Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 6
Pathophysiological processes in multiple sclerosis: focus on nuclear factor erythroid-2-related factor 2 and emerging pathways
Authors Arnold P ![]() , Mojumder D, DeToledo J, Lucius R, Wilms H
, Mojumder D, DeToledo J, Lucius R, Wilms H
Received 12 December 2013
Accepted for publication 14 January 2014
Published 24 February 2014 Volume 2014:6 Pages 35—42
DOI https://doi.org/10.2147/CPAA.S35033
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Philipp Arnold,1,* Deb Mojumder,2,* John DeToledo,2 Ralph Lucius,1 Henrik Wilms2
1Institute of Anatomy, Christian-Albrechts-University Kiel, Kiel, Germany; 2Department of Neurology, Texas Tech University Health Science Center, Lubbock, TX, USA
*These authors contributed equally to this work
Abstract: Multiple sclerosis (MS) is a disease of the central nervous system that is characterized by the demyelination of neuronal axons. Four different patterns of demyelination have been described, showing the heterogeneity in the immunopathologic processes involved in the demyelination. This review will focus on reactive oxygen species (ROS)-related inflammation in MS. Special emphasis will be placed on the nuclear factor erythroid-2-related factor 2 (Nrf2) as it regulates the transcription of ROS-protective genes. In the cytosol, Nrf2 binds to Keap1 (Kelch-like ECH-associated protein 1), and together they are degraded by the 26S proteasome after ubiquitination. If challenged by ROS Nrf2, binding to Keap1 is abrogated, and it translocates into the nucleus. Here it binds to the antioxidant response element and to a small protein termed Maf (musculoaponeurotic fibrosarcoma oncogene homolog). This leads to an enhanced transcription of ROS protective genes and represents the physiological answer against ROS challenge. It has been shown that dimethyl fumarate (DMF) has the same effect and leads to an enhanced transcription of ROS-protective genes. This response is mediated through a reduced binding of Nrf2 to Keap1, thus resulting in a higher level of free Nrf2 in the cytosol. Consequently, more Nrf2 translocates to the nucleus, promoting transcription of its target genes. DMF has been used for the treatment of psoriasis for many years in Germany without the occurrence of major side effects. In psoriasis, DMF reduces ROS-related inflammation in skin. A DMF analog, BG-12, was recently approved for the treatment of relapsing-remitting MS by the European Union and the US Food and Drug Administration. As an oral formulation, it gives patients a convenient and effective alternative to the injectable immune modulators in the long-term treatment of MS.
Keywords: MS, fumaric acid ester, ROS
Multiple sclerosis – a “paradigm shift”
The classic description of multiple sclerosis (MS) indicated a central nervous system (CNS) disease with the main qualifiers being T-cell mediated process, demyelination, relapsing-remitting nature, and inflammation. Immunopathological evidence in MS suggests a variety of pathological types disputing a T-cell “only” mediated disease.1 In fact, what was thought to be the description of an MS lesion involving activated T-cells, macrophage- and cytokine-mediated multifocal damage of myelin is now considered a category of MS (type I), which surprisingly affects less than half of all MS patients. Other pathological types include: type II, caused by complement-mediated multifocal damage via activated B-cells; type III, involving loss of myelin-associated glycoprotein and apoptosis of oligodendrocytes, resulting in diffuse low grade inflammation; and type IV, characterized by non-inflammation-induced degeneration of oligodendroglial cells. Although demyelination remains the most conspicuous pathology of MS, neurons, especially their axonal compartment, are also prominently affected.2–4 Serial magnetic resonance imaging disputes the notion of sharply defined relapses and remissions in MS, with mounting evidence that progressive changes can occur without clinical worsening.5 Finally, the traditional concept of MS being an inflammatory disease is under scrutiny, with evidence suggesting that neurodegeneration initiated by non-inflammatory oligodendroglia cell death resulting in secondary inflammation may contribute to the pathology of MS in some cases.6 Neurodegeneration, likely triggered by microglial activation, is also prominent in secondary-progressive MS after the inflammation of relapsing-remitting MS subsides.7 Damage by free radicals has been associated with many neurodegenerative diseases including Alzheimer’s disease, amyotrophic lateral sclerosis, Parkinson’s disease, and Huntington’s disease. In this review, we present evidence for free-radical damage being a part of the pathogenesis of MS, and we describe the possible involvement of transcription factor Nrf2 (nuclear factor erythroid-2-related factor 2) and its downstream pathways in MS. Finally, therapies which may target Nrf2, implications for disease management, and future directions will be reviewed.
Free radicals: their role, sources, and endogenous antioxidant systems
Free radicals in the human body are ions that have unpaired valence electrons and therefore are “electron acceptors” for other molecules. The process of accepting an electron oxidizes the acceptor molecule, making the donor known as “oxidizing agent.” Free radicals usually originate from oxygen (reactive oxygen species [ROS]) or nitrogen (reactive nitrogen species [RNS]). ROS include the following radicals: O2•−, H2O2, •OH, RO2•, RO•, whereas RNS include: •NO, •NO2, and ONOO−. Free radicals are routinely generated under physiological conditions and are very important in different regulatory functions of the body. Mechanisms involving free radicals include oxidative phosphorylation, detoxification of xenobiotics by cytochrome P450, apoptosis of defective cells, destruction of microorganisms and cancer cells by macrophages and cytotoxic lymphocytes, use by various oxygenases for production of prostaglandins and leukotrienes, use as second messengers and regulation of redox-sensitive transcription factors (for details, see Devasagayam et al8). Reactive oxidants are produced by different mechanisms, in multiple compartments within the cell (for reviews, see Ma9 and Finkel10). When deregulated, free radicals are produced in excess in the cellular milieu, where they may oxidize lipids, proteins, and deoxyribonucleic acid (DNA) and may result in degenerative diseases.
Molecular oxygen is used by mitochondria for the oxidative metabolism of glucose and other substrates, leading to the production of adenosine triphosphate (ATP), the universal energy currency of cells. The process of using oxygen to oxidize glucose to produce ATP is known as oxidative phosphorylation. During oxidative phosphorylation, 0.4%–4.0% of oxygen consumed goes into production of free radical superoxide (•O2−).11–16 Superoxide can then be converted into ROS and RNS. Inside the mitochondria, superoxide molecules are converted to H2O2 by superoxide dismutase (SOD)2.11,12,17 H2O2 is then converted to H2O and O2 by glutathione (GSH) peroxidase in mitochondria, or diffuses into the cytosol and is converted by catalase in peroxisomes to H2O and O2, also. In the presence of reduced transition metals such as copper or iron (Cu or Fe), H2O2 can be converted to the highly reactive •OH radical. Although mitochondria are the primary site of ROS production from aerobic respiration, nearly all enzymes that use molecular oxygen (eg, plasma membrane–bound NADPH [reduced nicotinamide adenine dinucleotide phosphate] oxidase [NOX], microsomal cytochrome P450, and cytoplasmic xanthine oxidase) produce ROS directly or indirectly (for details, see Ma18). Especially in the brain, ROS are mainly derived from activated macrophages and microglia,19 in which plasma membrane–bound NOX, particularly NOX2, are mainly responsible for the oxidative burst of these cells. Importantly, during a phagocyte’s, such as a macrophage’s, respiratory burst, myeloperoxidase activity generates hypochlorous acid (HOCl) from H2O2 and chloride anion (Cl−), serving as a feature of free-radical damage.18 RNS are formed in cells, starting with the synthesis of nitric oxide (•NO) by NO synthase, which is present in different cell types expressing NO synthases 1–3 in the brain.20 Examples of RNS include •NO, •NO2, and ONOO−. NO• reacts with superoxide (O2•−) to form a stronger oxidant peroxynitrite anion (ONOO−). ONOO− reacts with other molecules to generate other RNS such as •NO2 and N2O3.18
Endogenous antioxidant systems exist within cells, with the purpose of neutralizing excess ROS. This is critical for proper cellular function. Reduced GSH, a major cellular antioxidant, is regenerated by GSH reductase and reduced nicotinamide adenine dinucleotide phosphate (NADP) plus (NADPH)21 and other mechanisms along with other antioxidants.22,23 Reduced GSH levels fall with increased oxidative stress. Other major antioxidants include: low-molecular-weight compounds such as vitamins C and E, bilirubin, and urate; non-catalytic antioxidant proteins, such as thioredoxin (Trx), glutaredoxin, and metallothioneins; enzymes, such as superoxide dismutase 2, catalase, peroxiredoxin, and GSH peroxidase. Ultimately, redox reactions in cells are enabled by nicotinamide pairs NADP+/NADPH and NAD+/NADH. NADPH is used to reduce oxidized Trx and GSH (GSSG) through Trx reductase and GSH reductase, respectively. Sulfiredoxin reduces oxidized peroxiredoxin from sulfinic (inactive) to sulfenic (active) acid in an ATP- and GSH-dependent manner. Antioxidant systems are controlled by regulators at multiple levels that respond to oxidative stress.18 One is via the important transcription factor Nrf2 in concert with the antioxidant response element (ARE) in promoters of target genes (see under the heading “Nrf2 pathways and oxidative damage”).
Mechanisms and evidence for free-radical damage in MS
Excess free-radical activity is detected using biochemical methods including assay of key enzymes or by immunocytochemical identification of oxidized nucleotides, proteins, and lipids.24 Several lines of evidence indicate that free radicals are involved in the pathogenesis of MS either directly or indirectly via mitochondrial damage and activation of excitotoxic mechanisms. Several studies indicate the relationship of free-radical damage associated with neurodegeneration in demyelinating disease by promoting transendothelial leukocyte migration and contributing to oligodendrocyte damage and axonal degeneration,25–27 but there is more evidence specific to MS. One pattern of tissue injury in MS is similar to initial white-matter stroke lesions showing microglia activation, distal oligodendrogliopathy, oligodendrocyte apoptosis, and acute axonal injury, after which demyelination and reactive gliosis occurs, suggesting that energy deficiency may play an important role in the pathophysiology of neurodegenerative processes in MS patients.28,29 These hypoxia-like tissue injury type lesions have been found to have ~8.2 times higher expression of oxidized lipids and oxidized DNA compared with complement-associated demyelinating lesions in MS.29 Additionally, the numerous axonal spheroids and end bulbs seen in these lesions are reactive for oxidized phospholipids.29 Oxidative stress as a contributor to early MS pathology is indicated by increased levels of lipid peroxidation markers in the cerebrospinal fluid of probable MS patients.30 Antioxidant enzymes such as CuZnSOD, MnSOD, catalase, NAD(P)H:quinineoxidoreductase 1 are elevated in astrocytes, as well as in perivascular and myelin-laden macrophages in active lesions.31 The surrogate marker for ROS produced by macrophages, myeloperoxidase, is upregulated in actively demyelinating white matter and cortical lesions,32–34 indicating oxidative burst of macrophages and microglia via the NOX2 NADPH oxidase. In addition, there is increased expression of nitric oxide synthase (NOS) in macrophages and microglia at the edge of active lesions.35,36 Mitochondrial injury in neurons is also a potentially important source of ROS.
Mitochondrial dysfunction produces free radicals causing neurodegenerative changes. This dysfunction is not a consequence of energy failure or a consequence of free-radical damage because NO and ROS target respiratory chain complexes at different levels, thereby inducing mitochondria transition and consecutively triggering apoptotic cascades26,37 via liberation of apoptosis-induced factor or cytochrome C.38 Mitochondrial disturbance in MS is indicated by microarray-based gene-expression analysis and histological approaches.39–42 The remarkable similarity of the tissue alterations in MS lesions to the lesions seen in acute ischemia of white matter due to stroke28 bears testament to mitochondrial injury associated with energy failure. In active MS lesions, the initial mitochondrial changes are characterized by loss of COX1 (cytochrome C oxidase 1) and loss of complex IV activity in the mitochondrial respiratory chain.42 In chronic MS lesions and re-myelinating axons, compensatory increase in complex IV densities have been noted, indicating initial dysfunction of the respiratory chain.43 In chronic inactive lesions, expression of mitochondrial numbers and activity is increased, possibly indicating the higher energy demand of demyelinated axons as compared with myelinated axons.44 Finally, we will briefly review the findings in experimental autoimmune encephalomyelitis (EAE), the most commonly used experimental model for MS. In the EAE model, intra-axonal mitochondrial dysfunction is known to occur before axonal degeneration or local increase in macrophages and prior to any overt demyelination, further supporting the notion that mitochondrial dysfunction may be an early event in the pathophysiology of MS. The EAE model is also relevant to this discussion because it showed that axonal degeneration is reduced by the administration of exogenous antioxidants.45
Oxidative stress can reduce the efficiency of glutamate transporters, resulting in raised glutamate concentration in the extracellular space,46 contributing to excitotoxic damage. The excitotoxic mechanism for damage in MS is supported by the fact that demyelinating lesions are histologically similar to those in animal models of excitotoxicity.47–49 Moreover, oligodendrocytes, which are the myelin-producing cells of the CNS, are highly vulnerable to glutamate excitotoxicity, mainly via activation of AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)/kainate receptors.46,47 Yet more evidence for excitotoxic damage in MS comes from the EAE model of MS, where the deleterious effect of excess glutamate can be decreased via blockade of AMPA/kainate.50
Nrf2 pathways and oxidative damage
Under the influence of ROS, Phase II detoxifying enzymes and antioxidant proteins are produced by cells.51 This upregulation of proteins has to be accounted for on the DNA level. Here, Nrf2 is an important transcription factor to mediate the altered protein expression. There are many pathways involving Nrf2 regulation, and numerous interaction partners have been described (Figure 1). Under physiological conditions, Nrf2 is located in the cytosol and binds to Keap1 (Kelch-like ECH-associated protein 1).52 Through Cullin-3 binding to ring-box 1, an E3 ubiquitin-ligase complex is formed. After ubiquitination, the entire Keap1/Nrf2 complex is prone to proteasomal degradation. Here, Nrf2 has a half live of 10–40 minutes.53,54 Under the influence of electrophilic oxidative stress, as caused by ROS, Nrf2 is released from Keap1 and translocates into the nucleus. There, after dimerization with small Maf proteins, it binds to ARE and thereby initializes the transcription of genes encoding for ROS-protective proteins.54–57 Of the different small Maf proteins, MafG especially seems to be a primary interaction partner.58
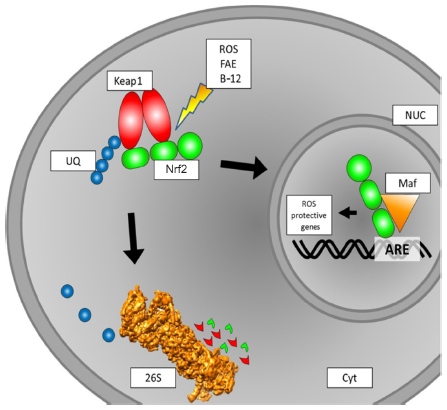 | Figure 1 Schematic overview of intracellular Nrf2 turnover. Under physiological conditions, Nrf2 binds to a Keap1 dimer. Keap1 is ubiquitinated and then prone to degradation by the 26S proteasome. If subjected to ROS, the binding of Nrf2 to Keap1 is inhibited, and Nrf2 translocates to the nucleus. This binding to Keap1 can also be inhibited by FAE or BG-12, leading to the same effect of more Nrf2 translocation to the nucleus. In the nucleus, Nrf2 binds to a small Maf protein. The Nrf2/Maf complex binds to the ARE box and initiates the transcription of ROS-protective genes. |
The upregulated proteins belong to two major enzymatic systems responsible for ROS detoxification. These systems are the heme oxygenase system and enzymes mediating GSH synthesis and utilization. Therefore, Nrf2 activation can lead to lower or even no damage related to ROS.59 In CNS lesions, as found in MS patients, ROS and NOS are mainly produced by astrocytes, macrophages, and microglia. The ROS/NOS then damage various parts of the CNS, such as myelin, axons, neurons, and oligodendrocytes. This may lead to the damage of mitochondria and subsequent further accumulation of ROS/NOS. As tissue alterations found in MS patients are similar to those found in white-matter stroke patients,28 the energy supply failure in mitochondria may play an important role in the pathophysiology of MS.39,42,60
Review of therapies which may target Nrf2
Acute attacks (relapses) of MS that result in neurologic symptoms and increased disability or impairments in vision, strength, or cerebellar function are typically treated with glucocorticoids.61 In patients with acute CNS inflammatory demyelinating disease, who do not respond to glucocorticoid therapy, plasma exchange may be beneficial.62,63 Patients with relapsing-remitting MS who manifest current disease activity by either clinical symptoms or recent lesions on magnetic resonance imaging (MRI) should be offered treatment with disease-modifying therapy. Certain immunomodulatory agents, including interferon beta preparations, glatiramer acetate, natalizumab, fingolimod, and teriflunomide, have shown important beneficial effects for patients with relapsing-remitting MS: 1) a decreased relapse rate and 2) a slower accumulation of brain lesions on MRI.
In long-term treatment, interferon beta and glatiramer acetate are commonly used. Whereas interferon beta is a naturally occurring body compound responsible for the regulation of pro- and anti-inflammatory processes,64 glatiramer acetate is an artificial polypeptide. While the exact mode of action for glatiramer acetate is still unknown, clinical studies have proven its beneficial potential in MS.65
As ROS/NOS may play an important role in MS pathogenesis, Nrf2 is an interesting therapeutic target, as reduction of ROS may directly lead to lower inflammation without the necessity of immune suppressors. In vitro studies and in vivo studies in murine systems have shown Nrf2 modulation by various compounds.66–74 Nevertheless, these are not suitable for use in clinical trials, as they are potentially toxic. For fumaric acid esters (FAEs), there is a comprehensive safety profile, and the compound is used to treat psoriasis in German-speaking countries. As in MS, patients diagnosed with psoriasis suffer from (skin) inflammation induced by ROS.75 The most important FAE, namely dimethyl fumarate (DMF), has an immediate metabolite, monomethyl fumarate (MMF).76 Using EAE mice to study MS has revealed the beneficial effects of FAEs. Application of FAEs in the chronic phase of EAE has shown preserving effects on myelin, axons, and neurons.77,78
This preserving effect is due to an alteration of the Keap1 ability to interact with Nrf2, most likely mediated by MMF. The free Nrf2 then translocates to the nucleus, as described, and leads to the transcription of genes encoding for ROS protective proteins. In two large trials, an oral formulation of DMF (BG-12) significantly reduced relapse rates and the development of new brain lesions on MRI in patients with active MS,77,78 and results from one of these trials, suggest that BG-12 reduces the rate of disability progression. DMF was recommended for approval in the European Union as a perioral treatment for MS by the European Medicines Agency in March 2013 under the name Tecfidera® (Biogen Idec, Inc., Weston, MA, USA). On March 27, 2013, the US Food and Drug Administration (FDA) approved Tecfidera capsules to treat adults with relapsing forms of MS in the US. Therefore, treatment with FAEs (especially DMF) is the first approach to actually target the inflammation-causing ROS by activation of an inherent antioxidant response.
Implications for disease management
Based on our previous discussion, the current knowledge of the immunopathology of demyelination justifies two different pharmacologic approaches to MS. One directed against acute relapse and inflammation and one aiming to slow down MS progression.
Based on our current understanding, the treatment of chronic ROS-related human diseases with agents such as FAEs would require long-term use of these agents. The ultimate goal is to maintain constant elevated levels of antioxidant proteins and to lower the absolute amount of ROS, helping to protect myelin, neurons, and axons as seen in the EAE mouse model. This could help to reduce the severity and frequency of inflammatory relapses. As compared with other injectable immunomodulatory drugs used in MS such as interferon beta preparations and glatiramer acetate, DMF has as an orally active agent, which has the benefit of increasing patients’ compliance with the therapy. In comparison with other FDA-approved oral disease-modifying MS drugs such as fingolimod (Gilenya®; Novartis International AG, Basel, Switzerland) and teriflunomide (Aubagio®; Sanofi-Aventis, Bridgewater, NJ, USA), DMF has been used for decades in the treatment of psoriasis. Long-term studies have established the safety of DMF without an increased risk of infections, malignancies, or significant long-term toxic effects.
Future directions
FAEs leads to the release of Nrf2 from the Keap1 complex and the subsequent upregulation of a wide variety of oxidative stress response genes. It would be interesting to see which pathway is responsible for the beneficial effects in MS patients. For primary culture CNS cells, it was found that the FAEs DMF and MMF lead to higher redox potentials in cells and an increase in GSH as well as in ATP levels. Moreover, the mitochondrial membrane potential can be influenced, too. The cell viability in ROS-challenged neurons and astrocytes is increased after DMF or MMF treatment and drastically reduced in Nrf2-deficient cells, thus indicating the role of an Nrf2-dependent oxidative stress response pathway, possibly induced by a better mitochondrial function.79 This is supported by the fact that malfunctioning mitochondria accumulate in axons of MS patients.80
On the other hand, in vitro data suggest reduced levels of GSH in astrocytes after treatment with DMF.81 This unexpected finding illustrates the complexity of the CNS cellular network and the potential and need for future research.
For cardiovascular and renal injury, MG132 was reported to have activating effects on the Nrf-ARE signaling pathway.51 MG132 is an inhibitor for the 26S proteasome reversely binding the chymotrypsin-like active site, thus being a competitive inhibitor. Its activating properties are thought to come from the inhibition of the proteasome and the reduced proteolysis of Nrf2. Unfortunately, MG132 is only for use in in-vitro studies, but there are other proteasome inhibitors that are in clinical trial. One being salinosporamide A, which inhibits the trypsin-like domain of the proteasome core particle.82,83 The first positive results on patients with multiple myeloma were reported in 2011,84 with an unfortunate downside for use in MS patients. The side effects observed were severe, and treatment was only possible for a short period of 21 days. This makes the use of salinosporamide A in long-term medication impossible.
Carfilzomib, a proteasome inhibitor, targeting the chymotrypsin-like catalytic site of the 26S proteasome, was approved by the FDA on July 20, 2012. Nevertheless, there is only data for 22 patients treated with the compound for more than 1 year. Carfilzomib was generated on the basis of the natural antitumor product epoxomicin. This proteasome-inhibiting compound was found to have anti-inflammatory properties in vitro and in vivo in the murine ear edema assay.85 Unfortunately, it was not reported whether or not the Nrf2 system is involved in this response. Finding a “mild” (reversely binding) proteasome inhibitor (possibly for the chymotrypsin-like catalytic domain) that is usable in patients would be highly desirable and could be beneficial for MS patients.
Conclusion and discussion
Multiple subtypes of MS have been described over the last few years, involving different cell types and factors. The most prominent pathophysiological features observed in these various subtypes are demyelination of axons and oligodendrocyte cell death. Inflammation itself can no longer be considered the common feature among MS types, as type IV seems to be a primary form of degeneration without any evidence of inflammation. The very broad description of MS pathophysiology helps to explain why existing treatment options aimed specifically at inflammation and immunological factors have had limited success.
In this review, a specific characteristic of MS is targeted, namely the occurrence of ROS in CNS inflammation. As it was shown that reduction of ROS has a positive impact on the duration and severity of inflammatory processes, the transcription factor Nrf2 came into the focus of research. After binding to ARE, it elevates the transcription of antioxidant response genes, thereby enhancing the oxidative stress response. Increasing Nrf2 levels can be obtained in two ways. Either Keap1, the cytosolic interaction partner of Nrf2 has to be blocked, or the degradation via the 26S proteasome has to be abolished. The inhibition of the proteasomal degradation is limited, as many proteins use the 26S proteasome. A more specific approach is the blockade of Nrf2 binding to Keap1. This blockade can be mediated by FAEs and has shown positive effects in vitro and in vivo. The benefits of reduction of ROS in the management of human diseases is not without precedent. The clinical efficacy of FAEs in reducing ROS-related inflammation is supported by years of experience in psoriasis patients in German-speaking countries. Recently, BG-12, a DMF analog, was approved by the European Union and the FDA for relapsing-remitting MS. Relapse rates were lower, and relapses were less severe in the clinical studies, having a positive effect on patients’ lives. Nevertheless, a long-term treatment in a large number of MS patients has to show the validity of the new treatment. To further improve the understanding of ROS-related MS, a deeper understanding of the ROS-forming cell processes is necessary. This could lead to treatment options preventing the initial formation of ROS and possibly preventing the inflammatory process.
Acknowledgment
This work was supported by a grant from the CH-Foundation (to HW).
Disclosure
The authors report no conflicts of interest in this work.
References
Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47(6):707–717. | |
Bjartmar C, Trapp BD. Axonal degeneration and progressive neurologic disability in multiple sclerosis. Neurotox Res. 2003;5(1–2):157–164. | |
Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Bruck W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. 2002;125(Pt 10):2202–2212. | |
Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120 (Pt 3):393–399. | |
Simon J. Magnetic resonance imaging in the diagnosis of multiple sclerosis, elucidation of disease, course, and determining prognosis. In: In: Burks JS, Johnson KP, editors. Multiple Sclerosis: Diagnosis, Medical Management, and Rehabilitation. New York: Demos Medical Publishing; 2000:99–126. | |
Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55(4):458–468. | |
Gonsette RE. Neurodegeneration in multiple sclerosis: the role of oxidative stress and excitotoxicity. J Neurol Sci. 2008;274(1–2):48–53. | |
Devasagayam TP, Tilak JC, Boloor KK, Sane KS, Ghaskadbi SS, Lele RD. Free radicals and antioxidants in human health: current status and future prospects. J Assoc Physicians India. 2004;52:794–804. | |
Ma Q. Transcriptional responses to oxidative stress: pathological and toxicological implications. Pharmacol Ther. 2010;125(3):376–393. | |
Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287(7):4434–4440. | |
Boveris A. Determination of the production of superoxide radicals and hydrogen peroxide in mitochondria. Methods Enzymol. 1984;105:429–435. | |
Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59(3):527–605. | |
Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr. 1997;29(1):89–95. | |
Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191(2):421–427. | |
Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91(23):10771–10778. | |
Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29(3–4):222–230. | |
Boveris A, Cadenas E. Mitochondrial production of hydrogen peroxide regulation by nitric oxide and the role of ubisemiquinone. IUBMB Life. 2000;50(4–5):245–250. | |
Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. | |
Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. | |
Love S. Oxidative stress in brain ischemia. Brain Pathol. 1999;9(1):119–131. | |
Lu SC. Regulation of glutathione synthesis. Curr Top Cell Regul. 2000;36:95–116. | |
Biewenga GP, Haenen GR, Bast A. The pharmacology of the antioxidant lipoic acid. Gen Pharmacol. 1997;29(3):315–331. | |
Packer L, Witt EH, Tritschler HJ. alpha-Lipoic acid as a biological antioxidant. Free Radic Biol Med. 1995;19(2):227–250. | |
van Horssen J, Witte ME, Schreibelt G, de Vries HE. Radical changes in multiple sclerosis pathogenesis. Biochim Biophys Acta. 2011;1812(2):141–150. | |
Hendriks JJ, Alblas J, van der Pol SM, van Tol EA, Dijkstra CD, de Vries HE. Flavonoids influence monocytic GTPase activity and are protective in experimental allergic encephalitis. J Exp Med. 2004;200(12):1667–1672. | |
Smith KJ, Kapoor R, Felts PA. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 1999;9(1):69–92. | |
van Meeteren ME, Hendriks JJ, Dijkstra CD, van Tol EA. Dietary compounds prevent oxidative damage and nitric oxide production by cells involved in demyelinating disease. Biochem Pharmacol. 2004;67(5):967–975. | |
Aboul-Enein F, Rauschka H, Kornek B, et al. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol. 2003;62(1):25–33. | |
Haider L, Fischer MT, Frischer JM, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134(Pt 7):1914–1924. | |
Naidoo R, Knapp ML. Studies of lipid peroxidation products in cerebrospinal fluid and serum in multiple sclerosis and other conditions. Clin Chem. 1992;38(12):2449–2454. | |
van Horssen J, Schreibelt G, Drexhage J, et al. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic Biol Med. 2008;45(12):1729–1737. | |
Gray E, Thomas TL, Betmouni S, Scolding N, Love S. Elevated myeloperoxidase activity in white matter in multiple sclerosis. Neurosci Lett. 2008;444(2):195–198. | |
Gray E, Thomas TL, Betmouni S, Scolding N, Love S. Elevated activity and microglial expression of myeloperoxidase in demyelinated cerebral cortex in multiple sclerosis. Brain Pathol. 2008;18(1):86–95. | |
Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain. 2007;130(Pt 11):2800–2815. | |
Cross AH, Manning PT, Keeling RM, Schmidt RE, Misko TP. Peroxynitrite formation within the central nervous system in active multiple sclerosis. J Neuroimmunol. 1998;88(1–2):45–56. | |
Liu JS, Zhao ML, Brosnan CF, Lee SC. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am J Pathol. 2001;158(6):2057–2066. | |
Smith KJ, Lassmann H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002;1(4):232–241. | |
Veto S, Acs P, Bauer J, et al. Inhibiting poly(ADP-ribose) polymerase: a potential therapy against oligodendrocyte death. Brain. 2010;133(Pt 3):822–834. | |
Witte ME, Bo L, Rodenburg RJ, et al. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol. 2009;219(2):193–204. | |
Dutta R, McDonough J, Yin X, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59(3):478–489. | |
Dutta R, Trapp BD. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog Neurobiol. 2011;93(1):1–12. | |
Mahad D, Ziabreva I, Lassmann H, Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain. 2008;131(Pt 7):1722–1735. | |
Zambonin JL, Zhao C, Ohno N, et al. Increased mitochondrial content in remyelinated axons: implications for multiple sclerosis. Brain. 2011;134(Pt 7):1901–1913. | |
Mahad DJ, Ziabreva I, Campbell G, et al. Mitochondrial changes within axons in multiple sclerosis. Brain. 2009;132(Pt 5):1161–1174. | |
Nikic I, Merkler D, Sorbara C, et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med. 2011;17(4):495–499. | |
Volterra A, Trotti D, Tromba C, Floridi S, Racagni G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J Neurosci. 1994;14(5 Pt 1):2924–2932. | |
Matute C, Sanchez-Gomez MV, Martinez-Millan L, Miledi R. Glutamate receptor-mediated toxicity in optic nerve oligodendrocytes. Proc Natl Acad Sci U S A. 1997;94(16):8830–8835. | |
Matute C. Characteristics of acute and chronic kainate excitotoxic damage to the optic nerve. Proc Natl Acad Sci U S A. 1998;95(17):10229–10234. | |
McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. 1998;4(3):291–297. | |
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938–952. | |
Cui W, Bai Y, Luo P, Miao L, Cai L. Preventive and therapeutic effects of MG132 by activating Nrf2-ARE signaling pathway on oxidative stress-induced cardiovascular and renal injury. Oxid Med Cell Longev. 2013;2013:306073. | |
Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34(4):176–188. | |
Alam J, Killeen E, Gong P, et al. Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am J Physiol Renal Physiol. 2003;284(4):F743–F752. | |
Stewart D, Killeen E, Naquin R, Alam S, Alam J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J Biol Chem. 2003;278(4):2396–2402. | |
Dinkova-Kostova AT, Holtzclaw WD, Cole RN, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99(18):11908–11913. | |
Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. | |
Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8(4):379–391. | |
Hirotsu Y, Katsuoka F, Funayama R, et al. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012;40(20):10228–10239. | |
Lee JM, Li J, Johnson DA, et al. Nrf2, a multi-organ protector? FASEB J. 2005;19(9):1061–1066. | |
Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009;8(3):280–291. | |
Filippini G, Brusaferri F, Sibley WA, et al. Corticosteroids or ACTH for acute exacerbations in multiple sclerosis. Cochrane Database Syst Rev. 2000;(4):CD001331. | |
Weiner HL, Dau PC, Khatri BO, et al. Double-blind study of true vs sham plasma exchange in patients treated with immunosuppression for acute attacks of multiple sclerosis. Neurology. 1989;39(9):1143–1149. | |
Weinshenker BG, O’Brien PC, Petterson TM, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol. 1999;46(6):878–886. | |
Kieseier BC. The mechanism of action of interferon-beta in relapsing multiple sclerosis. CNS Drugs. 2011;25(6):491–502. | |
Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45(7):1268–1276. | |
Chen PC, Vargas MR, Pani AK, et al. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: critical role for the astrocyte. Proc Natl Acad Sci U S A. 2009;106(8):2933–2938. | |
Ellrichmann G, Petrasch-Parwez E, Lee DH, et al. Efficacy of fumaric acid esters in the R6/2 and YAC128 models of Huntington’s disease. PloS One. 2011;6(1):e16172. | |
Kappos L, Gold R, Miller DH, et al. Effect of BG-12 on contrast-enhanced lesions in patients with relapsing – remitting multiple sclerosis: subgroup analyses from the phase 2b study. Mult Scler. 2012;18(3):314–321. | |
Kappos L, Gold R, Miller DH, et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;372(9648):1463–1472. | |
Bittner S, Meuth SG, Gobel K, et al. TASK1 modulates inflammation and neurodegeneration in autoimmune inflammation of the central nervous system. Brain. 2009;132(Pt 9):2501–2516. | |
Khodagholi F, Eftekharzadeh B, Maghsoudi N, Rezaei PF. Chitosan prevents oxidative stress-induced amyloid beta formation and cytotoxicity in NT2 neurons: involvement of transcription factors Nrf2 and NF-kappaB. Mol Cell Biochem. 2010;337(1–2):39–51. | |
Lewerenz J, Albrecht P, Tien ML, et al. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem. 2009;111(2):332–343. | |
Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem Res Toxicol. 2008;21(3):705–710. | |
Thiessen A, Schmidt MM, Dringen R. Fumaric acid dialkyl esters deprive cultured rat oligodendroglial cells of glutathione and upregulate the expression of heme oxygenase 1. Neurosci Lett. 2010;475(1):56–60. | |
Kavanagh GM, Burton JL, Donnell VO. Effects of dithranol on neutrophil superoxide generation in patients with psoriasis. Br J Dermatol. 1996;134(2):234–237. | |
Litjens NH, Burggraaf J, van Strijen E, et al. Pharmacokinetics of oral fumarates in healthy subjects. Br J of Clin Pharmacol. 2004;58(4):429–432. | |
Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–1097. | |
Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. | |
Scannevin RH, Chollate S, Jung MY, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341(1):274–284. | |
Lee DH, Gold R, Linker RA. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: therapeutic modulation via fumaric acid esters. Int J Mol Sci. 2012;13(9):11783–11803. | |
Schmidt MM, Dringen R. Fumaric acid diesters deprive cultured primary astrocytes rapidly of glutathione. Neurochem Int. 2010;57(4):460–467. | |
Chauhan D, Catley L, Li G, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005;8(5):407–419. | |
Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew Chem Int Ed Engl. 2003;42(3):355–357. | |
Potts BC, Albitar MX, Anderson KC, et al. Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr Cancer Drug Targets. 2011;11(3):254–284. | |
Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci U S A. 1999;96(18):10403–10408. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.