–anti-TNF combination. This has not been documented with the use of vedolizumab alone. The risk of infection with anti-TNF agents is reported to occur in 3.6% of patients versus 1.7% of controls.22
In our single-center experience of vedolizumab treatment in ten patients for UC and CD, two patients experienced possibly drug-related adverse reactions. A 59-year-old man with fistulizing CD who had failed multiple anti-TNF agents was treated with vedolizumab. After the second loading dose at week 2, he experienced painful large upper body lymphadenopathy. The patient was hospitalized, and serological testing for HIV, Epstein-Barr virus, cytomegalovirus, and hepatitis was negative. The patient was treated symptomatically with IV hydration and pain management with complete resolution of lymphadenopathy after 1 day. Vedolizumab was resumed, and lymphadenopathy had not recurred during 3 months of follow-up. A second patient, 66-year-old female with UC and poor prior response to thiopurines and anti-TNF therapy, was admitted with acute appendicitis 1 day following second vedolizumab loading dose. She underwent appendectomy with no further sequela and is currently receiving vedolizumab as maintenance therapy and has achieved good clinical response. Given the mechanism of action of vedolizumab, it is plausible that it may make patients more susceptible to gastrointestinal infections. To our knowledge, there have been no other reported cases of appendicitis or transient lymphadenopathy.
Of note, while the safety profile of vedolizumab appears to be favorable in clinical studies, it should be a caveat that this drug is still relatively new. Longer clinical data are still needed to evaluate other unknown potential adverse effects, such as in our two patients. While the absolute rate of these events remains low, it should be weighed against the substantial benefits associated with treatment.
Vedolizumab versus other biological agents
Currently, there are no head-to-head trials between various biological agents assessing their efficacy and safety in the management of the UC, thus making it difficult for the treating physician to choose the “right” biological agent for individual patients. The best evidence to date comes from the systematic review and network meta-analysis done by Danese et al. Their study included the randomized controlled trials (RCTs) assessing the efficacy of four biological agents currently available for the management of UC – IFX, adalimumab, golimumab, and vedolizumab. It is important to note that they excluded patients previously exposed to biologics in their analysis. All four biological agents were superior to placebo in inducing clinical response, remission, and mucosal healing (ten RCTs, 2,282 biological-naïve patients). IFX was superior to adalimumab in inducing clinical response (odds ratio 2.79, 95% CI 0.95–8.83). Though none of the other comparisons between the four agents reached statistical significance, the study did demonstrate that IFX had the strongest data for induction therapy in UC followed by vedolizumab, golimumab, and adalimumab.23 Given the differences in patients, recruitment criteria, definitions of response, and remission between the trials, the findings may not reflect the induction results of a true head-to-head study of these agents.
For assessing the efficacy of the four biologics in maintenance therapy, six RCTs were included (1,502 biological-naïve patients). Again, all the four biological agents were superior to placebo in the maintenance of clinical response, remission, and mucosal healing. The authors did not compare between the four biologics because of the difference in the study designs of the maintenance-of-remission trials.23
Patient-focused perspectives such as quality of life, patient satisfaction, and acceptability
Since current treatments for UC are used to control inflammation, rather than provide a cure, maximizing the patients’ quality of life is an important goal.
Studies by Feagan et al showed an improvement in the mean IBD quality-of-life questionnaire (IBDQ) scores, which suggest an improved global quality of life in terms of their underlying disease. GEMINI 1 study showed a mean 20% improvement in the IBDQ at week 6 in the vedolizumab group compared to placebo. Furthermore, in the study group that had treatment extended to 52 weeks, there was further improvement in the IBDQ scores at 52 weeks compared to 30 weeks. This is conceivable as the clinical remission rates increased from 16.9% at week 6 to 41.8% at week 52.24 The time to clinical response suggest that vedolizumab, unlike anti-TNF agents, may not demonstrate an immediate effect in clinical symptoms. Sands et al demonstrated that the effects of vedolizumab on clinical remission may not become evident until between weeks 6 and 10 among CD patients. Patients should be counseled on this prior to initiating treatment.
Patient acceptability is directly related to the adverse effect profile. There is no significant difference in adverse effects between vedolizumab and placebo, except for upper respiratory tract infections. In our experience, patients have a very favorable acceptance to the drug, but these patients were often doing poorly on anti-TNF or combination therapy at baseline. At our institution, even the two patients (out of the ten exposed) who experienced potentially related adverse drug reactions elected to continue therapy with vedolizumab. Table 4 details our institutional experience using vedolizumab since it became available in July 2014.
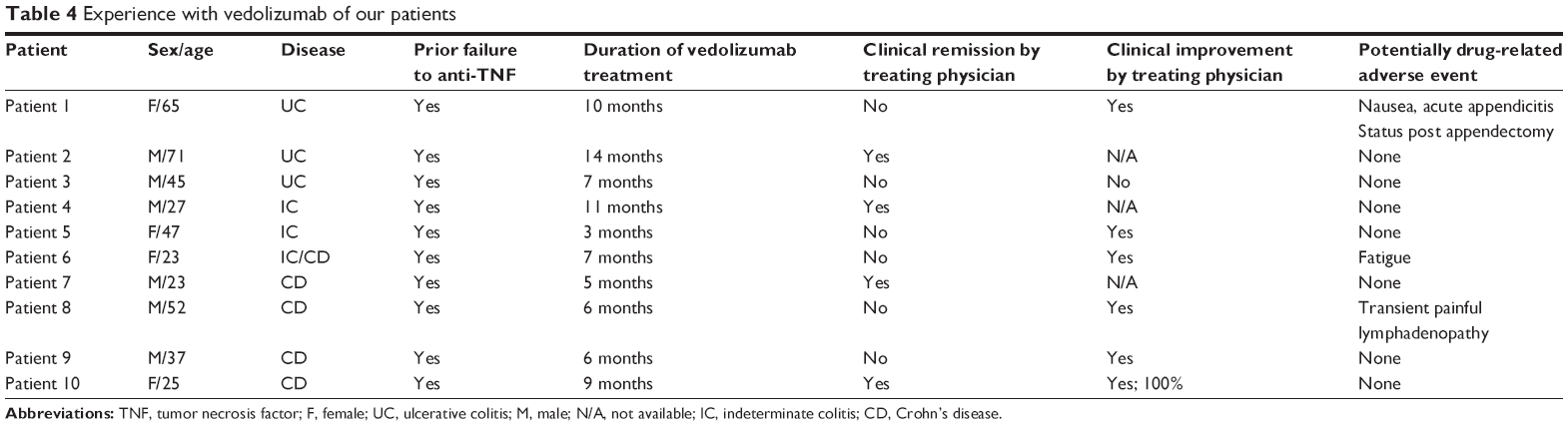 | Table 4 Experience with vedolizumab of our patients |
There are several limitations to vedolizumab therapy. It has to be administered intravenously. In contrast, subcutaneous biological (like adalimumab and golimumab) can be administered at home by self-injection. Another major limitation is cost. A study by Liu et al showed that adalimumab would cost much less than vedolizumab per responder and remitter at 1 year in IBD patients (both UC and CD combined). Adalimumab cost at 1 year per responder and remitter was $197,902 and $197,874, respectively. The cost of vedolizumab at 1 year per responder and remitter was $406,629 and $336,332, respectively.25 Given its short experience in “real life”, the probability that IFX has the most potency in UC induction, a potentially slower time to clinical improvement, and higher payor costs, vedolizumab is currently reserved for those having failed one anti-TNF therapy at our institution.
Conclusion and place in therapy
Vedolizumab shows promise in the future of managing moderate-to-severe UC.
As we gain more experience, the inevitable question about the positioning of vedolizumab in the treatment algorithm will arise – will it remain a second-line agent once one or even two anti-TNF agents have failed, or whether vedolizumab in moderate-to-severe UC may be considered a first-line therapy in specific circumstances. Industry is unlikely to conduct head-to-head studies, so we will need to look at how the drug will be adopted by clinicians in the years to come.
Further research to determine if there are phenotypic, genetic, or biomarker predictors of response would be beneficial in choosing an agent, particularly to reduce cost and improve patient outcomes. Additionally, no data are currently available regarding combination use of immunomodulators and vedolizumab or anti-TNF agents and vedolizumab in terms of safety and efficacy. The combination use of these medications needs to be studied, as we have learned from the SONIC trial that combination therapy with anti-TNF plus thiopurines was more likely to induce a corticosteroid-free clinical remission.26
Disclosure
Arun Swaminath is in the advisory board of Janssen and Abbvie. Megha Kothari and Prashant Mudireddy report no conflicts of interest in this work.
References
Danese S, Fiocchi C. Ulcerative colitis. N Engl J Med. 2011;365(18):1713–1725. | ||
Sands BE, Feagan BG, Rutgeerts P, et al. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology. 2014;147(3):618e3–627e3. | ||
Gilroy L, Allen PB. Is there a role for vedolizumab in the treatment of ulcerative colitis and Crohn’s disease? Clin Exp Gastroenterol. 2014;7:163–172. | ||
Sandborn WJ, Rutgeerts P, Enns R, et al. Adalimumab induction therapy for Crohn disease previously treated with infliximab: a randomized trial. Ann Intern Med. 2007;146(12):829–838. | ||
Deleporte A, Viennot S, Dupont B, et al. Efficacy of anti-TNF-alpha monoclonal antibodies in inflammatory bowel disease treatment. Int J Infereron Cytokine Mediator Res. 2013;2013(5):11–31. | ||
Rosario M, Dirks NL, Gastonguay MR, et al. Population pharmacokinetics-pharmacodynamics of vedolizumab in patients with ulcerative colitis and Crohn’s disease. Aliment Pharmacol Ther. 2015;42(2):188–202. | ||
Feagan BG, Rutgeerts P, Sands BE, et al; GEMINI 1 Study Group. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699–710. | ||
Raine T. Vedolizumab for inflammatory bowel disease: changing the game, or more of the same? United European Gastroenterol J. 2014;2(5):333–344. | ||
Bressler B, Marshall JK, Bernstein CN, et al; Toronto Ulcerative Colitis Consensus Group. Clinical practice guidelines for the medical management of nonhospitalized ulcerative colitis: the toronto consensus. Gastroenterology. 2015;148(5):1035e3–1058e3. | ||
Danese S, Panes J. Development of drugs to target interactions between leukocytes and endothelial cells and treatment algorithms for inflammatory bowel diseases. Gastroenterology. 2014;147(5):981–989. | ||
Soler D, Chapman T, Yang LL, Wyant T, Egan R, Fedyk ER. The binding specificity and selective antagonism of vedolizumab, an anti-alpha4beta7 integrin therapeutic antibody in development for inflammatory bowel diseases. J Pharmacol Exp Ther. 2009;330(3):864–875. | ||
Milch C, Wyant T, Xu J, et al. Vedolizumab, a monoclonal antibody to the gut homing alpha4beta7 integrin, does not affect cerebrospinal fluid T-lymphocyte immunophenotype. J Neuroimmunol. 2013;264(1–2):123–126. | ||
Wyant T, Leach T, Sankoh S, et al. Vedolizumab affects antibody responses to immunisation selectively in the gastrointestinal tract: randomised controlled trial results. Gut. 2015;64(1):77–83. | ||
Feagan BG, Feagan BG, McDonald JWD, et al. An ascending dose trial of a humanized A4B7 antibody in ulcerative colitis (UC). Gastroenterology. 2000;118(4):A874. | ||
Feagan BG, Greenberg GR, Wild G, et al. Treatment of ulcerative colitis with a humanized antibody to the alpha4beta7 integrin. N Engl J Med. 2005;352(24):2499–2507. | ||
Parikh A, Leach T, Wyant T, et al. Vedolizumab for the treatment of active ulcerative colitis: a randomized controlled phase 2 dose-ranging study. Inflamm Bowel Dis. 2012;18(8):1470–1479. | ||
Bickston SJ, Behm BW, Tsoulis DJ, et al. Vedolizumab for induction and maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. 2014;8:CD007571. | ||
Feagan B, Kaser A, Smyth M, Panaccione R, Sankoh S, Abhyankar B. Long-term efficacy of vedolizumab therapy for patients with ulcerative colitis. Am J Gastroenterol. 2014;109(suppl 2):S477. | ||
Sands BE, Shafran I, Farraye FA, et al. Efficacy and Safety of Retreatment with Vedolizumab in Patients with Ulcerative Colitis. Vienna: European Crohns and Colitis Organization; 2015. | ||
Govani SM, Waljee AK, Higgins PD. Anti-adhesion therapies and the rule of 3 for rare events. Am J Gastroenterol. 2013;108(12):1831–1832. | ||
Mantzaris GJ. Previous cancer and/or lymphoma in patients with refractory IBD–con: anti-TNF or conventional immunosuppressive treatment. Dig Dis. 2014;32(suppl 1):122–127. | ||
Lakatos PL, Miheller P. Is there an increased risk of lymphoma and malignancies under anti-TNF therapy in IBD? Curr Drug Targets. 2010;11(2):179–186. | ||
Danese S, Fiorino G, Peyrin-Biroulet L, et al. Biological agents for moderately to severely active ulcerative colitis: a systematic review and network meta-analysis. Ann Intern Med. 2014;160(10):704–711. | ||
Feagan B, Colombel J-F, Rubin D, Mody R, Sankoh S, Lasch K. Improvements in Health-Realted Quality of Life in Patients with UC Treated with Vedolizumab. Vienna: European Crohns and Colitis foundation; 2014. | ||
Liu Y, Ozbay AB, Skup M, Reichmann W, Diener M, Chao J. Comparison of cost per responder and remitter in patients with inflammatory bowel disease in the United States: an indirect comparison of adalimumab and vedolizumab. Am Coll Gastroenterol. 2014;109(suppl 2):S479. | ||
Colombel JF, Sandborn WJ, Reinisch W, et al; SONIC Study Group. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N Engl J Med. 2010;362(15):1383–1395. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.