Back to Archived Journals » Gastrointestinal Cancer: Targets and Therapy » Volume 4
Pathology, genetic alterations, and targets of differentially expressed microRNAs in pancreatic cancer
Authors Azevedo-Pouly AC, Schmittgen T
Received 30 January 2014
Accepted for publication 5 April 2014
Published 18 June 2014 Volume 2014:4 Pages 75—87
DOI https://doi.org/10.2147/GICTT.S38297
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Ana Clara P Azevedo-Pouly, Thomas D Schmittgen
Division of Pharmaceutics and Pharmaceutical Chemistry, the Ohio State University College of Pharmacy, Columbus, OH, USA
Abstract: Since their discovery in mammals in 2001, the field of microRNA (miRNA) research has grown exponentially. miRNAs regulate protein translation following binding to conserved sequences within the 3' untranslated region of messenger RNAs. miRNAs are found to regulate nearly all biological processes, and their expression has been shown to differentially regulate a large number of diseases including cancer. Pancreatic ductal adenocarcinoma (PDAC) was one of the initial groups of cancers to demonstrate differential miRNA expression. Since then, there have been numerous studies linking differential miRNA expression to PDAC. Translational extrapolation of these studies has been done linking diagnostic, prognostic, and therapeutic applications, and multiple review articles and book chapters have been written on these subjects. The intent here is to provide an overview of pancreatic cancer and review the current state of the validated and published findings on the messenger RNA targets of differentially expressed miRNAs in PDAC. We then attempt to summarize these findings to extrapolate them in the hopes of better understanding how altered miRNA expression in PDAC may alter the phenotype of this disease.
Keywords: microRNA, pancreatic cancer, pancreatic ductal adenocarcinoma, target
Endocrine pancreatic cancers
Although pancreatic cancer is the tenth most common type of cancer in terms of incidence, it is the fourth leading cause of cancer-related death in the USA. The annual death rate is approximately 35,000, with a 5-year survival rate of less than 6%.1 While multiple types of cancers of the pancreas exist, the term “pancreatic cancer” most often refers to pancreatic ductal adenocarcinoma (PDAC) since it comprises 75%–90% of pancreatic tumors.2,3 Cancers of the pancreas are often classified based on their origin.
The pancreas is subdivided into the endocrine pancreas, the exocrine pancreas, and the surrounding stroma.4 The endocrine pancreas is responsible for regulation of carbohydrate metabolism.5 Cells of the endocrine pancreas form the islets of Langerhans, which are spherical centers containing alpha, beta, and delta cells surrounded by reticular fibers and blood capillaries.6 These cells compose roughly 1% of the pancreas and are mostly localized in the tail end of the organ.4 Islet cells as a group secrete insulin, glucagon, somatostatin, and pancreatic polypeptide.4,7,8 Tumors of the endocrine pancreas comprise less than 2% of all pancreatic tumors.9 They are known as pancreatic neuroendocrine tumors and are classified based on the hormone-secreting properties of the cells. Types of pancreatic neuroendocrine tumors include insulinomas, gastrinomas, glucagonomas, VIPomas, and somatostatinomas. Pancreatic neuroendocrine tumors are associated with loss of the multiple endocrine neoplasia type 1 gene (MEN1) and can range from less aggressive to highly malignant disease.10,11
Exocrine pancreatic cancers
Acinar cell carcinoma, pancreatoblastoma, and PDAC all arise from the exocrine part of the pancreas, which is composed of acinar cells, centroacinar cells, and ductal cells. Acinar cells are pyramidal-shaped cells that comprise the acini lobules. Acini make up approximately 84% of the pancreas and are responsible for secreting digestive proenzymes that are later activated in the duodenum.2,12 These cells have prominent Golgi apparatus to accommodate the secretion of over 20 digestive enzymes packaged in the zymogen granules. The enzymes secreted include proteases (ie, trypsin, chymotrypsins, carboxypeptidases, aminopeptidase, and elastase), amylase, lipases, and nucleases.12 Centroacinar cells are present within the acini and are located at the origin of the draining ducts. These cells are much smaller and lack the enzyme containing zymogen granules present in acinar cells. Centroacinar cells precede ductal cells in the formation of the ductal system, which gradually increases in size from intercalated and intralobular ducts to major ducts.13 Cells forming the ductal system (cubic, goblet, and cylinder epithelial cells) contribute to 10% of the total pancreas. The ductal system guarantees the delivery of the acini enzymes to the intestine, and serves a secretory function by contributing to the pancreatic juice via secretion of bicarbonate.4,12
The exocrine pancreas gives rise to multiple types of malignancies. Serous cystadenocarcinomas appear to have a centroacinar origin and lack mucinous differentiation. Although serous cystadenocarcinomas tend to form multiple fluid-filled cysts that are fairly benign, a subset shows recurrence and metastasis.14 Acinar cells can give rise to acinar cell carcinoma.15 These carcinomas are very rare and sometimes secrete digestive enzymes that lead to hypersecretion syndrome. Acinar cell carcinomas have early metastasis to the liver, are highly cellular, and lack the KRAS mutation present in other types of pancreatic cancer.16–18 Although very rare, tumors of mixed lineages also occur in the pancreas. Pancreatoblastoma is a prime example of such tumors. Pancreatoblastomas are well demarcated, consist of acinar, ductal, and endocrine cell types, and are more commonly found in early childhood.19 Other subtypes of mixed and/or unknown origin also exist.2
Exocrine tumors of the pancreas can also be of ductal cell origin. Adenosquamous, squamous carcinoma, colloid carcinoma, medullary carcinoma, and signet ring cell carcinoma are all examples of exocrine tumors of ductal origin. These carcinomas, however, are very rare in the pancreas.20–23 As previously mentioned, PDAC has the highest incidence among the exocrine tumors of the pancreas. As the name suggests, PDAC shows ductal-like differentiation with expression of cytokeratins 7, 8, 13, 18, and 1924–26 and mucin-positive cells that are typically cuboidal-shaped.27 For these reasons, PDAC has been historically classified as a ductal tumor of the exocrine pancreas. Another feature of this carcinoma is the desmoplastic stroma that the host tissue produces around the tumor. This desmoplastic reaction is composed of pancreatic stellate cells, fibroblasts, and extracellular matrix proteins like fibronectin 1 and collagens I and III.28 This stroma is very similar to the fibrosis caused by chronic pancreatitis (CP), which makes proper pathological diagnosis which can confound proper pathological diagnosis.29–31 PDAC is also very aggressive and highly metastatic to the liver and lymph nodes, with only 10%–20% of cases qualifying for resection at the time of diagnosis.32–34 These features, along with high stromal content, impose a barrier to effective drug delivery35 and contribute to the high mortality of the disease.
Precursor lesions to PDAC
Before the full-blown development of PDAC, a number of precursor lesions can be observed in human patients and mouse models of the disease. Pancreatic intraepithelial lesions (PanINs) are the most closely recapitulated lesions in mouse models of PDAC. Subclassification of these lesions depends on the amount of cellular atypia that is found. PanIN-1 lesions are the lowest grade lesions, and are marked by small nuclei and tall ductal “columnar” cells.36 As the lesions and atypia progress, the amount of Ki67 immunostaining (or cellular proliferation) increases.37,38 PanIN-2 lesions have more papillary architecture to the lesion and also contain nuclear abnormalities, such as expanded nuclei and loss of polarity. PanIN-3 lesions are strictly papillary or micropapillary, and are characterized by luminal necrosis, mitosis, macronucleoli, and loss of polarity.36 In terms of mutations, cyclin-dependent kinase inhibitor 2A (p16/CDKN2A) p16 loss is widely found in PanIN-2 lesions (16%–30%);39,40 p53 and SMAD family member 4 (SMAD4) loss are late mutations that appear to be exclusive to carcinoma in situ (PanIN-3).38,41 The oncogenic Kras is already present in 36% of PanIN-1a lesions, 44% of PanIN-1b lesions, and 87% of cases with PanIN-2 grade or higher.42 It is important to note that the early prevalence of this mutation demonstrates the necessity of this oncogenic change for cancer progression.
Risk factors for PDAC
Risk factors for PDAC include family history of certain cancer syndromes, cigarette smoking, obesity, and pancreatitis.43–49 Familial atypical multiple mole melanoma and Peutz-Jeghers syndrome have been linked with a risk for PDAC.50–54 Cigarette smoking is the only preventable risk factor identified so far and accounts for up to 25% of PDAC cases,44–46,49,55 although alcohol consumption has also been independently linked.56 Studies have reported up to an 18-fold increase in the incidence of pancreatic cancer in patients suffering from CP,57,58 but whether CP is a precursor or a result of PDAC is controversial. Not only is there an increased incidence of PDAC in patients with CP,59 but also the desmoplastic reaction mentioned above is histologically similar if not identical to pancreatitis. This implies that PDAC will always happen in the presence of something that looks like CP.60 However, the confounding factor of PDAC arising in the ductal cells and CP arising in acinar cells has inspired debate. Logsdon and Ji argue that CP is in fact a precursor of PDAC for various reasons.60 First, the precursor PanIN lesions of PDAC are found in almost all patients with CP.61,62 Second, the early Kras oncogenic mutation is found in approximately 30% of CP cases (although reports vary widely).63–66 Third, increased Kras activity can lead to PDAC as well as the inflammatory responses that lead to CP.60 Finally, a new mouse model that expresses the oncogenic Kras exclusively in the acinar cells can reiterate the PanIN progression to PDAC when combined with experimentally induced CP.67 This recent evidence questions the cell of origin of PDAC and has strengthened the paradigm that CP could be a precursor to PDAC.60
Genetic alterations in PDAC
Like all human cancers, PDAC is a disease consisting of multiple genetic changes that are either inherited or accumulate throughout the patient’s lifetime. PDAC is marked by a series of mutations in tumor suppressor genes and oncogenes. Tumor suppressor genes promote tumor growth upon their inactivation and oncogenes upon their activation. The most prevalent altered genes in PDAC are KRAS, receptor tyrosine-protein kinase erbB-2 (HER-2/neu, ERBB2), CDKN2A, TP53, SMAD4, and breast cancer 2, early onset (BRCA2).68–73 In a landmark paper, Jones et al used next-generation DNA sequencing on PDAC from 24 patients to demonstrate that PDAC stems from mutations of primarily four different genes, ie, KRAS, SMAD4, TP53, and CDKN2A.74 It has also been estimated that up to 70% of PDAC overexpresses HER-2/neu.68,75,76 HER-2/neu is part of the epidermal growth factor receptor family, is not expressed in normal ducts, but has an 80%–90% rate of expression in low-grade and high-grade PanINs.68 CDKN2A protein regulates the cell cycle by controlling the G1/S transition, a major check-point for cell division. CDKN2A protein is encoded in the CDKN2A gene in humans and is lost in 95% of pancreatic cancers by deletion, mutation, or hypermethylation.77 The TP53 gene encodes for the p53 protein, which is responsible for regulation of various points of the cell cycle and induction of apoptosis. Unregulated cell proliferation due to loss of p53 occurs in 55%–75% of pancreatic cancers.72,78 Inactivation of SMAD4 is present in about 50% of PDAC cases.79 SMAD4 is a downstream target of the transforming growth factor-beta (TGF-β) pathway, and upon activation of the signaling cascade, it travels to the nucleus to transcriptionally regulate cellular growth genes.80 Lastly, BRCA2 mutations have also been linked to PDAC. BRCA2 mutations lead to a defective BRCA2 protein that is unable to repair double strand breaks in DNA.80 The mutated gene is present in around 10% of PDAC cases and may be acquired or inherited, and individuals with this germline mutation can have up to a 10-fold increased chance of developing PDAC.69,81–83 The risk is even greater for other types of cancers.
In terms of oncogenic mutations, KRAS is central to PDAC. KRAS is part of the gene family that also contains neuroblastoma RAS viral (v-ras)oncogene homologue (NRAS) and Harvey rat sarcoma viral oncogene homologue (HRAS). The proteins encoded by these genes are G-like regulatory proteins that are involved in the control of cell growth. The Ras family of proteins comprises GTPases that convert guanosine-5′-triphosphate (GTP) into guanosine-5′-diphosphate (GDP). While bound to GTP, they are active and can trigger downstream pathways, but are turned off with the conversion to GDP. As a family, these proteins differ in their C-terminus and in the downstream pathways that they activate.84 KRAS is mutated in more than 90% of PDAC cases.85,86 Mutations in KRAS almost exclusively occur in codons 12, 13, and 61.87 KRAS and other Ras proteins are GTP-bound. Hydrolysis of GTP to GDP by GTPase-activating proteins turns the protein to an “off” state. Mutations in these codons cause KRAS to be resistant to GTPase-activating protein hydrolysis and are therefore in the constitutively active state. Constitutive KRAS activity leads to continuous induction of the mitogen-activated protein kinase and phosphatidylinositide-3 kinase-mammalian target of rapamycin (mTOR) pathways, which in turn leads to increased cell proliferation, angiogenesis, migration, and increased survival.88 In addition to the high prevalence and strong oncogenic activity of the KRAS mutations, this oncogenic event takes place very early in development of PDAC (PanIN stage 1), and appears to set the stage for disease progression.42 The few cases of PDAC that are wild-type for the KRAS mutation often possess the v-raf murine sarcoma viral oncogene homologue B (BRAF) mutation.89 These two oncogenic mutations of pancreatic cancer may be mutually exclusive in patients. Both, however, lead to activation of the mitogen-activated protein kinase pathway and highlight the importance of this pathway for the development of the disease.80
MicroRNA expression and PDAC
MicroRNAs (miRNAs) are small noncoding RNAs that are processed from larger precursor RNAs. The active, mature miRNA is roughly 21 nucleotides in length and functions by binding through partial complementarity to conserved sequences, typically within the 3′ untranslated region of protein coding messenger RNA (mRNA). Nucleotides 2–8 of the mature miRNA (ie, the so-called 5′ seed sequence) must interact with complete complementarity for the miRNA to be functional. Interaction of the miRNA with the mRNA most often results in reduced translation; sometimes with degradation of the mRNA but other times without mRNA degradation. Discovery and validation of miRNA target genes vary from study to study. One approach involves mining gene expression profiling or RNA sequencing data to look for inverse expression levels of miRNA and mRNA.90 Published target prediction programs are used to identify putative targets of the miRNA.91 Studies are validated using luciferase reporter assays to examine reduced luciferase expression following binding of the synthetic pre-miRNA to the target sequence of miRNA cloned downstream of the luciferase reporter gene.92 Also, Western blotting is a convenient tool to look for reduced expression of the protein in cell lines transfected with pre-miRNA oligo.93
PDAC was among the first groups of cancers to be linked to altered miRNA expression.94–96 In addition to pancreas tissue,94–96 differentially expressed miRNAs have been identified in PanINs,97,98 plasma,99,100 stool,101 and cystic fluid102 of patients with pancreatic disease. Differentially expressed miRNA is defined as those that are either increased or decreased in the tumor compared with normal pancreas or pancreas tissue that is benign and adjacent to the tumor. The definition of the fold change varies, but is commonly a 1.5–2-fold difference in expression. Differential miRNA expression in PDAC103,104 and in cystic pancreatic lesions105 has been reported in a number of excellent, recent review articles,103,104 and that information will not be repeated here. Instead, the remainder of this review focuses on published, validated target genes of differentially expressed miRNAs in PDAC. The data are summarized in Table 1. The table lists the differentially expressed miRNA, if it is increased or decreased in PDAC, the validated mRNA target gene from the study, the function of the target gene, and finally the citation. We will review what we believe to be the most important targets of differentially expressed miRNAs in PDAC. Due to space limitations, we will not report on each of the studies listed in Table 1.
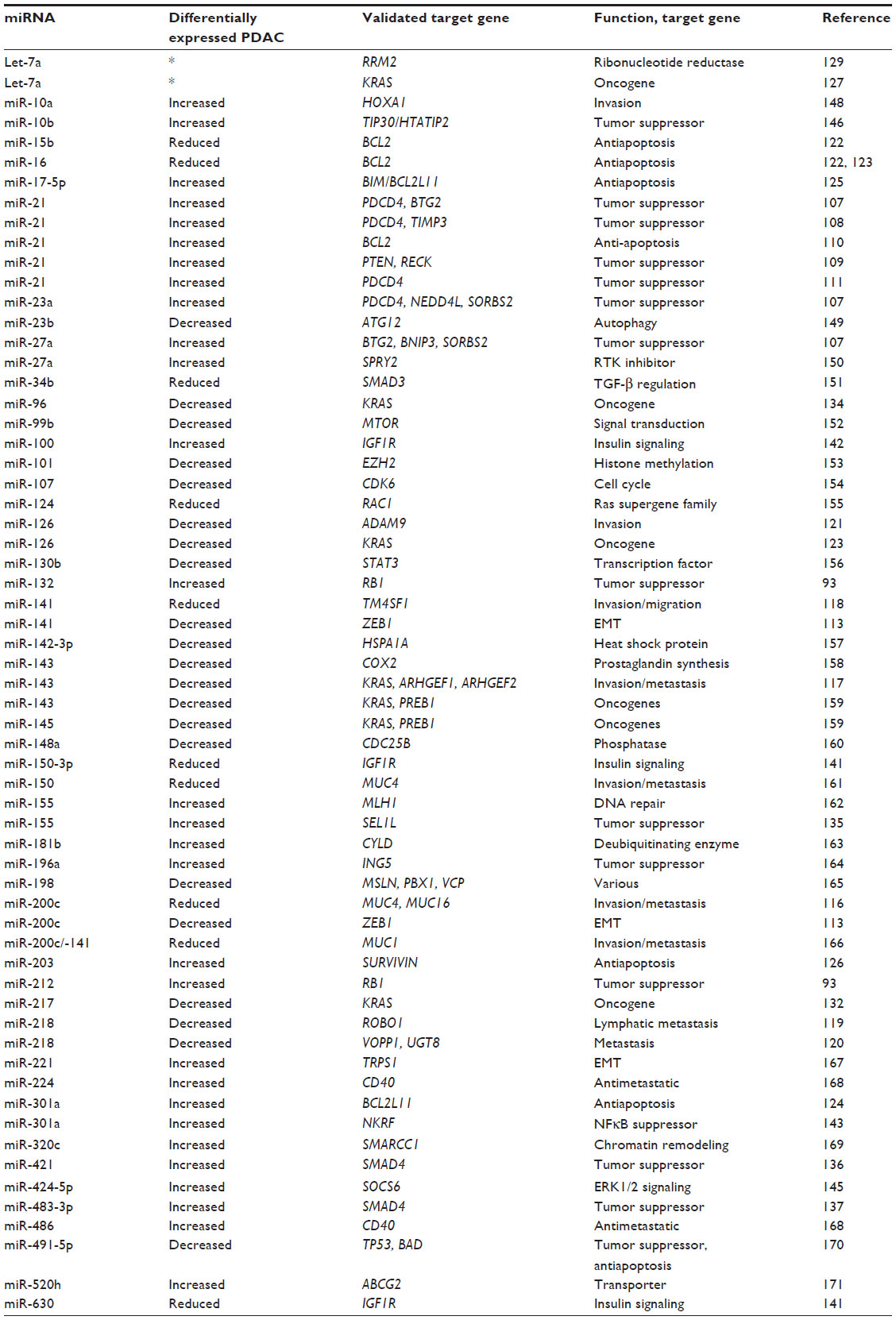 | Table 1 mRNA targets of differentially expressed miRNA in pancreatic ductal carcinoma |
miR-21
miR-21 is perhaps the most studied, differentially expressed miRNA in PDAC. The expression of miR-21 is increased in nearly all solid tumors, especially those of epithelial origin, as reviewed in Selcuklu et al.106 The MIR21 gene is found on human chromosome 17q23.1 and is located within exon 1 of the vacuole membrane protein 1 (VMP1) gene. Validated target mRNAs of miR-21 in PDAC include the tumor suppressors programmed cell death 4 (PDCD4),107,108 B-cell translocation gene 2 (BTG2),107 tissue inhibitor metallopeptidase 3,108 reversion-inducing-cysteine-rich protein with kazal motifs (RECK),109 and B-cell chronic lymphocytic leukemia/lymphoma 2 (BCL2).110 miR-21, along with miR-23a and miR-27a, acted synergistically to suppress a group of five tumor suppressors in PDAC.107 Inhibition of these miRNA with anti-miRNAs resulted in reduced cell proliferation in vitro and reduction in tumor burden in vivo.107 miR-21-mediated repression of PDCD4 resulted in increased cell proliferation and reduced cell death in vitro.111 Anti-miR-21 reduced cell proliferation in PDAC cell lines and there was a direct correlation between exposure to anti-miR-21 and increased expression of the tumor suppressors phosphatase and tensin homologue and RECK.109 A curcumin analog reduced miR-21 expression in a gemcitabine-resistant PDAC cell line; however, no miR-21 target gene associated with the resistance was noted.112
miR-200 family
The miR-200 family consists of five miRNAs, ie, miR-200a, miR-200b, miR-200c, miR-141, and miR-429. miR-200c and miR-141 are located on human chromosome 12p13.31, and miR-200a, miR-200b, and miR-429 are present on human chromosome 1p36.33. The miR-200 family has been widely implicated in modulating the epithelial mesenchymal transition in pancreatic and other types of cancer.113–115 Epithelial mesenchymal transition is the process by which cells lose their epithelial morphology and become more mesenchymal-like, with increased invasiveness and metastatic potential. Epithelial cells express high levels of E-cadherin (CDH1) while mesenchymal cells have high expression in zinc finger E-box binding homeobox 1 (ZEB1), vimentin, and N-cadherin. ZEB1 suppresses CDH1 and the miR-200 family also suppresses ZEB1. Therefore, a feedback loop exists in which low levels of the miR-200c family in mesenchymal cells result in increased levels of ZEB1 and therefore reduced CDH1.113,114
Reduced levels of the miR-200 family and increased expression of miR-21 was observed in gemcitabine-resistant cells compared with gemcitabine-sensitive cells.112 In addition to the epithelial mesenchymal transition markers, the miR-200 family has also been shown to target the coding sequences of mucin 4 (MUC4) and MUC16.116 MUC4 and MUC16 are highly overexpressed in PDAC and are involved in the metastatic phenotype of pancreatic and ovarian cancer cells.
miRNAs associated with invasion and metastasis of PDAC
In addition to the miR-200 family, a number of differentially expressed miRNAs have been shown to regulate invasion and metastasis in PDAC. miR-143 targets several metastasis-regulating genes, including Rho guanine nucleotide exchange factors 1 and 2 and KRAS.117 Transfection of miR-143 mimic into pancreatic cancer cell lines reduced migration and invasion by direct targeting of these genes as well as indirect effects on CDH1, matrix metallopeptidase 2 and matrix metallopeptidase 9.117 miR-141 reduced in vitro invasion and metastasis by targeting TM4SF1, but had no effect on cell proliferation, cell cycle, or apoptosis.118 miR-218 was reported to regulate three prometastatic genes, ie, roundabout axon guidance receptor 1, vesicular overexpressed in cancer prosurvival protein 1, and UDP glycosyltransferase 8.119,120 Enhancer of zeste homologue 2 suppression of miR-218 in PDAC cells reduced in vitro and in vivo cell proliferation as well as metastasis in nude mice through miR-218 targeting of UDP glycosyltransferase 8.120 miR-10a targeting of the homeobox A1 gene increased in vitro invasion, suggesting that increased levels of miR-10a in PDAC patients could increase metastasis. Reduced expression of miR-126 in PDAC cell lines resulted in increased migration, invasion, and reduction of CDH1 levels by targeting ADAM metallopeptidase domain 9.121
Apoptosis in PDAC
A variety of miRNAs that are deregulated in PDAC have been shown to regulate apoptosis. BCL2 is a well-known suppressor of apoptosis. The miRNAs miR-15b, miR-16, and miR-21, each of which is increased in PDAC, target BCL2.110,122 miR-21 resulted in increased levels of BCL2 via direct targeting of the BCL2 3′ untranslated region.110 Apoptosis in MIA PaCa-2 cells was increased or reduced by miR-21 inhibitor or miR-21 mimic, respectively.110 miR-15b and miR-16 are reduced in PDAC tissues.123 Activation of pancreatic stellate cells resulted in reduced miR-15b and miR-16 expression and reduced apoptosis by the miRNAs regulating BCL2.122 Bcl2-Like 11 (BCL2L11 or BIM) is a proapoptotic factor that interacts with other members of the BCL2 family. Apoptosis was reduced in PDAC through direct targeting of BIM by increased levels of miR-17-5p and miR-301a.124,125 When the PDAC cell line CFPAC-1 (cycstic fibrosis pancreatic adenocarcinoma cell line) was treated with survivin siRNA or miR-203, in vitro cell proliferation and in vivo tumor growth was reduced and apoptosis was increased in vitro.126
miRNAs regulating oncogenes in PDAC
A number of miRNAs have been discovered to regulate critical oncogenes in PDAC. Among these oncogenes, KRAS has been reported to be the target of the most differentially expressed miRNAs in PDAC. Let-7a was shown to regulate KRAS in AsPC-1 pancreatic cancer cells.127 Let-7 has already been implicated in the regulation of KRAS in a number of other solid tumors, including lung cancer.128 Let-7 is a family of isoforms that contain the identical 5′ seed sequence and thus would be expected to regulate many of the same genes. Examination of miRNA expression profiling data on the Gene Expression Omnibus show that some members of the let-7 family are reduced in PDAC specimens while others are increased in PDAC. Therefore, it is difficult to draw a conclusion on a let-7-KRAS axis in PDAC. Let-7a was reduced in a variety of PDACs that were resistant to gemcitabine.129
The miRNAs miR-143 and miR-145 were also shown to target KRAS along with Ras responsive element binding protein 1. Low levels of miR-143 and miR-145 in PDAC result in increased levels of Ras responsive element binding protein 1 which then suppress the miR-143 and miR-145 promoter, maintaining a feed forward mechanism that potentiates KRAS signaling.130 miR-126 targets KRAS, resulting in increased KRAS protein levels but no change in mRNA.123 Interestingly, the interaction between miR-126 and the KRAS 3′ untranslated region did not occur through the traditional 5′ seed interaction but instead by G·U wobbles within this region.123
miR-217 is another miRNA that has been reported to target KRAS. miR-217 along with miR-216a/-216b forms a cluster of three miRNAs with enriched expression in the pancreas.131 Of interest is that while the 5′ seed sequence of miR-217 differs from miR-216a/-216b, both miRNAs target KRAS; miR-217 in PDAC132 and 216b in nasopharyngeal carcinoma.133 Finally, miR-96 is another miRNA that is reduced in PDAC and has been linked to KRAS regulation through direct targeting of KRAS.134 Reduced expression of miRNAs, such as miR-217, miR-216a, miR-216b, miR-143, and miR-145, in PDAC will result in increased KRAS expression. As KRAS is mutated in over 90% of human PDAC, it is difficult to separate the contributions, if any, between the activated mutated form of KRAS and any additional bystander effects from miRNA-related increase in KRAS expression.
miRNA regulating tumor suppressors in PDAC
As a class, tumor suppressors are targeted by the greatest number of differentially expressed miRNAs in PDAC. As mentioned previously, PDCD4 (a tumor suppressor that inhibits translation initiation) is a direct target of miR-21111 and miR-23a.107 These miRNAs also target the tumor suppressors BTG2 and E3 ubiquitin-protein ligase NEDD4-like, reducing both in vitro and in vivo cell growth.107
Other tumor-suppressive targets of miRNAs in PDAC include the miR-132/-212 cluster that targets retinoblastoma.93 These miRNAs were increased in PDAC specimens and were shown to suppress RB1 mRNA and protein. Sel-1 suppressor of lin-12-like (SEL1L) is a putative tumor suppressor that is downregulated in PDAC. SEL1L functions by displacing misfolded proteins from the endoplasmic reticulum to the cytoplasm and is believed to play a role in Notch signaling. Liu et al identified miR-155, an miRNA often upregulated in PDAC, to directly target SEL1L mRNA.135
SMAD4, also known as DPC4 (ie, deleted in pancreatic cancer), is an important tumor suppressor that functions as a coactivator and mediator of TGF-β signaling. The trimeric complex formed between SMADs 2, 3, and 4 stimulates transcription of a number of critical downstream genes in the TGF-β pathway. As previously mentioned, mutations in SMAD4 lead to its inactivation. Another possibility that contributes to reduced SMAD4 activation in PDAC is direct targeting by miRNA. miR-421, an miRNA with increased expression in PDAC, was identified to target SMAD4.136 Ectopic expression of miR-421 reduced SMAD4 protein levels in PDAC cell lines, stimulating cell proliferation and colony formation.136 Another miRNA with increased expression in PDAC, miR-483-3p, was also found to target SMAD4.137 Like miR-421, forced expression of miR-483-3p increased cell proliferation in PDAC cell lines.137
miRNAs targeting IGF1R
miRNAs have also been implicated in targeting of insulin-like growth factor-1 receptor (IGF1R). IGF1R is mainly involved in differentiation, mitogenesis, and antiapoptotic activities. Activation of the receptor by insulin-like growth factor-1 (IGF1) or IGF2 leads to autophosphorylation of the tyrosine kinase domain and downstream activation of phosphatidylinositol 3-kinase/AKT and mitogen-activated protein kinase pathways. Many cancer types present with elevated expression of IGFI, IGFII, or IGF1R,138 including PDAC,139 and inhibition of IGF1/IGF1R have been demonstrated to enhance gemcitabine sensitivity in xenografts in mice.140 By investigating the effects of adamantyl retinoid-related molecules in PDAC, Farhana et al demonstrated the regulation of IGF1R by miR-150* and miR-630.141 Their study showed that adamantyl retinoid-related molecules induce apoptosis in PDAC cell lines by upregulation of miR-150* and miR-630. Pre-miR-150* mimics reduced both v-myb avian myeloblastosis viral oncogene homologue and IGF1R protein levels, while miR-630 mimic reduced IGF1R protein alone. Treatment with the precursors also leads to increased apoptosis and a decrease in stem cell-like sphere formation in the PDAC cell lines.141 IGF1R has also been implicated as a target of miR-100 in the pancreas.142 Modulation of IGF1R protein levels was reported with miR-100 inhibitor by immunohistochemistry. These studies suggest a tumor-suppressive role for miRNAs targeting IGF1R in PDAC.
Other miRNA/targets of interest in PDAC
Other miRNAs have oncogenic potential by targeting important signaling pathways in PDAC. In addition to direct targeting of tumor suppressors or oncogenes, miRNAs may exert their malignant phenotype by targeting activators or repressors of certain signaling pathways. For example, an miRNA that is overexpressed in cancer can exert its oncogenic effect by direct targeting of a repressor of an active signaling pathway in cancer. Such is the case for the nuclear factor kappa-B (NFκB) signaling pathway and miR-301a,143 an miRNA with increased expression in PDAC.95 The NFκB pathway is constitutively activated in most PDACs.144 Increased levels of miR-301a in PDAC suppress NFκB repressing factor, a repressor of NFκB.143 Since NFκB promotes transcription of miR-301a, these interactions create a feed forward loop in which high levels of miR-301a repress NFκB repressing factor, which in turn activates NFκB signaling in PDAC.
The expression of miR-424-5p was increased in PDAC and the adjacent benign pancreas when compared with normal pancreatic tissue.145 Suppressor of cytokine-induced signaling 6 (SOCS6) is negatively regulated by miR-424-5p through direct binding of the miRNA to the SOCS6 3′ untranslated region.145 SOCS6 is involved in regulating ERK1/2 signaling, and treatment of pancreatic cancer cell lines with an miR-424-5p inhibitor resulted in increased mRNA levels of two ERK1/2 signaling pathway target genes, ie, BCL2 and myeloid cell leukemia sequence 1.145
miR-10b has increased expression in both PDAC tissues and in the plasma of PDAC patients.146 Gene profiling identified Tat-interacting protein 30 (TIP30) as a putative target of miR-10b; this was validated using reporter assays and immunoblotting. TIP30 is an oxidoreductase with tumor-suppressive activity. Knockdown of TIP30 in PDAC cell lines using either pre-miR-10b oligo or siRNA to TIP30 resulted in enhanced epidermal growth factor-dependent invasion.146 Epidermal growth factor receptor kinase inhibitors attenuated the miR-10b-induced invasiveness in vitro.
Contribution of differentially expressed miRNAs to development and progression of PDAC
PDAC is a disease of genetic alterations that occur throughout the patient’s lifetime. These include genetic (ie, mutations, deletions) and epigenetic (miRNA, chromatin remodeling, DNA methylation) insults. As mentioned previously, the work of Jones et al identified four genes that are primarily mutated in human PDAC.74 It should be noted that this study focused on protein coding genes and not noncoding RNAs. Many studies have demonstrated altered expression of miRNAs in PDAC and have linked these differentially expressed non-coding RNAs to various target genes (Table 1). However, it has never been demonstrated that altered expression of miRNAs is causative of PDAC or contributes to its progression.
The present article overviews validated mRNAs that are targeted by differentially expressed miRNAs in PDAC. These include tumor suppressors, oncogenes, and mRNAs coding for proteins that are involved in epithelial mesenchymal transition, invasion, metastasis, various signaling pathways, and apoptosis. The critical question is whether these epigenetic modifications of cellular functions are the driver or passenger events in the formation or progression of PDAC. This is a difficult question to answer because association of a gene with a pathway causing PDAC is generally proven by overexpressing or knocking down the gene in a transgenic mouse model of PDAC. For example, Medina et al demonstrated the oncogenic potential of miR-21 by overexpressing the miRNA in mice, leading to a pre-B malignant lymphoid-like phenotype.128 To our knowledge, no one has published on a mouse model of PDAC that overexpresses or knocks down an miRNA. Given that over 90% of PDACs have KRAS mutations, it is difficult to envisage other genes that are causative of PDAC in the absence of mutated KRAS. Key experiments then are to overexpress oncogenic miRNAs or knockdown tumor-suppressive miRNAs in the pancreas using conditional transgenic approaches. The ability of these changes to induce PDAC in the mice may be studied independently or through enhanced means, such as combining with experimentally induced CP or by crossing with mice harboring a PDAC-causing mutation, as in KRAS G12D mice.147
Translational application of differential expressed miRNAs in PDAC includes diagnostic markers and perhaps therapeutic targets. Additional experiments must be completed to determine if the large number of mRNA target genes that are presently known and those that will be discovered in the future contribute to the development of PDAC in humans.
Acknowledgment
This work was supported by grant 2U01CA111294 and an F31 predoctoral fellowship to ACPAP.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. | |
Basturk O, Coban I, Adsay NV. Pathologic classification and biological behavior of pancreatic neoplasia. In: Neoptolemos JP, Urrutia RA, Abbruzzese J, Büchler MW, editors. Pancreatic Cancer. New York, NY, USA: Springer; 2010. | |
Morohoshi T, Held G, Kloppel G. Exocrine pancreatic tumours and their histological classification. A study based on 167 autopsy and 97 surgical cases. Histopathology. 1983;7(5):645–661. | |
Rovasio R. Development and structure of the pancreas. In: Neoptolemos JP, Urrutia RA, Abbruzzese J, Büchler MW, editors. Pancreatic Cancer. New York, NY, USA: Springer; 2010. | |
Randle PJ, Ashcroft SJ. Carbohydrate metabolism in pancreatic islets and the release of insulin. Biochem J. 1969;112(1):1P–2P. | |
Adsay NV, Zamboni G. Paraduodenal pancreatitis: a clinico-pathologically distinct entity unifying “cystic dystrophy of heterotopic pancreas”, “para-duodenal wall cyst”, and “groove pancreatitis”. Semin Diagn Pathol. 2004;21(4):247–254. | |
Baetens D, Malaisse-Lagae F, Perrelet A, Orci L. Endocrine pancreas: three-dimensional reconstruction shows two types of islets of Langerhans. Science. 1979;206(4424):1323–1325. | |
Ishihara H, Maechler P, Gjinovci A, Herrera PL, Wollheim CB. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat Cell Biol. 2003;5(4):330–335. | |
Yao JC, Eisner MP, Leary C, et al. Population-based study of islet cell carcinoma. Ann Surg Oncol. 2007;14(12):3492–3500. | |
Muniraj T, Vignesh S, Shetty S, Thiruvengadam S, Aslanian HR. Pancreatic neuroendocrine tumors. Dis Mon. 2013;59(1):5–19. | |
Oberg K. Pancreatic endocrine tumors. Semin Oncol. 2010;37(6):594–618. | |
Pandol SJ. Digestive enzymes. In: The Exocrine Pancreas. San Rafael, CA, USA: Morgan and Claypool Life Sciences; 2010. | |
Washington MK. Gross and Microscopic Anatomy of the Pancreas. Sudbury, MA, USA: Jones and Bartlett; 2005. | |
King JC, Ng TT, White SC, Cortina G, Reber HA, Hines OJ. Pancreatic serous cystadenocarcinoma: a case report and review of the literature. J Gastrointest Surg. 2009;13(10):1864–1868. | |
Webb JN. Acinar cell neoplasms of the exocrine pancreas. J Clin Pathol. 1977;30(2):103–112. | |
de Wilde RF, Ottenhof NA, Jansen M, et al. Analysis of LKB1 mutations and other molecular alterations in pancreatic acinar cell carcinoma. Mod Pathol. 2011;24(9):1229–1236. | |
Hoorens A, Lemoine NR, McLellan E, et al. Pancreatic acinar cell carcinoma. An analysis of cell lineage markers, p53 expression, and Ki-ras mutation. Am J Pathol. 1993;143(3):685–698. | |
Hruban RH, Pitman MB, Klimstra DS. Tumors of the pancreas. In: Atlas of Tumor Pathology, Fascicle 6. Washington, DC, USA: Armed Forces Institute of Pathology; 2007. | |
Riesener KP, Hofer M, Virnich N, Fuzesi L, Schumpelick V. [Pancreatoblastoma – a rare pancreatic malignancy in childhood]. Langenbecks Arch Chir Suppl Kongressbd. 1996;113:1040–1042. German. | |
Adsay NV, Pierson C, Sarkar F, et al. Colloid (mucinous noncystic) carcinoma of the pancreas. Am J Surg Pathol. 2001;25(1):26–42. | |
Kardon DE, Thompson LD, Przygodzki RM, Heffess CS. Adenosquamous carcinoma of the pancreas: a clinicopathologic series of 25 cases. Mod Pathol. 2001;14(5):443–451. | |
Tracey KJ, O’Brien MJ, Williams LF, et al. Signet ring carcinoma of the pancreas, a rare variant with very high CEA values. Immunohistologic comparison with adenocarcinoma. Dig Dis Sci. 1984;29(6):573–576. | |
Wilentz RE, Goggins M, Redston M, et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: a newly described and characterized entity. Am J Pathol. 2000;156(5):1641–1651. | |
Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology. 2008;55(88):2016–2027. | |
Hamidov Z, Altendorf-Hofmann A, Chen Y, Settmacher U, Petersen I, Knosel T. Reduced expression of desmocollin 2 is an independent prognostic biomarker for shorter patients survival in pancreatic ductal adenocarcinoma. J Clin Pathol. 2011;64(11):990–994. | |
Schussler MH, Skoudy A, Ramaekers F, Real FX. Intermediate filaments as differentiation markers of normal pancreas and pancreas cancer. Am J Pathol. 1992;140(3):559–568. | |
Klimstra DS, Adasy NV. Benign and malignant tumors of the pancreas. In: Odze RD, editor. Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas. 1st ed. Philadelphia, PA, USA: Elsevier; 2004. | |
Rasheed ZA, Matsui W, Maitra A. Pathology of pancreatic stroma in PDAC. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum, India: Transworld Research Network; 2012. | |
Apte MV, Park S, Phillips PA, et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas. 2004;29(3):179–187. | |
Kong X, Li L, Li Z, Xie K. Targeted destruction of the orchestration of the pancreatic stroma and tumor cells in pancreatic cancer cases: molecular basis for therapeutic implications. Cytokine Growth Factor Rev. 2012;23(6):343–356. | |
Phillips P. Pancreatic stellate cells and fibrosis. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum, India: Transworld Research Network; 2012. | |
Geer RJ, Brennan MF. Prognostic indicators for survival after resection of pancreatic adenocarcinoma. Am J Surg. 1993;165(1):68–72. | |
Matsuno S, Egawa S, Fukuyama S, et al. Pancreatic Cancer Registry in Japan: 20 years of experience. Pancreas. 2004;28(3):219–230. | |
Sener SF, Fremgen A, Menck HR, Winchester DP. Pancreatic cancer: a report of treatment and survival trends for 100,313 patients diagnosed from 1985–1995, using the National Cancer Database. J Am Coll Surg. 1999;189(1):1–7. | |
Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. | |
Hruban RH, Adsay NV, Albores-Saavedra J, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25(5):579–586. | |
Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis accelerates initiation and progression to pancreatic cancer in mice expressing oncogenic Kras in the nestin cell lineage. PLoS One. 2011;6(11):e27725. | |
Klein WM, Hruban RH, Klein-Szanto AJ, Wilentz RE. Direct correlation between proliferative activity and dysplasia in pancreatic intraepithelial neoplasia (PanIN): additional evidence for a recently proposed model of progression. Mod Pathol. 2002;15(4):441–447. | |
Rosty C, Geradts J, Sato N, et al. p16 Inactivation in pancreatic intraepithelial neoplasias (PanINs) arising in patients with chronic pancreatitis. Am J Surg Pathol. 2003;27(12):1495–1501. | |
Wilentz RE, Geradts J, Maynard R, et al. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res. 1998;58(20):4740–4744. | |
Luttges J, Galehdari H, Brocker V, et al. Allelic loss is often the first hit in the biallelic inactivation of the p53 and DPC4 genes during pancreatic carcinogenesis. Am J Pathol. 2001;158(5):1677–1683. | |
Lohr M, Kloppel G, Maisonneuve P, Lowenfels AB, Luttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7(1):17–23. | |
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N Engl J Med. 2003;348(17):1625–1638. | |
Doll R, Peto R, Wheatley K, Gray R, Sutherland I. Mortality in relation to smoking: 40 years’ observations on male British doctors. BMJ. 1994;309(6959):901–911. | |
Lin Y, Tamakoshi A, Kawamura T, et al. A prospective cohort study of cigarette smoking and pancreatic cancer in Japan. Cancer Causes Control. 2002;13(3):249–254. | |
Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006;20(2):197–209. | |
Lowenfels AB, Maisonneuve P, Lankisch PG. Chronic pancreatitis and other risk factors for pancreatic cancer. Gastroenterol Clin North Am. 1999;28(3):673–685. | |
Michaud DS, Giovannucci E, Willett WC, Colditz GA, Stampfer MJ, Fuchs CS. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA. 2001;286(8):921–929. | |
Silverman DT, Dunn JA, Hoover RN, et al. Cigarette smoking and pancreas cancer: a case-control study based on direct interviews. J Natl Cancer Inst. 1994;86(20):1510–1516. | |
Bergman W, Watson P, de Jong J, Lynch HT, Fusaro RM. Systemic cancer and the FAMMM syndrome. Br J Cancer. 1990;61(6):932–936. | |
Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119(6):1447–1453. | |
Korsse SE, Harinck F, van Lier MG, et al. Pancreatic cancer risk in Peutz-Jeghers syndrome patients: a large cohort study and implications for surveillance. J Med Genet. 2013;50(1):59–64. | |
Lynch HT, Fusaro RM. Pancreatic cancer and the familial atypical multiple mole melanoma (FAMMM) syndrome. Pancreas. 1991;6(2):127–131. | |
Vasen HF, Gruis NA, Frants RR, van Der Velden PA, Hille ET, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int J Cancer. 2000;87(6):809–811. | |
Nakamura K, Nagata C, Wada K, et al. Cigarette smoking and other lifestyle factors in relation to the risk of pancreatic cancer death: a prospective cohort study in Japan. Jpn J Clin Oncol. 2011;41(2):225–231. | |
Gapstur SM, Jacobs EJ, Deka A, McCullough ML, Patel AV, Thun MJ. Association of alcohol intake with pancreatic cancer mortality in never smokers. Arch Intern Med. 2011;171(5):444–451. | |
Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24(3):349–358. | |
Talamini G, Bassi C, Falconi M, et al. Early detection of pancreatic cancer following the diagnosis of chronic pancreatitis. Digestion. 1999;60(6):554–561. | |
Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89(6):442–446. | |
Logsdon CD, Ji B. Ras activity in acinar cells links chronic pancreatitis and pancreatic cancer. Clin Gastroenterol Hepatol. 2009;7(Suppl 11):S40–S43. | |
Hermanova M, Nenutil R, Kren L, Feit J, Pavlovsky Z, Dite P. Proliferative activity in pancreatic intraepithelial neoplasias of chronic pancreatitis resection specimens: detection of a high-risk lesion. Neoplasma. 2004;51(5):400–404. | |
Volkholz H, Stolte M, Becker V. Epithelial dysplasias in chronic pancreatitis. Virchows Arch A Pathol Anat Histol. 1982;396(3):331–349. | |
Berthelemy P, Bouisson M, Escourrou J, Vaysse N, Rumeau JL, Pradayrol L. Identification of K-ras mutations in pancreatic juice in the early diagnosis of pancreatic cancer. Ann Intern Med. 1995;123(3):188–191. | |
Hsiang D, Friess H, Buchler MW, Ebert M, Butler J, Korc M. Absence of K-ras mutations in the pancreatic parenchyma of patients with chronic pancreatitis. Am J Surg. 1997;174(3):242–246. | |
Lohr M, Maisonneuve P, Lowenfels AB. K-Ras mutations and benign pancreatic disease. Int J Pancreatol. 2000;27(2):93–103. | |
Nakaizumi A, Uehara H, Takenaka A, et al. Diagnosis of pancreatic cancer by cytology and measurement of oncogene and tumor markers in pure pancreatic juice aspirated by endoscopy. Hepatogastroenterology. 1999;46(25):31–37. | |
Guerra C, Schuhmacher AJ, Canamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11(3):291–302. | |
Day JD, Digiuseppe JA, Yeo C, et al. Immunohistochemical evaluation of HER-2/neu expression in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasms. Hum Pathol. 1996;27(2):119–124. | |
Goggins M, Schutte M, Lu J, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56(23):5360–5364. | |
Lynch HT, Brand RE, Hogg D, et al. Phenotypic variation in eight extended CDKN2A germline mutation familial atypical multiple mole melanoma-pancreatic carcinoma-prone families: the familial atypical mole melanoma-pancreatic carcinoma syndrome. Cancer. 2002;94(1):84–96. | |
Ottenhof NA, de Wilde RF, Maitra A, Hruban RH, Offerhaus GJ. Molecular characteristics of pancreatic ductal adenocarcinoma. Patholog Res Int. 2011;2011:620601. | |
Redston MS, Caldas C, Seymour AB, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994;54(11):3025–3033. | |
Wilentz RE, Su GH, Dai JL, et al. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol. 2000;156(1):37–43. | |
Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. | |
Dugan MC, Dergham ST, Kucway R, et al. HER-2/neu expression in pancreatic adenocarcinoma: relation to tumor differentiation and survival. Pancreas. 1997;14(3):229–236. | |
Tsiambas E, Karameris A, Dervenis C, et al. HER2/neu expression and gene alterations in pancreatic ductal adenocarcinoma: a comparative immunohistochemistry and chromogenic in situ hybridization study based on tissue microarrays and computerized image analysis. JOP. 2006;7(3):283–294. | |
Schutte M, Hruban RH, Geradts J, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57(15):3126–3130. | |
Scarpa A, Capelli P, Mukai K, et al. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol. 1993;142(5):1534–1543. | |
Schutte M, Hruban RH, Hedrick L, et al. DPC4 gene in various tumor types. Cancer Res. 1996;56(11):2527–2530. | |
Maitra A, Kern SE, Hruban RH. Molecular pathogenesis of pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006;20(2):211–226. | |
Lucas AL, Shakya R, Lipsyc MD, et al. High prevalence of BRCA1 and BRCA2 germline mutations with loss of heterozygosity in a series of resected pancreatic adenocarcinoma and other neoplastic lesions. Clin Cancer Res. 2013;19(13):3396–3403. | |
Schutte M, da Costa LT, Hahn SA, et al. Identification by representational difference analysis of a homozygous deletion in pancreatic carcinoma that lies within the BRCA2 region. Proc Natl Acad Sci U S A. 1995;92(13):5950–5954. | |
van der Heijden MS, Brody JR, Gallmeier E, et al. Functional defects in the Fanconi anemia pathway in pancreatic cancer cells. Am J Pathol. 2004;165(2):651–657. | |
McCormick F. Ras-related proteins in signal transduction and growth control. Mol Reprod Dev. 1995;42(4):500–506. | |
Hruban RH, van Mansfeld AD, Offerhaus GJ, et al. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol. 1993;143(2):545–554. | |
Thomas RK, Baker AC, Debiasi RM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39(3):347–351. | |
Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17(24):3112–3126. | |
Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2(3):359–372. | |
Calhoun ES, Jones JB, Ashfaq R, et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am J Pathol. 2003;163(4):1255–1260. | |
Manzardo AM, Gunewardena S, Butler MG. Over-expression of the miRNA cluster at chromosome 14q32 in the alcoholic brain correlates with suppression of predicted target mRNA required for oligodendrocyte proliferation. Gene. 2013;526(2):356–363. | |
Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105. | |
Baccarini A, Brown BD. Monitoring microRNA activity and validating microRNA targets by reporter-based approaches. Methods Mol Biol. 2010;667:215–233. | |
Park JK, Henry JC, Jiang J, et al. miR-132 and miR-212 are increased in pancreatic cancer and target the retinoblastoma tumor suppressor. Biochem Biophys Res Commun. 2011;406(4):518–523. | |
Bloomston M, Frankel WL, Petrocca F, et al. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297(17):1901–1908. | |
Lee EJ, Gusev Y, Jiang J, et al. Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer. 2007;120:1046–1054. | |
Szafranska AE, Davison TS, John J, et al. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene. 2007;26(30):4442–4452. | |
du Rieu MC, Torrisani J, Selves J, et al. MicroRNA-21 is induced early in pancreatic ductal adenocarcinoma precursor lesions. Clin Chem. 2010;56(4):603–612. | |
Yu J, Li A, Hong SM, Hruban RH, Goggins M. MicroRNA alterations of pancreatic intraepithelial neoplasias. Clin Cancer Res. 2012;18(4):981–992. | |
Schultz NA, Dehlendorff C, Jensen BV, et al. MicroRNA biomarkers in whole blood for detection of pancreatic cancer. JAMA. 2014;311(4):392–404. | |
Wang J, Chen J, Chang P, et al. MicroRNAs in plasma of pancreatic ductal adenocarcinoma patients as novel blood-based biomarkers of disease. Cancer Prev Res (Phila). 2009;2(9):807–813. | |
Ren Y, Gao J, Liu JQ, et al. Differential signature of fecal microRNAs in patients with pancreatic cancer. Mol Med Rep. 2012;6(1):201–209. | |
Ryu JK, Matthaei H, Dal Molin M, et al. Elevated microRNA miR-21 levels in pancreatic cyst fluid are predictive of mucinous precursor lesions of ductal adenocarcinoma. Pancreatology. 2011;11(3):343–350. | |
Khan S, Ansarullah, Kumar D, Jaggi M, Chauhan SC. Targeting microRNAs in pancreatic cancer: microplayers in the big game. Cancer Res. 2013;73(22):6541–6547. | |
Tang S, Bonaroti J, Unlu S, et al. Sweating the small stuff: microRNAs and genetic changes define pancreatic cancer. Pancreas. 2013;42(5):740–759. | |
Frampton AE, Gall TM, Giovannetti E, et al. Distinct miRNA profiles are associated with malignant transformation of pancreatic cystic tumors revealing potential biomarkers for clinical use. Expert Rev Mol Diagn. 2013;13(4):325–329. | |
Selcuklu SD, Donoghue MT, Spillane C. miR-21 as a key regulator of oncogenic processes. Biochem Soc Trans. 2009;37 Pt 4:918–925. | |
Frampton AE, Castellano L, Colombo T, et al. MicroRNAs cooperatively inhibit a network of tumor suppressor genes to promote pancreatic tumor growth and progression. Gastroenterology. 2014;146(1):268–277. | |
Nagao Y, Hisaoka M, Matsuyama A, et al. Association of microRNA-21 expression with its targets, PDCD4 and TIMP3, in pancreatic ductal adenocarcinoma. Mod Pathol. 2012;25(1):112–121. | |
Park JK, Lee EJ, Esau E, Schmittgen TD. Antisense inhibition of miR-21 or miR-221 arrests cell cycle, induces apoptosis and sensitizes the effects of gemcitabine in pancreatic adenocarcinoma. Pancreas. 2009;38(7):e190–e199. | |
Dong J, Zhao YP, Zhou L, Zhang TP, Chen G. Bcl-2 upregulation induced by miR-21 via a direct interaction is associated with apoptosis and chemoresistance in MIA PaCa-2 pancreatic cancer cells. Arch Med Res. 2011;42(1):8–14. | |
Bhatti I, Lee A, James V, et al. Knockdown of microRNA-21 inhibits proliferation and increases cell death by targeting programmed cell death 4 (PDCD4) in pancreatic ductal adenocarcinoma. J Gastrointest Surg. 2011;15(1):199–208. | |
Ali S, Ahmad A, Banerjee S, et al. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70(9):3606–3617. | |
Burk U, Schubert J, Wellner U, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9(6):582–589. | |
Li Y, VandenBoom TG 2nd, Kong D, et al. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69(16):6704–6712. | |
Wellner U, Schubert J, Burk UC, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–1495. | |
Radhakrishnan P, Mohr AM, Grandgenett PM, Steele MM, Batra SK, Hollingsworth MA. MicroRNA-200c modulates the expression of MUC4 and MUC16 by directly targeting their coding sequences in human pancreatic cancer. PLoS One. 2013;8(10):e73356. | |
Hu Y, Ou Y, Wu K, Chen Y, Sun W. miR-143 inhibits the metastasis of pancreatic cancer and an associated signaling pathway. Tumour Biol. 2012;33(6):1863–1870. | |
Xu L, Li Q, Xu D, et al. hsa-miR-141 downregulates TM4SF1 to inhibit pancreatic cancer cell invasion and migration. Int J Oncol. 2014;44(2):459–466. | |
He H, Di Y, Liang M, et al. The microRNA-218 and ROBO-1 signaling axis correlates with the lymphatic metastasis of pancreatic cancer. Oncol Rep. 2013;30(2):651–658. | |
Li CH, To KF, Tong JH, et al. Enhancer of zeste homolog 2 silences microRNA-218 in human pancreatic ductal adenocarcinoma cells by inducing formation of heterochromatin. Gastroenterology. 2013;144(5):1086–1097. | |
Hamada S, Satoh K, Fujibuchi W, et al. MiR-126 acts as a tumor suppressor in pancreatic cancer cells via the regulation of ADAM9. Mol Cancer Res. 2012;10(1):3–10. | |
Shen J, Wan R, Hu G, et al. miR-15b and miR-16 induce the apoptosis of rat activated pancreatic stellate cells by targeting Bcl-2 in vitro. Pancreatology. 2012;12(2):91–99. | |
Jiao LR, Frampton AE, Jacob J, et al. MicroRNAs targeting oncogenes are down-regulated in pancreatic malignant transformation from benign tumors. PLoS One. 2012;7(2):e32068. | |
Chen Z, Chen LY, Dai HY, Wang P, Gao S, Wang K. miR-301a promotes pancreatic cancer cell proliferation by directly inhibiting Bim expression. J Cell Biochem. 2012;113(10):3229–3235. | |
Yan HJ, Liu WS, Sun WH, et al. miR-17-5p inhibitor enhances chemosensitivity to gemcitabine via upregulating Bim expression in pancreatic cancer cells. Dig Dis Sci. 2012;57(12):3160–3167. | |
Xu D, Wang Q, An Y, Xu L. MiR203 regulates the proliferation, apoptosis and cell cycle progression of pancreatic cancer cells by targeting Survivin. Mol Med Rep. 2013;8(2):379–384. | |
Oh JS, Kim JJ, Byun JY, Kim IA. Lin28-let7 modulates radiosensitivity of human cancer cells with activation of K-Ras. Int J Radiat Oncol Biol Phys. 2010;76(1):5–8. | |
Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467(7311):86–90. | |
Bhutia YD, Hung SW, Krentz M, et al. Differential processing of let-7a precursors influences RRM2 expression and chemosensitivity in pancreatic cancer: role of LIN-28 and SET oncoprotein. PLoS One. 2013;8(1):e53436. | |
Kent OA, Chivukula RR, Mullendore M, et al. Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev. 2010;24(24):2754–2759. | |
Liang Y, Ridzon D, Wong L, Chen C. Characterization of microRNA expression profiles in normal human tissues. BMC Genomics. 2007;8(1):166. | |
Zhao WG, Yu SN, Lu ZH, Ma YH, Gu YM, Chen J. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting KRAS. Carcinogenesis. 2010;31(10):1726–1733. | |
Deng M, Tang H, Zhou Y, et al. miR-216b suppresses tumor growth and invasion by targeting KRAS in nasopharyngeal carcinoma. J Cell Sci. 2011;124 Pt 17:2997–3005. | |
Yu S, Lu Z, Liu C, et al. miRNA-96 suppresses KRAS and functions as a tumor suppressor gene in pancreatic cancer. Cancer Res. 2010;70(14):6015–6025. | |
Liu Q, Chen J, Wang J, et al. Putative tumor suppressor gene SEL1L was downregulated by aberrantly upregulated hsa-mir-155 in human pancreatic ductal adenocarcinoma. Mol Carcinog. Epub 2013 May 9. | |
Hao J, Zhang S, Zhou Y, Liu C, Hu X, Shao C. MicroRNA 421 suppresses DPC4/Smad4 in pancreatic cancer. Biochem Biophys Res Commun. 2011;406(4):552–557. | |
Hao J, Zhang S, Zhou Y, Hu X, Shao C. MicroRNA 483-3p suppresses the expression of DPC4/Smad4 in pancreatic cancer. FEBS Lett. 2011;585(1):207–213. | |
Zhang L, Zhou W, Velculescu VE, et al. Gene expression profiles in normal and cancer cells. Science. 1997;276(5316):1268–1272. | |
Bergmann U, Funatomi H, Yokoyama M, Beger HG, Korc M. Insulin-like growth factor I overexpression in human pancreatic cancer: evidence for autocrine and paracrine roles. Cancer Res. 1995;55(10):2007–2011. | |
Awasthi N, Zhang C, Ruan W, Schwarz MA, Schwarz RE. BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Mol Cancer Ther. 2012;11(12):2644–2653. | |
Farhana L, Dawson MI, Murshed F, Das JK, Rishi AK, Fontana JA. Upregulation of miR-150* and miR-630 induces apoptosis in pancreatic cancer cells by targeting IGF-1R. PLoS One. 2013;8(5):e61015. | |
Huang JS, Egger ME, Grizzle WE, McNally LR. MicroRNA-100 regulates IGF1-receptor expression in metastatic pancreatic cancer cells. Biotech Histochem. 2013;88(7):397–402. | |
Lu Z, Li Y, Takwi A, et al. miR-301a as an NF-kappaB activator in pancreatic cancer cells. EMBO J. 2011;30(1):57–67. | |
Ling J, Kang Y, Zhao R, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(1):105–120. | |
Wu K, Hu G, He X, et al. MicroRNA-424-5p suppresses the expression of SOCS6 in pancreatic cancer. Pathol Oncol Res. 2013;19(4):739–748. | |
Ouyang H, Gore J, Deitz S, Korc M. microRNA-10b enhances pancreatic cancer cell invasion by suppressing TIP30 expression and promoting EGF and TGF-beta actions. Oncogene. Epub 2013 October 7. | |
Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–450. | |
Ohuchida K, Mizumoto K, Lin C, et al. MicroRNA-10a is overexpressed in human pancreatic cancer and involved in its invasiveness partially via suppression of the HOXA1 gene. Ann Surg Oncol. 2012;19(7):2394–2402. | |
Wang P, Zhang J, Zhang L, et al. MicroRNA 23b regulates autophagy associated with radioresistance of pancreatic cancer cells. Gastroenterology. 2013;145(5):1133–1143. e12. | |
Ma Y, Yu S, Zhao W, Lu Z, Chen J. miR-27a regulates the growth, colony formation and migration of pancreatic cancer cells by targeting Sprouty2. Cancer Lett. 2010;298(2):150–158. | |
Liu C, Cheng H, Shi S, et al. MicroRNA-34b inhibits pancreatic cancer metastasis through repressing Smad3. Curr Mol Med. 2013;13(4):467–478. | |
Wei F, Liu Y, Guo Y, et al. miR-99b-targeted mTOR induction contributes to irradiation resistance in pancreatic cancer. Mol Cancer. 2013;12(1):81. | |
Nakahara O, Takamori H, Iwatsuki M, et al. Carcinogenesis of intraductal papillary mucinous neoplasm of the pancreas: loss of microRNA-101 promotes overexpression of histone methyltransferase EZH2. Ann Surg Oncol. 2012;19 Suppl 3:S565–S571. | |
Lee KH, Lotterman C, Karikari C, et al. Epigenetic silencing of microRNA miR-107 regulates cyclin-dependent kinase 6 expression in pancreatic cancer. Pancreatology. 2009;9(3):293–301. | |
Wang P, Chen L, Zhang J, et al. Methylation-mediated silencing of the miR-124 genes facilitates pancreatic cancer progression and metastasis by targeting Rac1. Oncogene. 2014;33(4):514–524. | |
Zhao G, Zhang JG, Shi Y, et al. MiR-130b is a prognostic marker and inhibits cell proliferation and invasion in pancreatic cancer through targeting STAT3. PLoS One. 2013;8(9):e73803. | |
MacKenzie TN, Mujumdar N, Banerjee S, et al. Triptolide induces the expression of miR-142-3p: a negative regulator of heat shock protein 70 and pancreatic cancer cell proliferation. Mol Cancer Ther. 2013;12(7):1266–1275. | |
Pham H, Ekaterina Rodriguez C, Donald GW, et al. miR-143 decreases COX-2 mRNA stability and expression in pancreatic cancer cells. Biochem Biophys Res Commun. 2013;439(1):6–11. | |
Kent OA, Fox-Talbot K, Halushka MK. RREB1 repressed miR-143/145 modulates KRAS signaling through downregulation of multiple targets. Oncogene. 2013;32(20):2576–2585. | |
Liffers ST, Munding JB, Vogt M, et al. MicroRNA-148a is down-regulated in human pancreatic ductal adenocarcinomas and regulates cell survival by targeting CDC25B. Lab Invest. 2011;91(10):1472–1479. | |
Srivastava SK, Bhardwaj A, Singh S, et al. MicroRNA-150 directly targets MUC4 and suppresses growth and malignant behavior of pancreatic cancer cells. Carcinogenesis. 2011;32(12):1832–1839. | |
Liu WJ, Zhao YP, Zhang TP, et al. MLH1 as a direct target of MiR-155 and a potential predictor of favorable prognosis in pancreatic cancer. J Gastrointest Surg. 2013;17(8):1399–1405. | |
Takiuchi D, Eguchi H, Nagano H, et al. Involvement of microRNA-181b in the gemcitabine resistance of pancreatic cancer cells. Pancreatology. 2013;13(5):517–523. | |
Liu M, Du Y, Gao J, et al. Aberrant expression miR-196a is associated with abnormal apoptosis, invasion, and proliferation of pancreatic cancer cells. Pancreas. 2013;42(7):1169–1181. | |
Marin-Muller C, Li D, Bharadwaj U, et al. A tumorigenic factor interactome connected through tumor suppressor microRNA-198 in human pancreatic cancer. Clin Cancer Res. 2013;19(21):5901–5913. | |
Mohr AM, Bailey JM, Lewallen ME, et al. MUC1 regulates expression of multiple microRNAs involved in pancreatic tumor progression, including the miR-200c/141 cluster. PLoS One. 2013;8(10):e73306. | |
Su A, He S, Tian B, Hu W, Zhang Z. MicroRNA-221 mediates the effects of PDGF-BB on migration, proliferation, and the epithelial-mesenchymal transition in pancreatic cancer cells. PLoS One. 2013;8(8):e71309. | |
Mees ST, Mardin WA, Sielker S, et al. Involvement of CD40 targeting miR-224 and miR-486 on the progression of pancreatic ductal adenocarcinomas. Ann Surg Oncol. 2009;16(8):2339–2350. | |
Iwagami Y, Eguchi H, Nagano H, et al. miR-320c regulates gemcitabine-resistance in pancreatic cancer via SMARCC1. Br J Cancer. 2013;109(2):502–511. | |
Guo R, Wang Y, Shi WY, Liu B, Hou SQ, Liu L. MicroRNA miR-491-5p targeting both TP53 and Bcl-XL induces cell apoptosis in SW1990 pancreatic cancer cells through mitochondria mediated pathway. Molecules. 2012;17(12):14733–14747. | |
Wang F, Xue X, Wei J, et al. hsa-miR-520h downregulates ABCG2 in pancreatic cancer cells to inhibit migration, invasion, and side populations. Br J Cancer. 2010;103(4):567–574. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.