Back to Journals » International Journal of General Medicine » Volume 15
Pathological and Evolutive Correlations in Steroid Resistant Nephrotic Syndrome in Children
Authors Starcea IM, Bogos RA, Scurtu G, Munteanu M, Russu R, Lupu VV ![]() , Lupu A, Trandafir L, Miron IC, Mocanu MA
, Lupu A, Trandafir L, Miron IC, Mocanu MA
Received 18 November 2021
Accepted for publication 12 April 2022
Published 19 April 2022 Volume 2022:15 Pages 4187—4193
DOI https://doi.org/10.2147/IJGM.S348346
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Iuliana Magdalena Starcea,1,2 Roxana Alexandra Bogos,1 Georgiana Scurtu,1 Mihaela Munteanu,2 Radu Russu,2 Vasile Valeriu Lupu,1 Ancuta Lupu,1 Laura Trandafir,1 Ingrith Crenguta Miron,1 Maria Adriana Mocanu1,2
1Pediatrics Department, “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania; 2Nephrology Division, St. Mary’s Emergency Children Hospital, Iasi, Romania
Correspondence: Iuliana Magdalena Starcea, Pediatrics Department, “Grigore T. Popa” University of Medicine and Pharmacy, 16 University St, Iasi, 700115, Romania, Tel +40726704612, Email [email protected]; [email protected]
Background: Nephrotic syndrome (NS) is the term used for the association of edema and massive proteinuria. From a therapeutic point of view, it is important to distinguish between primitive and secondary kidney damage. The clinical evolution, prognosis and therapeutic response in the NS in children are directly determined by the anatomopathological aspect. Steroid resistant nephrotic syndrome was diagnosed in patients with idiopathic NS based on lack of complete remission despite treatment with steroids.
Purpose: To analyse the anatomopathological aspects of steroid resistant nephrotic syndrome (SRNS) and their correlation with evolution.
Materials and Methods: We made a retrospective study with the aim to analyze the anatomo-pathological aspects and their correlations with evolution in 68 cases of steroid resistant nephrotic syndrome (SRNS) hospitalized in the Pediatric Nephrology Department in Iaşi, Romania. We defined SRNS in all cases without response to corticosteroids after the first month of therapy. For all the cases selected, the period of follow-up was the minimal 6 months.
Results and Discussions: A 36% case of nephrotic syndrome was corticoresistant, with the mean age at onset of patients with SRNS being 9.18 years, compared to KDIGO studies in which the corticosteroid resistance is 10– 20%. Renal biopsy was performed in 80.88% children with SRNS and was allowed the evaluation of the activity and chronicity index. Total remission was obtained in 44.01% children with SRNS. The correlation of the anatomopathological aspects with the evolution is not statistically significant (p = 0.76), observing different therapeutic responses to all the analyzed histological types.
Conclusion: Almost half of NS in children are cortico resistant. Remission was obtained in 44% of cases of SRNS. Predicting the response to long-term treatment in SRNS is difficult using only renal biopsy; it is necessary to introduce genetic molecular analyses to establish a judicious therapeutic attitude.
Keywords: nephrotic syndrome, children, steroid resistance, evolution
Introduction
The term nephrotic syndrome (NS) is used for the association of edema and massive proteinuria. According to the last KDIGO guidelines (Nov. 2021), the SSNS is the NS with the complete remission after 4 weeks of therapy with prednisone or prednisolone at standard dose. SRNS is defined as a lack of complete remission at 4 weeks of therapy with prednisone or prednisolone at standard dose.1 From a therapeutic point of view, it is important to distinguish between primitive and secondary kidney damage, secondary to systemic diseases. Most children (90%) develop primitive nephrotic syndromes, characterized histologically by minimal glomerular lesions (MCNS), mesangial proliferation (MezPGN), or focal segmental glomerulosclerosis (FSGS). 10% of children have nephrotic syndromes secondary to systemic diseases (infections, vasculitis, neoplasms, etc.), hereditary diseases (Alport syndrome), or treatments with various drugs (gold salts, D-penicillamine, mercury, nonsteroidal anti-inflammatory drugs, conversion enzyme inhibitors, etc.). Focal segmental glomerulosclerosis (FSGS) has been reported in patients with mitochondrial cytopathy, in Galloway-Mowat syndrome (microcephaly, hiatal hernia and nephrotic syndrome) and in Schimke syndrome (spondyloepiphyseal dysplasia, immunodeficiency and nephrotic syndrome). The clinical evolution, the prognosis and the therapeutic response to nephrotic syndrome in children are directly determined by the anatomopathological aspect.
Materials and Methods
We made a retrospective study with the aim to analyze the anatomo-pathological aspects and their correlations with evolution in 68 cases of steroid resistant nephrotic syndrome (SRNS) hospitalized in the Pediatric Nephrology Department in Iaşi, Romania. We defined SRNS in all cases without response to corticosteroids after the first month of therapy. For all the cases selected, the period of follow-up was the minimal 6 months. NS was defined as the presence of edema, proteinuria higher than 40 mg/ m2/h, serum albumin less than 2.5 gr/dl, and hypercholesterolemia.2,3 SRNS was diagnosed in patients with idiopathic NS based on lack of complete remission despite treatment with steroids.4 Complete remission was defined as 24 h protein excretion <4 mg/m2/h. Partial remission was defined when proteinuria continued between 4 mg/ m2/hour and 40 mg/m2/hour or between 30 and 300 mg/dl by multistic. Non-response was defined as a reduction in the basal proteinuria value <50%. Relapse was defined as three consecutive days of ≥3+ proteinuria, returning to the nephrotic range after achieving remission.5 The present study aims to analyze the cases of steroid resistant nephrotic syndrome (SRNS) in terms of anatomopathological aspects and their correlation with evolution. Between 2010 and 2019, 187 children with the initial diagnosis of primitive NS were hospitalized in the Pediatric Nephrology Department in Iaşi. One hundred and twenty-one cases were MCNS and 68 steroid-resistant (SRNS) cases. We excluded children under 6 months of age (for exclusion of congenital nephrotic syndrome), the cases with a follow-up of less than 6 months, cases of non-nephrotic proteinuria, nephrotic syndrome secondary to metabolic, infectious, vascular, malignant and cardiac diseases, as well as cases sent late, or cases already treated (to avoid biases of referral and selection). The following were analyzed: age at the onset of nephrotic syndrome, distribution of cases according to the anatomopathological aspect identified by percutaneous renal biopsy and assessment of prognostic factors by calculating the activity and chronicity index, the response to treatment evaluated by obtaining total clinical remission or absence of remission, correlations between the anatomopathological aspects identified in the studied patients and the response to treatment. In all the cases biopsied, we performed light microscopy and immunofluorescence, with minimum 15 glomeruli by each bioptic sample.
Results
Of the total cases of nephrotic syndrome hospitalized in our clinic, 68 developed corticosteroid resistance, representing 36%. Of the 68 children with SRNS, 65 children (95.5%) were primary SRNS, after 4 weeks of standard therapy with prednisone and 3 children with secondary SRNS, after first relapse of NS. The distribution by sex is approximately equal (B: F = 36:32). The mean age at the onset of patients with SRNS was 9.18 years, with limits between 1 year and 7 months and 16 years. Renal biopsy was performed in 55 children with SRNS (80.88%). The distribution of cases according to age and AP lesions is shown in Table 1. Renal biopsy allowed the evaluation of the activity index (AI) and chronicity index (CI) in the analyzed cases. AI is calculated by summing the scores given for mesangial proliferation (0, 1, 2 or 3), the number of glomeruli with segmental necrosis (0 for 0% affected glomeruli, 1 for 1–20% affected glomeruli, 2 for 21–50% affected glomeruli and 3 for more than 50% affected glomeruli) and for interstitial edema and infiltrate with mononuclear (0, 1, 2 or 3). AI is maximum 9. Among the patients studied, AI varied between 1 and 8. There are significant correlations between AI and the anatomopathological aspects (p = 0.05). Patients with MezPGN are more likely than other histological types to have lower levels of activity (Figure 1).
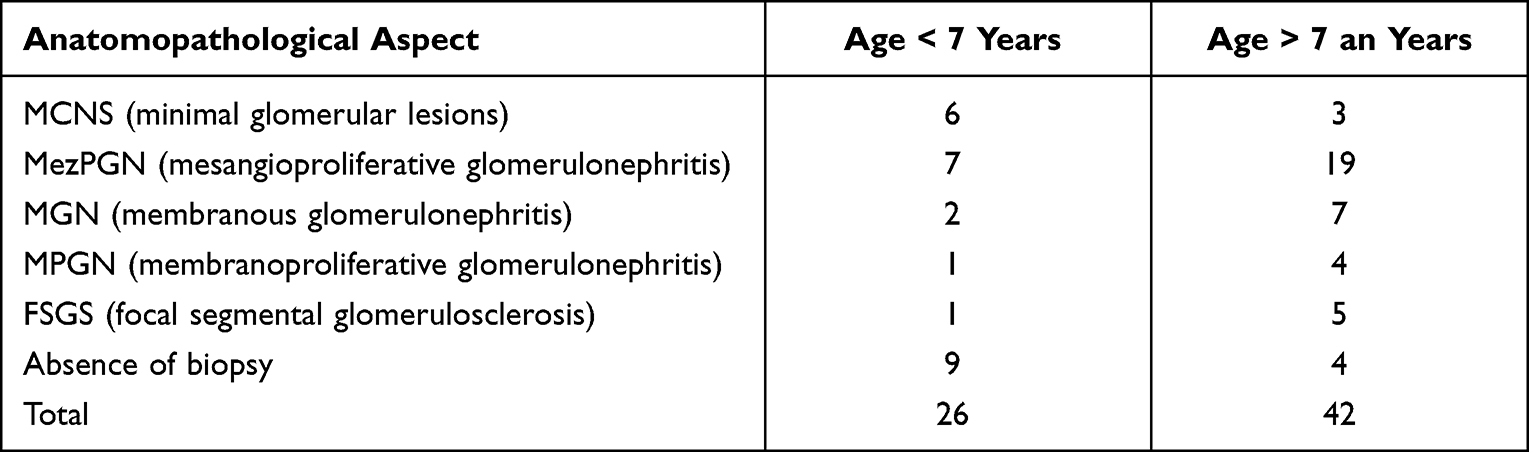 |
Table 1 Distribution of Cases According to Age and Anatomopathological Lesions |
 |
Figure 1 Renal biopsy – evaluation of the activity index. |
CI is calculated by summing the scores given for glomeruli with fibrous crescents or synechiae, hyalinosis, segmental sclerosis or global sclerosis (0, 1, 2 or 3) and for tubular atrophy and interstitial fibrosis or the presence of inflammatory cells (0, 1, 2 or 3). The CI is maximum 6. In the patients in the analyzed group the IC varied between 0 and 5. There are significant correlations between CI and the anatomopathological aspects (p = 0.048). Of the 65 children with primary SRNS, 20 children were followed for less than 1 year. Of these 20 cases, 4 died shortly after onset, 2 children from septic complications, 1 child from thromboembolic complications and 1 child from hypertensive encephalopathy. The analysis of the evolution of SRNS cases was performed only in 48 children (70.58%), at which the follow-up exceeded 12 months. Patients were treated with various treatment regimens, using in the second line of treatment Methylprednisolone in pulses, 1g/sqm, Cyclophosphamide 0.5g/sqm, Cyclosporine A 3–5mg/kg, MFM 800–1200mg/sqm, Enalapril and plasmapheresis. Total duration of treatment was minimum 6 months, but in general, we gave in medium 18 months of therapy at each patient. We did not have access to use Rituximab in our patients. Total remission was obtained in 30 children from the studied group (44.11%), clinical remission in 19 children and absence of remission in 19 children (27.94%). So, 72.05% of cases went into remission. The correlation of the anatomopathological aspects with the evolution is not statistically significant (p = 0.76), observing different therapeutic responses to all the analyzed histological types (Figure 2).
 |
Figure 2 Renal biopsy – evaluation of the evolution of SRNS cases. |
Discussion
The analysis of our group shows an increased incidence of corticosteroid resistance (36%), compared to KDIGO studies in which the percentage of corticosteroid resistance is 10–20%.6 The criteria for performing a kidney biopsy in a child with idiopathic nephrotic syndrome have been restricted over time, in order to avoid unnecessary biopsies in the nephrotic syndrome with minimal changes. However, there is still a diversity of views on biopsy criteria.7 The various histopathological aspects represent different stages of evolution of the nephrotic syndrome.8
MCNS and FSGS do not differ in clinical presentation, but the prognosis is different; FSGS is completed by the onset of chronic renal failure, while MCNS rarely reaches this stage. Juxtaglomerularly located glomeruli are preferentially affected by sclerosis in primary FSGS; therefore, it must be analyzed whether the biopsy was performed in the cortical depth, especially in cases apparently without lesions in light microscopy. It should also be noted that any area of tubular atrophy is significant in renal biopsy, raising the suspicion of segmental and focal sclerotic lesions, therefore serial sections should be examined.9
The variety of anatomopathological and evolutionary aspects, in relation to a variety of therapeutic means, raises the question: are there different pathogenic factors that act on the same target or is there a single trigger that causes different responses? The structure of the glomerular filtration barrier includes an inner layer of endothelial cells along the capillary loops, the glomerular basement membrane (GBM) and the outer podocytes. The exact structure of the filter slit is still incompletely known.10
In NS, it has been found in electron microscopy the retraction of podocyte processes inside the cell body, with the appearance of a flat epithelial layer. Numerous studies have been conducted in recent decades to identify genetic aspects of SRNS. NS usually occurs sporadically, but familial cases with autosomal dominant (AD) or recessive (RA) transmission have been reported. AD-transmitting genes were identified on chromosomes 19q13, 11q21-22 and 11q24, causing mutations in ACTN4 (the gene encoding α-actinin-4) and in TRPC6 (ion channel for calcium, present in the diaphragmatic slit). For AR transmission of NS 4 loci were identified: 19q13, 1q25-31, 14q24.2, 2p12-p13.2.11
Diseases caused by the NPHS1 and NPHS2 genes, which encode nephrine and podocin, have been described in families with changes on 19q13 and 1q25-31. The therapeutic response to cortisone in nephrotic syndrome appears to be genetically encoded. Three variants of the MDR1 gene (1236T, 2677T and 3435T) have been described in patients with a slow response to cortisone therapy.12 In our group, we identified 3 cases of nephrotic syndrome with genetic determinism. The first two were of the Finish type, and a third was associated with Schimke syndrome. For example, we present the case of patient G.B., diagnosed in the clinic with SRNS Finish type. Molecular testing identified a deletion in intron 21 of the NPHS1 gene, in homozygous form, confirming the diagnosis of Finnish-type SN. The genetic analysis of the parents specified that both are heterozygous for the mutation in the NPHS1 gene. The first biopsy, performed at 4 months, identified 15 optically unmodified glomeruli (Figure 3), and immunofluorescence for IgA, IgG, IgM, and fibrinogen was negative.
 |
Figure 3 MCNS patient G.B. (St. Mary’s Emergency Children Hospital, Iasi). |
Unilateral nephrectomy was necessary, being performed at 6 months, and which showed glomerular cystic degeneration, periglomerular inflammation and glomerular sclerosis (Figure 4). The patient died at the age of 2, from sepsis with pulmonary starting point.
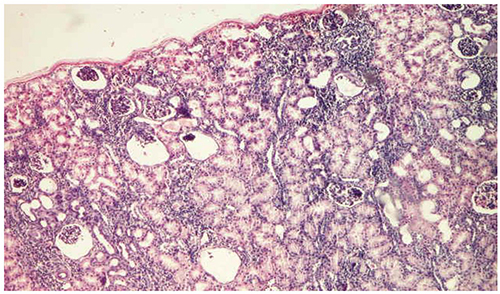 |
Figure 4 Glomerular cystic degeneration in patient G.B. (St. Mary’s Emergency Children Hospital, Iasi). |
In our statistics, it is observed that 25 of the cases of SRNS (36.8%) started under 7 years old, which contradicts the idea that young age is a favorable prognostic factor. It is discussed whether the report of FSGS frequency increase reflects the real increase in the prevalence of this histological type, or is only the consequence of changing biopsy indications or increasing access to renal biopsy.9 Most published studies draw attention to the predominance of MCNS at an early age (under 7 years), while FSGS has been reported at older ages. In the analyzed group, the relationship between the age of onset and the histological subtypes is consistent with the data reported in the literature, in terms of the age of onset of cases with minimal lesions and cases of FSGS. Renal biopsy allowed the evaluation of the activity index (AI) and chronicity index (CI) in the analyzed cases, in accordance with the studies in the literature.13
Immunofluorescence may alter the histological classification of nephrotic syndrome. In MCNS, immunofluorescence is usually negative. However, some biopsies have shown deposits of IgM and C1q in the mesangium, requiring a description of the entity of IgM nephropathy, the prognosis of which is controversial. IgM nephropathy (IgMN) is an important but rather neglected pathology responsible for renal morbidity in children and adults. According to some authors, this anatomopathological form does not respond to cortisone treatment and therefore has a reserved prognosis,14 while other authors claim that there is no clinical significance.15 About one-third of patients are steroid-responsive, and the other two-thirds are resistant or addicted to steroids.16 In our study, from the cases with minimal lesions and corticosteroid-resistant allure, 3 cases with positive immunofluorescence for IgM were identified (Figure 5), representing 13.23% of the total, in accordance with data from the literature, which shows an incidence of 2–18.5%.14
 |
Figure 5 Patient M.N., with MCNS, immunofluorescence with positive IgM (St. Mary’s Emergency Children Hospital, Iasi). |
The total remission was obtained in 30 children from the studied group (44.11%), 27% being in partial remission, without being able to identify a correlation with the anatomopathological aspect. The data are consistent with the literature that notes remission of up to 70%, partial or total in the case of SRNS, in children (17).
Conclusion
The onset of nephrotic syndrome under 7 years is not a guarantee of favorable evolution, with patients in this age group representing 36.8% of cases of SRNS. MezPGN represents the dominant anatomopathological aspect in children with SRNS, but remission was obtained in 44% of cases of SRNS, without being able to establish a statistically significant correlation with the anatomopathological subtypes. It is well known that mesangial proliferation is at an early stage or may accompany FSGS, which can explain the evolution of steroids resistance in children under the age of 7 years who associate this with histological aspect. Predicting the response to long-term treatment in SRNS is difficult using only renal biopsy; it is necessary to introduce genetic molecular analyses to establish a judicious therapeutic attitude.
Ethics Approval
The study was approved by the ethics committee of the Ethics Commission of the St. Mary’s Emergency Children Hospital, Iasi. Due to the retrospective nature of the study and the anonymity of all patient information, the ethics committee concluded that written informed consent was not necessary and gave a favorable opinion for study result publication. We confirm that the study itself complied with the Declaration of Helsinki.
Author Contributions
All authors contributed equally to this paper. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.\.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rovin BH, Adler SG, Barratt J, et al. KDIGO 2021 Clinical practice guideline for the management of glomerular diseases, Chapter 4: nephrotic syndrome in children. Kidney Int. 2021;100:57–60.
2. Gipson DS, Massengill SF, Yao L, et al. Management of childhood onset nephrotic syndrome. Pediatrics. 2009;124(2):747–757. doi:10.1542/peds.2008-1559
3. Pokrajac D, Kamber AH, Karasalihovic Z. Children with steroid-resistant nephrotic syndrome: a single –center experience. Mater Sociomed. 2018;30(2):84–88. doi:10.5455/msm.2018.30.84-88
4. NKF-KDIGO guideline development staff. KDIGO clinical practice guideline for glomerulonephritis. Kidney Int Suppl. 2012;2(2):139–274.
5. Levine SA. Nephrotic Syndrome. In: Brenner BM, Floyd C, editors. Brenner & Rector’s the Kidney. Philadelphia: WB Saunders; 2003:297–313.
6. NKF-KDIGO guideline development staff. KDIGO clinical practice guideline for glomerulonephritis. Kidney Int Suppl. 2012;2(2):163–171.
7. Gulati S, Sharma AP, Sharma RK, Gupta A, Gupta RK. Do current recommendations for kidney biopsy in nephrotic syndrome need modifications? Pediatr Nephrol. 2002;17(6):404–408. doi:10.1007/s00467-002-0840-3
8. Pal A, Kaskel F. History of nephrotic syndrome and evolution of its treatment. Front Pediatr. 2016;4(56). doi:10.3389/fped.2016.00056
9. Agarwal SK, Sethi S, Dinda AK. Basics of kidney biopsy: a nephrologist’s perspective. Indian J Nephrol. 2013;23(4):243–252. doi:10.4103/0971-4065.114462
10. Jalanko H. Congenital nephrotic syndrome. Pediatr Nephrol. 2009;24(11):2121–2128. doi:10.1007/s00467-007-0633-9
11. Caridi G, Trivelli A, Sanna-Cherchi S, Perfumo F, Ghiggeri GM. Familial forms of nephrotic syndrome. Pediatr Nephrol. 2010;25(2):241–252. doi:10.1007/s00467-008-1051-3
12. Schijvens AM, Ter Heine R, de Wildt SN, Schreuder MF. Pharmacology and pharmacogenetics of prednisone and prednisolone in patients with nephrotic syndrome. Pediatr Nephrol. 2019;34(3):389–403. doi:10.1007/s00467-018-3929-z
13. Vanikar A. IgM nephropathy; can we still ignore it. J Nephropathol. 2013;2(2):98–103. doi:10.5812/nephropathol.9872
14. Mubarak M. IgM nephropathy; time to act. J Nephropathol. 2014;3(1):22–25. doi:10.12860/jnp.2014.05
15. Connor TM, Aiello V, Griffith M, et al. The natural history of immunoglobulin M nephropathy in adults. Nephrol Dial Transplant. 2017;32(5):823–829. doi:10.1093/ndt/gfw063
16. Tullus K, Webb H, Bagga A. Management of steroid-resistant nephrotic syndrome in children and adolescents. Lancet Child Adolesc Health. 2018;2(12):880–890. doi:10.1016/S2352-4642(18)30283-9
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.